

Abstract
The success of Streptococcus pneumoniae (the pneumococcus) as a pulmonary pathogen is related to its restriction of innate immune responses by respiratory epithelial cells. The mechanisms used to overcome this restriction are incompletely elucidated. Pulmonary chemokine expression involves complex cellular and molecular networks, involving the pulmonary epithelium, but the specific cellular interactions and the cytokines that control them are incompletely defined. We show that serotype 2 or 4 pneumococci induce only modest levels of CXCL8 expression from epithelial cell lines, even in the absence of a polysaccharide capsule. In contrast, coculture of A549 cells with the macrophage-like THP-1 cell line, differentiated with vitamin D, or monocyte-derived macrophages enhanced CXCL8 release. Supernatants from the THP-1 cell line prime A549 cells to release CXCL8 at levels similar to cocultures. Interleukin-1Ra (IL-1Ra) inhibits CXCL8 release from cocultures and reduces the activity of macrophage-conditioned media, but inhibition of tumor necrosis factor alpha (TNF-α) had only a minimal effect on CXCL8 release. Release of IL-1β but not TNF-α was upregulated in cocultures. IL-1 type 1 receptor knockout C57BL/6 and BALB/c mice confirmed the importance of IL-1 signaling in CXC chemokine expression and neutrophil recruitment in vivo. In fulminant disease, increased IL-1 signaling resulted in increased neutrophils in the airway and more invasive disease. These results demonstrate that IL-1 is an important component of the cellular network involving macrophages and epithelial cells, which facilitates CXC chemokine expression and aids neutrophil recruitment during pneumococcal pneumonia. They also highlight a potential clinical role for anti-IL-1 treatment to limit excessive neutrophilic inflammation in the lung.
INTRODUCTION
Streptococcus pneumoniae (the pneumococcus) is a major cause of respiratory tract infection and invasive bacterial disease (6). S. pneumoniae is also a common commensal of the upper respiratory tract, but innate host defenses prevent disease in most colonized individuals (6, 40). The success of the innate response against pneumococcus is emphasized by the relatively low incidence of community-acquired pneumonia or invasive disease in comparison to the frequency of upper respiratory tract colonization (16, 39).
The respiratory tract epithelium plays a critical role in the recognition of bacterial pathogens and in the induction of the inflammatory response (20). Epithelial cells express a range of Toll-like receptors (TLR) and other pattern recognition receptors (13). The clinical importance of these pathways of innate recognition of S. pneumoniae has been highlighted by the identification of genetic mutations or polymorphisms in these signaling pathways, which confer altered susceptibility to invasive pneumococcal disease (29, 31). Engagement of a range of pattern recognition receptors, including TLRs, nucleotide-binding oligomerization domain (NOD) proteins, the NOD-like receptor (NLR) family, and pyrin domain-containing 3 (NALP3), by pneumococci activates NF-κB and induces CXCL8 (interleukin-8 [IL-8]) which leads to recruitment of polymorphonuclear leukocytes (neutrophils) (8, 25, 41, 49, 65). This is a key feature of pneumococcal infection when resident pulmonary defenses are overwhelmed (15).
Epithelial cell responses are enhanced by cooperative signaling with other cell types. Bronchoalveolar lavage fluid from lungs infected with pneumococci stimulates epithelial cell NF-κB activation (44), and alveolar macrophages enhance CXCL8 production by epithelial cells in lung explants (66). It is postulated that, in order to avoid excessive inflammatory responses to commensal organisms, airway epithelial cells express constitutively low levels of TLRs, but studies of TLR2 expression indicate that it may be upregulated when inflammation is present (35, 64). Previous studies have demonstrated that monocytes enhance epithelial cytokine responses to various TLR agonists (9, 37). Increases in proinflammatory cytokine and chemokine release in cocultures of pulmonary epithelial cells and monocytes have been noted in response to a range of lipopolysaccharides (9, 42, 52), staphylococcal exotoxins (30), Mycobacterium tuberculosis (63), and respiratory syncytial virus (RSV) (58). Thus, cell-cell communication between monocytes and epithelial cells is an important early step in the immune response to respiratory tract infections.
S. pneumoniae possesses a number of virulence factors which may confound front-line immune responses (28), including a polysaccharide capsule which limits bacterial phagocytosis by macrophages (2) and inhibits bacterial attachment to respiratory epithelial cells (1, 22, 51). In this study, we have characterized the cellular and molecular factors involved in the regulation of CXCL8 expression by respiratory tract epithelial cells in response to S. pneumoniae infection. We demonstrate that IL-1β secretion by a macrophage-like cell line is required for maximal secretion of CXCL8 by epithelial cells in response to S. pneumoniae in vitro. We also establish important roles for IL-1β in regulating the expression of CXC chemokines and rate IL-1 signaling to the pulmonary infiltration of polymorphonuclear cells in vivo in murine models of S. pneumoniae infection.
MATERIALS AND METHODS
Materials.
Gentamicin was purchased from Roussel Laboratories (Uxbridge, United Kingdom), vitamin D3 (1,25-dihydroxy-cholecalciferol) from Sigma-Aldrich (Poole, United Kingdom), and recombinant human IL-1β, recombinant human soluble tumor necrosis factor (TNF) receptor type 1, and recombinant human IL-1 receptor antagonist (IL-1Ra) from PeproTech EC Ltd. (London, United Kingdom).
Bacterial strains and growth conditions.
Streptococcus pneumoniae strains used in this study were serotype 2 strain D39 and its isogenic unencapsulated derivative FP22 (43) and serotype 4 strain TIGR4 and its unencapsulated derivative FP23 (2). D39 and TIGR4 were obtained from the American Type Culture Collection (ATCC) bacteriology collection, FP22 from Tim Mitchell (University of Glasgow), and FP23 from Gianni Pozzi and Franco Ianelli (University of Sienna). Nontypeable conjunctival clinical strains 08-1773 (MLST type 448) and 02-3522 (pre-MLST) were from Tim Mitchell and the Scottish Haemophilus, Legionella, Meningococcus, and Pneumococcus Reference Laboratory (SHLMPRL). Bacteria were grown to exponential phase in brain heart infusion broth (Oxoid Unipath, Basingstoke, United Kingdom) with 20% heat-inactivated fetal calf serum (FCS) (Autogen Bioclear, Wiltshire, United Kingdom), and aliquots were stored at −80°C. The concentration (CFU/ml) was determined by Miles Misra count for each strain as previously described (14). Freshly thawed aliquots were spun at 9,000 × g for 2 min, and the bacterial pellet was washed twice in phosphate-buffered saline (PBS) prior to use.
Human cell culture.
All cell lines were obtained from the European Collection of Animal Cell Cultures (ECACC), and all culture media were from BioWhittaker (Lonza, Belgium). A549 cells, a human lung carcinoma cell line, were maintained in Dulbecco's modified Eagle's medium (DMEM) with 4.5 g/liter glucose, 2 mmol/liter l-glutamine, and 10% FCS. Detroit 562 cells, a human pharyngeal carcinoma cell line, were maintained in Eagle's minimal essential medium (EMEM) with 2 mmol/liter l-glutamine and 10% FCS. Epithelial cells were seeded at 2 × 105 cells/ml in 24-well plates (Costar) and grown to confluence for 24 h prior to use.
THP-1 cells, a monocytic leukemic cell line, were maintained in RPMI 1640 containing 2 mmol/liter l-glutamine and 10% FCS. Differentiation to a macrophage phenotype was achieved by resuspending cells at 2 × 104 cells/ml in fresh medium with the addition of vitamin D3 (VD3) at 10−7 M and incubation for 72 h in 5% CO2 at 37°C (26, 50). This differentiation protocol produces a nonadherent immature macrophage phenotype more similar to an inflammatory macrophage than a highly differentiated tissue macrophage in terms of its response to microbial factors (12). Peripheral mononuclear cells were isolated from human blood as described previously (14), from healthy donors after informed consent and approval by the South Sheffield Research Ethics Committee. The day after isolation, nonadherent cells were removed by washing in RPMI, and adherent monocyte-derived macrophages (MDM) were collected by gentle scraping. Hemocytometer counts of all cell cultures were performed on the day of experimentation to determine approximate cell numbers per well. In cocultures, nonadherent VD3-differentiated THP-1 or 1-day-old MDM were added to adherent A549 epithelial cells at a ratio of 1:10 (using data for cell numbers derived from the hemocytometer counts of representative wells of A549 cells), and cells were cultured in RPMI with 10% FCS (with low endotoxin levels).
Infection of cell cultures.
Growth medium was removed from epithelial cell monolayers, and cells were washed twice with PBS. Fresh growth medium (1 ml per well) was applied, and cells were infected with bacterial strains at a multiplicity of infection (MOI) of 10 for epithelial cells and 100 for VD3-differentiated THP-1 cells or MDM (to ensure the same number of bacteria were added to each well). Pneumococci were opsonized in RPMI 1640 supplemented with 10% antipneumococcal immune serum and incubated on a rotating device for 30 min at 37°C prior to infections for all experiments involving THP-1 cells and in experiments investigating the effect of opsonization on epithelial cell responses (2). Infected cell cultures were centrifuged at 150 × g for 5 min prior to incubation at 37°C in 5% CO2. Culture medium (1 ml) was removed from wells at each time point, spun at 10,000 × g for 10 min, and stored at −80°C prior to cytokine assay. Epithelial cells showed significant loss of viability following overnight incubation with S. pneumoniae, and therefore gentamicin (100 μg/ml) was added to all wells at the 2-h time point to prevent bacterial overgrowth and loss of epithelial cell viability. As a positive control for CXCL8 production, some wells were treated with 10 ng/ml IL-1β for the indicated time periods.
Bacterial adherence assay.
Infected epithelial cell monolayers (MOI of 10) were centrifuged at 150 × g for 5 min prior to incubation for 2 h at 37°C in 5% CO2. In certain experiments, specific variables were altered to determine their effect on adherence. These included increasing the MOI to 50, centrifuging at 1,200 × g, using opsonized pneumococci, and incubating for different durations. At 2 h (or at 1 h or 4 h in experiments with altered incubation periods), the medium (containing nonadherent bacteria) was removed from each well, and nonadherent bacterial counts were determined by Miles Misra (36). Epithelial cells (including any adherent bacteria) were then removed from each well with trypsin and versine, and adherent bacterial counts were determined by Miles Misra. The proportion of adherent bacteria could then be calculated (62).
Investigation of the nature of proinflammatory mediators generated in cocultures.
To generate macrophage-conditioned media, VD3-differentiated THP-1 cells (2 × 104/well) were exposed to opsonized D39 S. pneumoniae (MOI of 100:1) (macrophage-conditioned medium D [MCM-D] or mock-infected MCM [MCM-MI]) for 24 h (100 μg/ml gentamicin was added at 2 h to all wells). MCM-D and MCM-MI were removed from all wells at 24 h and spun at 10,000 × g for 10 min, and the supernatants (1 ml) were then applied to epithelial cell monolayers. In blocking experiments, IL-1Ra (200 ng/ml) and/or soluble TNF receptor type 1 (sTNFR1; 50 ng/ml) were added to conditioned media after transfer onto epithelial cell monolayers at time zero. Plates were then incubated in 5% CO2 at 37°C, and media were removed from wells at 6 h, spun at 10,000 × g for 10 min, and then stored at −80°C prior to cytokine assay. Samples of conditioned media were saved at time zero (following centrifugation) in order to determine baseline levels of CXCL8 prior to transfer onto epithelial monolayers. These baseline values were subtracted from the total CXCL8 concentration at 6 h to calculate the amount produced by stimulated epithelial cells alone.
Blockade of candidate proinflammatory molecules.
Coculture wells were prepared as described above with A549 epithelial cells and THP-1 at a ratio of 10:1, and blocking agents (IL-1Ra, 200 ng/ml) (58, 63) and/or sTNFR1 (50 ng/ml) (38) were added at time zero. Wells were then infected with opsonized pneumococci (MOI of 10), spun at 150 × g for 5 min, and then incubated at 37°C in 5% CO2. Medium was removed at 6 h, spun at 10,000 × g for 10 min, and stored at −80°C. Gentamicin (100 μg/ml) was added to all wells at 2 h.
Role of soluble mediators.
To physically separate THP-1 and A549 cells, THP-1 was added to cell culture inserts, pore size of 1 μm (BD Falcon), with A549 cells in the well below the insert, at ratio of 10:1 of A549:THP-1 cells. Opsonized D39 pneumococci were added at equal doses to both the insert and lower well compartments to achieve an overall MOI of 10:1 pneumococci:A549 cells. Plates were then spun at 150 × g for 5 min and incubated at 37°C in 5% CO2, and then media were removed at 6 h, spun at 10,000 × g for 10 min, and stored at −80°C. Gentamicin (100 μg/ml) was added to all wells at 2 h.
Murine pneumonia models.
IL-1 type 1 receptor knockout mice (18), allele Il1r1Tm1Imx, were kindly provided by Immunex Corporation to M.J.H.N. and were backcrossed for at least 10 generations against both BALB/cOlaHsd and C57BL/6NHsd separately. Heterozygotes were bred to yield homozygotes, and working colonies of homozygotes were maintained. BALB/cOlaHsd or C57BL/6NHsd (Harlan) were used as wild-type controls. Adult mice of both sexes were used. All animal experiments were conducted in accordance with the Home Office Animals (Scientific Procedures) Act of 1986 and local ethical approval.
Pulmonary infection of mice was with 5 × 105 or 1 × 107 CFU serotype 4 S. pneumoniae prepared and delivered by direct tracheal instillation as described previously (15). At 24 h postinfection, mice were killed by overdose of sodium pentobarbitone, and bronchial alveolar fluid, lung, and blood were collected for determination of cell differential, cytokines, and viable bacteria in lung and blood as described previously (15).
Cytokine assays.
CXCL8, IL-1β, and TNF-α levels in the culture supernatants were measured using human CXCL8, IL-1β, and TNF-α DuoSet enzyme-linked immunosorbent assay (ELISA) kits, respectively (R&D Systems, Abingdon, United Kingdom) according to the manufacturer's guidelines. Murine CXCL1, CXCL2, and TNF-α were measured in bronchoalveolar lavage (BAL) fluid using murine CXCL1 and CXCL2 DuoSet ELISA kits (R&D Systems, Abingdon, United Kingdom) and murine TNF-α ELISA Ready-SET-Go! reagent set (eBioscience, Hatfield, United Kingdom), respectively, according to the manufacturers' guidelines.
Optical densities were determined using an Opsys MR microplate reader (Dynex Technologies). The lower limit of detection was approximately 30 pg/ml for CXCL8, 20 pg/ml for human IL-1β, 30 pg/ml for human TNF-α, 16 pg/ml for murine CXCL1, 32 pg/ml for murine CXCL, and 16 pg/ml for murine TNF-α.
Statistics.
All results are recorded as means ± standard errors of the means (SEM) unless otherwise stated. Statistical testing was performed using Prism 5.02 software (GraphPad Software Inc.). Unpaired t test, Mann-Whitney test, or one-way analysis of variance (ANOVA) (with Bonferroni's posttest) were used to analyze differences between groups, as appropriate. Significance was defined as P values of <0.05.
RESULTS
Epithelial cell responses to S. pneumoniae are limited by the polysaccharide capsule.
Epithelial cells contribute to innate host defense, but S. pneumoniae contains several adaptations, which can modulate its interaction with epithelial cells (13, 19, 32, 59, 62), in particular the expression of the polysaccharide capsule (1, 21, 22, 51, 57). The mechanisms used to integrate the epithelial cell response into the innate host response, despite microbiological restriction, are uncertain. We confirmed that there was minimal adherence of encapsulated serotype 2 or 4 S. pneumoniae to epithelial cell lines (see the supplemental material at http://www.shef.ac.uk/medicine/infectionandimmunity/staffprofiles/dockrell.html, Fig. S1A to S1D). Binding was not significantly enhanced by opsonization of bacteria, by increasing the MOI to 50:1, by increasing the incubation period, or by centrifuging the bacteria onto the cells at 1,200 × g rather than 150 × g (data not shown). Mutants lacking capsule, either on a serotype 2 or 4 background (2, 43), demonstrated significantly greater binding than the parental capsulated strains (see the supplemental material at http://www.shef.ac.uk/medicine/infectionandimmunity/staffprofiles/dockrell.html, Fig. S1A to S1D).
CXCL8 is critical for the effective recruitment and activation of polymorphonuclear cells at sites of inflammation (3, 54). Microbial factors stimulate epithelial cells via a range of receptors to produce CXCL8 (5, 35, 49). We investigated the ability of nonopsonized pneumococci to stimulate CXCL8 release from epithelial cell monolayers. There was no significant difference between the CXCL8 response of mock-infected epithelial cells and those exposed to encapsulated pneumococci, which was low in comparison with cells stimulated with IL-1β (11) (Fig. 1A and B). We demonstrated significantly greater CXCL8 production in response to unencapsulated pneumococci; however, when we compared the encapsulated serotype 2 and 4 strains to unencapsulated clinical strains, we did not note any significant difference in CXCL8 release (see the supplemental material at http://www.shef.ac.uk/medicine/infectionandimmunity/staffprofiles/dockrell.html, Fig. S2), although it was not clear whether these results reflected the marked genetic differences between these unencapsulated strains and other pneumooccal serotypes. Opsonization modestly enhanced the epithelial cell CXCL8 response to encapsulated pneumococci (see the supplemental material at http://www.shef.ac.uk/medicine/infectionandimmunity/staffprofiles/dockrell.html, Fig. S3). We therefore opsonized pneumococci in all subsequent experiments. We also observed significantly greater CXCL8 responses to the unencapsulated serotype 4 strain, FP23, than to other strains, possibly in keeping with enhanced proinflammatory cytokine responses to piliated pneumococci (4).
Fig 1.

Presence of macrophages enhances epithelial cell CXCL8 production. (A and B) A549 epithelial cells were either mock infected (white bars) or challenged with encapsulated (pale-gray bars) or unencapsulated (dark-gray bars) nonopsonized serotype 2 (A) and serotype 4 (B) S. pneumoniae (MOI of 10) or IL-1β (black bars). CXCL8 levels in the cell culture supernatants were measured by ELISA at the indicated time points (n = 4 to 6 independent experiments). (C and D) A549 epithelial cells alone (EC), THP-1 cells alone (MC), and A549-THP-1 cocultures (Co) were challenged with opsonized S. pneumoniae for up to 24 h. CXCL8 levels in the cell culture supernatants were measured by ELISA at 2-, 6-, and 24-h time points. (C) Encapsulated (D39) and unencapsulated (FP22) serotype 2 pneumococci (n = 7); (D) encapsulated (TIGR4) and unencapsulated (FP23) serotype 4 pneumococci (n = 5). Mock-infected (MI) and IL-1β data at the 24-h time point is shown for comparison. One-way ANOVA (all P values are <0.05) with Bonferroni's posttest: *, P < 0.05; **, P < 0.01; #, P < 0.05 for CXCL8 level in coculture versus the sum of CXCL8 levels from A549 and THP-1 cells alone).
Coculture of epithelial cells with a macrophage-like cell line enhances CXCL8 secretion following exposure to S. pneumoniae.
Cytokine and chemokine responses can be enhanced in epithelial cell cultures by monocyte- or macrophage-derived factors (9, 30, 37, 42, 52, 53, 58, 63, 66). CXCL8 expression by the THP-1 macrophage cell line or epithelial cells in response to pneumococci was consistently less than the epithelial cell response to IL-1β (Fig. 1C and D). In contrast, we demonstrated a marked enhancement of CXCL8 responses for macrophage-epithelial cell cocultures. The generation of CXCL8 by cocultures treated with pneumococci at the 6- and 24-h time points was significantly greater than the sum of the CXCL8 responses of A549 epithelial cells and THP-1 cells alone. The responses were apparent by 6 h after infection and were sustained up to 24 h after infection. We therefore used the 6-h time point in subsequent experiments. Similar results were obtained with the Detroit epithelial cell line, which also showed enhanced CXCL8 expression in coculture conditions (data not shown). Interestingly, although the macrophage-like cell line showed a significantly greater CXCL8 response to encapsulated bacteria than to unencapsulated bacteria, the presence of the polysaccharide capsule had little effect on the magnitude of the coculture response to pneumococci. We also noted that opsonization corrected the inhibitory effect of the capsule on the response of epithelial cell monocultures to pneumococci.
IL-1 production by the THP-1 macrophage-like cell line stimulates CXCL8 expression from epithelial cells following exposure to S. pneumoniae.
We next addressed whether cell contact between THP-1 cells and epithelial cells was a necessary or contributory factor. When epithelial cells were separated from monocytes using semipermeable transwells, we observed no significant reduction in CXCL8 secretion (Fig. 2), suggesting that direct cell contact was not required and that a soluble factor was responsible for priming the epithelial cells. Bronchial alveolar fluid from the lungs of mice infected with S. pneumoniae has been shown to stimulate NF-κB activation and resultant chemokine expression by epithelial cells, although the cellular source of the factors responsible are incompletely characterized (44). Previous work from our group has indicated in a number of models that IL-1 may play important roles in the induction of inflammatory responses (9, 38). When we transferred supernatants from the mock-infected THP-1 cells onto epithelial monolayers, we observed little release of CXCL8, while supernatants from the S. pneumoniae-exposed THP-1 cells induced levels of CXCL8 secretion comparable to infected cocultures (Fig. 3). We also investigated the mediators responsible for activation of A549 cells by bacterial-exposed THP-1 cells and focused on the two dominant early proinflammatory cytokines, TNF-α and IL-1 (24, 27, 45, 52). IL-1Ra, but not soluble TNF-α receptor 1, treatment blocked the ability of macrophage-conditioned media to stimulate CXCL8 production by the epithelial cell line.
Fig 2.
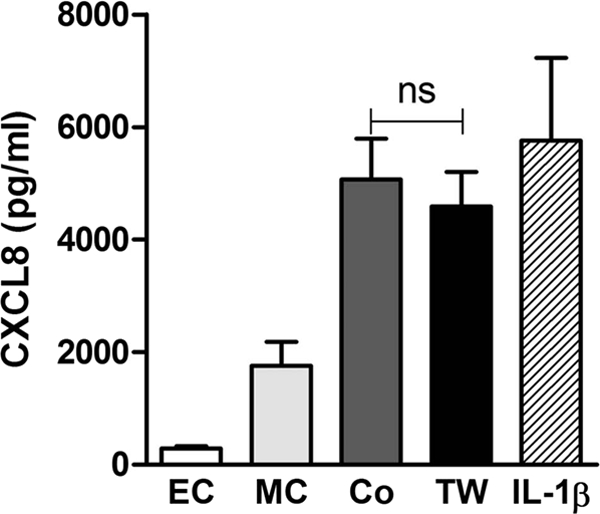
Physical separation of macrophages from epithelial cells does not impair priming of CXCL8 production. A549 epithelial cells alone (EC), THP-1 cells alone (MC), A549-THP-1 cocultures (Co), and A549 and THP-1 cells separated by transwells (TW) were challenged with D39 pneumococci (MOI of 10). CXCL8 levels in the cell culture supernatants were measured by ELISA at 6 h. IL-1β (positive control) data are shown for comparison (IL-1β). One-way ANOVA with Bonferroni's posttest (n = 6). ns, not significant.
Fig 3.

Conditioned media from THP-1 cells challenged with pneumococci primes epithelial cell CXCL8 production. THP-1 cells were exposed to either mock infection or D39 S. pneumoniae (MOI of 100) for 24 h. Mock-infected macrophage-conditioned media (MCM-MI) and D39-challenged macrophage-conditioned media (MCM-D) were collected at 24 h. Monolayers of A549 epithelial cells were then incubated with medium alone (EC), MCM-MI, THP-1 cells and D39 (MOI 10) (Co+D), MCM-D, MCM-D plus IL-1Ra (MCM-D/IL-1Ra), MCM-D plus sTNF type 1 receptor (MCM-D/TNFR1), MCM-D plus both inhibitors (MCM-D/Both), or IL-1β. CXCL8 levels in the cell culture supernatants were measured by ELISA at 6 h (n = 3); one-way ANOVA (P < 0.001) with Bonferroni's posttest: *, P < 0.05; **, P < 0.01.
IL-1Ra inhibits CXCL8 release from cocultures challenged with S. pneumoniae.
We next addressed whether the contribution of IL-1 and TNF-α altered with bacterial strain. IL-1Ra reduced CXCL8 secretion by cocultures in the presence of both encapsulated and unencapsulated strains and also the piliated type 4 strains (4). TNF-α blockade had no effect on CXCL8 secretion in the presence or absence of IL-1Ra at the 6-h time point (Fig. 4) and made only a minor contribution to CXCL8 release in cocultures at 24 h, which reached statistical significance only for the unencapsulated type 4 pneumococci (see the supplemental material at http://www.shef.ac.uk/medicine/infectionandimmunity/staffprofiles/dockrell.html, Fig. S4). Overall these findings suggested that IL-1 was the major factor enhancing CXCL8 release. We also confirmed that if we used primary monocyte-derived macrophages (MDM) 1 day after differentiation from monocytes, they were also able to enhance CXCL8 production from epithelial cells and that IL-1 also contributed to this release (Fig. 5).
Fig 4.

IL-1Ra blocks enhancement of CXCL8 production in cocultures. Monolayers of A549 epithelial cells alone (EC), THP-1 cells alone (MC), A549-THP-1 cocultures (Co), cocultures with IL-1Ra (Co/IL-1Ra), cocultures with sTNF type 1 receptor (Co/TNFR1), and cocultures with both inhibitors (Co/both) were challenged with encapsulated D39 (A) and unencapsulated FP22 serotype 2 (B) pneumococci and encapsulated TIGR4 (C) and unencapsulated FP23 serotype 4 (D) pneumococci. IL-1β-stimulated cocultures are also shown for comparison. CXCL8 levels in the cell culture supernatants were measured by ELISA at 6 h (n = 3 to 4); one-way ANOVA (all P values are <0.002) with Bonferroni's posttest: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Fig 5.

Enhanced epithelial cell CXCL8 production is also seen in cocultures containing primary monocyte-derived macrophages. A549 epithelial cells alone (EC), monocyte-derived macrophages alone (MDM), and A549-MDM cocultures (Co) were challenged with opsonized S. pneumoniae D39 (A) or TIGR4 (B) (MOI of 10), with or without IL-1 receptor antagonist (IL-1Ra) for 6 h, and CXCL8 levels in the cell culture supernatants were measured by ELISA and normalized for cell number (data show release/1 × 105 A549 cells). Mock-infected (MI) and IL-1β data are shown for comparison. One-way ANOVA (all P values are <0.05) with Bonferroni's posttest: *, P < 0.05; ***, P < 0.01. P values of <0.01 for CXCL8 levels in coculture versus the sum of CXCL8 levels from A549 cells and MDM alone (n = 3 to 5) from 3 independent experiments.
IL-1β release is enhanced in cocultures.
We next confirmed IL-1β and TNF-α release from the THP-1 cell line following challenge with D39 pneumococci, excluding the possibility that the lack of inhibition by TNF-α blockade was merely a consequence of lack of production (Fig. 6A and B). However, only IL-1β was enhanced in cocultures. The same pattern of cytokine release was observed in response to type 4 pneumococci (data not shown). We also showed that neither IL-1β nor TNF-α were induced following application of macrophage-conditioned media to epithelial cell monolayers, thus confirming the THP-1 cells as the major source of IL-1β and TNF-α (Fig. 6C and D).
Fig 6.

IL-1β but not TNF-α levels are enhanced in cocultures following challenge with pneumococci. A549 epithelial cells (EC) were exposed to media alone (−) or D39 pneumococci (+), as were THP-1 cells (MC) and A549-THP-1 cocultures (Co) and IL-1β (A) and TNF-α (B) levels measured by ELISA at 6 and 24 h (n = 3); one-way ANOVA (all P values are <0.02) with Bonferroni's post test: *, P < 0.05. A549 epithelial cells were exposed to conditioned media from D39-challenged THP-1 cells, and IL-1β (C) and TNF-α (D) levels were measured by ELISA at 0, 2, 6 and 24 h.
Modulation of IL-1 signaling alters production of CXC chemokines and polymorphonuclear cell recruitment during S. pneumoniae infection in vivo.
To confirm the relevance of these findings, we next investigated pneumonia models in mice with differing sensitivity to IL-1 signaling (23). C57BL/6 mice recruit polymorphonuclear cells only when resident host defenses against pneumococci become overwhelmed (17). BALB/c mice develop an early neutrophilic response against pneumococci, which is greater than that seen in C57BL/6 mice. Il1r1−/− C57BL/6 mice have normal leukocyte numbers but a reduction in early bacterial clearance in a model of pneumococcal pneumonia (18, 46), while Il1r1−/− BALB/c mice have never been studied in a model of pneumococcal pneumonia to our knowledge. We confirmed that polymorphonuclear cell recruitment was greater for BALB/c mice than for C57BL/6 mice following challenge with a similar dose of pneumococci (Fig. 7A and D). In each genetic background, Il1r1−/− mice recruited fewer polymorphonuclear cells.
Fig 7.

Modulation of IL-1 signaling alters production of CXC chemokines and polymorphonuclear cell (PMN) recruitment during S. pneumoniae infection in vivo. (A) Percentage of PMNs in bronchial alveolar lavage (BAL) fluid from C57BL/6 control mice and mice deficient in IL-1 type 1 receptor on a C57BL/6 background (IL-1R1−/−) 24 h after intratracheal instillation of 5 × 105 CFU type 4 pneumococci. Levels of CXCL1 (B) and CXCL2 (C) in BAL fluid from the same experiment as described for panel A. (D) Percentage of PMNs in BAL fluid from BALB/c control mice and mice deficient in IL-1 type receptor on a BALB/c background (IL-1R−/−) challenged with pneumococci as described for panel A. Levels of CXCL1 (E) and CXCL2 (F) in BAL fluid from the same experiment as that described for panel D. *, P < 0.05; **, P < 0.01, unpaired t test.
We then addressed the expression of CXCL1 (KC) and CXCL2 (MIP-2α), which are the murine paralogs of CXCL8 in each mouse strain. CXCL1 was significantly reduced in each Il1r1−/− mouse strain (Fig. 7B and E). There was a nonsignificant decrease in CXCL2 for Il1r1−/− C57BL/6 and no alteration in Il1r1−/− BALB/c mice (Fig. 7C and F). Although numbers of mice were small, the Il1r1−/− C57BL/6 mice had a trend toward increased bacterial numbers in lung and blood, in keeping with prior observations (46), but there was no difference in bacterial numbers in Il1r1−/− BALB/c mice (see the supplemental material at http://www.shef.ac.uk/medicine/infectionandimmunity/staffprofiles/dockrell.html, Fig. S5).
Modulation of IL-1 signaling can reduce the severity of invasive disease in fulminant pneumococcal infection.
We next increased the severity of infection in the BALB/c mice, which increased the number of polymorphonuclear cells in the BAL fluid. In this model, polymorphonuclear cell recruitment was again reduced in the Il1r1−/− mice (Fig. 8A), although levels of recruitment were much greater than with the lower-dose infection model (compare Fig. 8A with Fig. 7D). Interestingly CXCL1/2 expression was not lower in the lungs of Il1r1−/− mice with severe disease (Fig. 8B and C). This suggested other compensatory changes in the cytokine network might have occurred to enable neutrophil recruitment, and we observed that levels of TNF-α, a known stimulus for CXCL1 chemokine expression during pneumococcal infection (55), was increased in Il1r1−/− BALB/c mice (Fig. 8D). We found no alteration in colony counts in the lung but a significant decrease in colony counts in the blood of Il1r1−/− BALB/c mice (Fig. 8E and F).
Fig 8.

Modulation of IL-1 signaling can reduce the severity of invasive disease in fulminant pneumococcal infection. (A) Percentage of neutrophils (PMN) in bronchial alveolar lavage (BAL) fluid from BALB/c control mice and mice deficient in IL-1 type 1 receptor on a BALB/c background (IL-1R1−/−) 24 h after intratracheal instillation of 1 × 107 CFU type 4 pneumococci. Levels of CXCL1 (B), CXCL2 (C), and TNF-α (D) in BAL fluid from the same experiment as that described for panel A. CFU of bacteria in lung homogenates (E) and blood (F) in the same experiments as that described for panel A. *, P < 0.05, unpaired t test; **, P < 0.01, Mann-Whitney test.
DISCUSSION
In this study, we demonstrate a major role for macrophage-alveolar epithelial cell signaling during the initial phase of host defense against S. pneumoniae. We describe a critical role for IL-1 in stimulating CXCL8 release in response to S. pneumoniae and show that a macrophage cell line and primary macrophages are important sources of this cytokine. In vivo models have confirmed that IL-1 signaling stimulates CXCL1/2 chemokine expression in mice and facilitates recruitment of polymorphonuclear leukocytes to the lung during infection.
Respiratory epithelial cells are an important component of pulmonary host defense, possessing pattern recognition receptors, such as TLRs, which enable sensing of microorganisms and link recognition of bacteria to NF-κB activation and transcription of cytokines and chemokines (13, 20, 44). Hyporesponsiveness in this system, as recently demonstrated for an association between IRAK4 polymorphisms and susceptibility to Gram-positive infections in patients with critical illness, limit the effectiveness of the innate immune responses (56). We show that direct exposure of A549 cells to pneumococci induces only low-level CXCL8 secretion. Capsule limits the adherence of pneumococci to epithelial cells (21, 51), and this was confirmed again in our study. Nevertheless, cytokine expression correlates poorly with adherence; previous studies have found that the epithelial cell inflammatory response is not associated with the level of bacterial adherence (7), nor is it associated with the binding capacity of particular pneumococcal strains (48). Some bacteria can adhere and invade epithelial cell surfaces by downregulation of capsule, while some encapsulated strains can coat themselves in IgA fragments to overcome the electrostatic effects of a negatively charged capsule (21, 62).
Epithelial cell responses during inflammation are, however, dependent on a cellular network in which additional cell types prime epithelial cells to upregulate chemokine release (37, 42). These networks contain multiple potential cellular sources of a broad range of cytokines, including monocyte synthesis and secretion of IL-1β and TNF-α, which can prime alveolar epithelial cells to release CXCL8 (37, 42, 53). In the context of the early stages of pneumococcal infection, both IL-1β and TNF-α are important in stimulating NF-κB activation in pulmonary epithelium (44), but the sources of these cytokines are not fully defined. There is evidence that macrophages enhance CXCL8 expression in lung explants, but which cell types produce the cytokine is uncertain (66). We now show that a macrophage phenotype, resembling monocytes or inflammatory macrophages (12), releases IL-1 to stimulate CXCL8 release from a type II alveolar epithelial cell line. In contrast, macrophage-expressed TNF-α was not a significant factor in priming epithelial cells to produce CXCL8 in cocultures containing the macrophage cell line. The response induced by primary macrophages also involved IL-1, but the degree of inhibition by IL-1Ra was less marked, suggesting that other factors could also play a role.
These findings are consistent with data from previous studies which observed that IL-1Ra inhibits epithelial cell CXCL8 secretion in response to conditioned medium from M. tuberculosis-infected human monocytes (63) and in response to conditioned media from RSV-infected monocytes (58). Teichoic acid in the pneumococcal cell wall is a potent stimulus for IL-1 expression from monocytes (45). Teichoicated species in the cell wall could be the predominant microbial factor driving the enhanced CXCL8 response in cocultures, with an additional role played by strain variable factors, such as pili, a known stimulus for TNF-α production, at later stages of infection or during the fulminant mouse model when IL-1 signaling was impaired.
In interpreting the results of our study, it is necessary to bear in mind the complex nature of cell-cell signaling and cytokine networks. It is possible that the synergistic response in coculture conditions involves a component of bidirectional signaling between macrophages and epithelial cells (60). Although macrophage-conditioned media enhanced CXCL8 secretion from epithelial cells, signals could flow in the reverse direction. IL-1β expression was enhanced in cocultures. Activating signals from stimulated epithelial cells may potentiate the response of monocytes or macrophages (and possibly other immune cells in vivo), with inflammatory mediators acting in a paracrine fashion to stimulate production of other effector molecules. Once the initial recruitment of polymorphonuclear leukocytes has occurred, these cells can release IL-1β to fuel further chemokine release from macrophages and nonmyeloid cells, as demonstrated recently in a murine model of arthritis (10).
Cocultures of A549 and THP-1 cell lines provide a useful and well-recognized in vitro model of the alveolar space. We can confirm that the VD3-differentiated THP-1 cells have a differentiation state which resembles a monocyte or macrophage in the early stage of tissue differentiation (12). We verified findings with a primary macrophage that was also in the early stage of differentiation. Our confirmation in vivo, using two different strains of mice, that modulation of IL-1 signaling influences CXCL1/2 expression and polymorphonuclear cell numbers in the lung during pneumococcal pneumonia confirmed in vivo the validity of this approach. Other studies have supported our findings that IL-1 contributes to host defense in vivo against pneumococci, although in the case of the lung, the effects were most important in the first 24 to 48 h after infection (45, 67).
IL-1 dependent neutrophilic inflammation has improved bacterial clearance in a previous model of pneumococcal pneumonia (46), and our studies with moderate bacterial inocula were consistent with this. However, in fulminant disease, neutrophilic inflammation and reactive oxygen species generation by polymorphonuclear leukocytes may adversely affect clinical outcomes (33, 34), and we observed that IL-1-dependent neutrophilic inflammation enhanced levels of tissue invasion and bacteremia. In some settings, neutrophil depletion can improve outcomes in murine models of fulminant pneumonia (33). We have previously highlighted the potential importance of leukocyte-derived IL-1 in the initiation of airway inflammation (37, 38, 42). The data shown here provide further support for the concept that targeting IL-1 in airway infection may in some settings be able to limit damaging effects of excessive inflammation by suppressing signaling between monocytes/macrophages and airway epithelial cells. This may be more tractable than manipulation of other pathways, such as the TNF-α pathway, which may have more significant overall effects in regulating host responses to pneumococci (46). Alternatively, since TNF-α was raised in the absence of IL-1 signaling in the Il1r1−/− mice, it might be that the combined inhibition of IL-1 and TNF-α signaling could be more effective at inhibiting CXC chemokine generation. Any potential benefits would need to be weighed carefully against the possible harmful effects of inhibiting recruitment of the minimum number of polymorphonuclear leukocytes required to clear microorganisms. The timing of administration is likely to be key. Administration would need to be delayed until after the early stages of the host response and would ideally involve identifying a subgroup of patients with selective markers indicating that an excessive inflammatory response might develop and predispose the individual to complications, such as acute respiratory distress syndrome (ARDS), which may occur following pneumonia (61). Previous studies using IL-1Ra in the early stages of pneumonia evolution have not shown substantial alteration in inflammatory outcomes or survival in a murine model (47), but targeting such an intervention to select patients with markers of more severe inflammation might be tractable.
In conclusion, we present evidence for a synergistic relationship between alveolar epithelial cells and macrophages in the early inflammatory response to S. pneumoniae. We demonstrate a critical role for IL-1 signaling in CXC chemokine expression and neutrophil recruitment to the murine lung during pneumococcal pneumonia and suggest that in fulminant disease therapeutic modulation of this pathway, as could be achieved with IL-1Ra, could reduce the extent of invasive disease.
ACKNOWLEDGMENTS
This work is supported by a Wellcome Trust Senior Clinical Fellowship to D.H.D. (number 076945), a British Lung Foundation Fellowship (F05/7) and an MRC Discipline Hopping Grant (number G0902354) to H.M.M., and by grants from Wellcome Trust and Medical Research Council to T.J.M. I.S. and M.K.B.W. are supported by an MRC Research Grant (number G0801983).
Footnotes
Published ahead of print 12 December 2011
REFERENCES
- 1. Adamou JE, Wizemann TM, Barren P, Langermann S. 1998. Adherence of Streptococcus pneumoniae to human bronchial epithelial cells (BEAS-2B). Infect. Immun. 66:820–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ali F, et al. 2003. Streptococcus pneumoniae-associated human macrophage apoptosis after bacterial internalization via complement and Fcgamma receptors correlates with intracellular bacterial load. J. Infect. Dis. 188:1119–1131 [DOI] [PubMed] [Google Scholar]
- 3. Baggiolini M, Walz A, Kunkel SL. 1989. Neutrophil-activating peptide-1/interleukin 8, a novel cytokine that activates neutrophils. J. Clinical Invest. 84:1045–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Barocchi MA, et al. 2006. A pneumococcal pilus influences virulence and host inflammatory responses. Proc. Natl. Acad. Sci. U. S. A. 103:2857–2862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Beisswenger C, Lysenko ES, Weiser JN. 2009. Early bacterial colonization induces Toll-like receptor-dependent transforming growth factor beta signaling in the epithelium. Infect. Immun. 77:2212–2220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bogaert D, De Groot R, Hermans PW. 2004. Streptococcus pneumoniae colonisation: the key to pneumococcal disease. Lancet Infect. Dis. 4:144–154 [DOI] [PubMed] [Google Scholar]
- 7. Bootsma HJ, Egmont-Petersen M, Hermans PW. 2007. Analysis of the in vitro transcriptional response of human pharyngeal epithelial cells to adherent Streptococcus pneumoniae: evidence for a distinct response to encapsulated strains. Infect. Immun. 75:5489–5499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Calbo E, Garau J. 2010. Of mice and men: innate immunity in pneumococcal pneumonia. Int. J. Antimicrob. Agents 35:107–113 [DOI] [PubMed] [Google Scholar]
- 9. Chaudhuri N, et al. 2010. Diesel exhaust particles override natural injury-limiting pathways in the lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 299:L263–L271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chou RC, et al. 2010. Lipid-cytokine-chemokine cascade drives neutrophil recruitment in a murine model of inflammatory arthritis. Immunity 33:266–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Coulter KR, Wewers MD, Lowe MP, Knoell DL. 1999. Extracellular regulation of interleukin (IL)-1beta through lung epithelial cells and defective IL-1 type II receptor expression. Am. J. Resp. Cell. Mol. Biol. 20:964–975 [DOI] [PubMed] [Google Scholar]
- 12. Daigneault M, Preston JA, Marriott HM, Whyte MK, Dockrell DH. 2010. The identification of markers of macrophage differentiation in PMA-stimulated THP-1 cells and monocyte-derived macrophages. PLoS One 5:e8668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Diamond G, Legarda D, Ryan LK. 2000. The innate immune response of the respiratory epithelium. Immunol. Rev. 173:27–38 [DOI] [PubMed] [Google Scholar]
- 14. Dockrell DH, Lee M, Lynch DH, Read RC. 2001. Immune-mediated phagocytosis and killing of Streptococcus pneumoniae are associated with direct and bystander macrophage apoptosis. J. Infect. Dis. 184:713–722 [DOI] [PubMed] [Google Scholar]
- 15. Dockrell DH, et al. 2003. Alveolar macrophage apoptosis contributes to pneumococcal clearance in a resolving model of pulmonary infection. J. Immunol. 171:5380–5388 [DOI] [PubMed] [Google Scholar]
- 16. Eskola J, et al. 1992. Epidemiology of invasive pneumococcal infections in children in Finland. J. Am. Med. Assoc. 268:3323–3327 [PubMed] [Google Scholar]
- 17. Gingles NA, et al. 2001. Role of genetic resistance in invasive pneumococcal infection: identification and study of susceptibility and resistance in inbred mouse strains. Infect. Immun. 69:426–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Glaccum MB, et al. 1997. Phenotypic and functional characterization of mice that lack the type I receptor for IL-1. J. Immunol. 159:3364–3371 [PubMed] [Google Scholar]
- 19. Gosink KK, Mann ER, Guglielmo C, Tuomanen EI, Masure HR. 2000. Role of novel choline binding proteins in virulence of Streptococcus pneumoniae. Infect. Immun. 68:5690–5695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gribar SC, Richardson WM, Sodhi CP, Hackam DJ. 2008. No longer an innocent bystander: epithelial Toll-like receptor signaling in the development of mucosal inflammation. Mol. Med. 14:645–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hammerschmidt S, et al. 2005. Illustration of pneumococcal polysaccharide capsule during adherence and invasion of epithelial cells. Infect. Immun. 73:4653–4667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hathaway LJ, Battig P, Muhlemann K. 2007. In vitro expression of the first capsule gene of Streptococcus pneumoniae, cpsA, is associated with serotype-specific colonization prevalence and invasiveness. Microbiology 153:2465–2471 [DOI] [PubMed] [Google Scholar]
- 23. Herbold W, et al. 2010. Importance of CXC chemokine receptor 2 in alveolar neutrophil and exudate macrophage recruitment in response to pneumococcal lung infection. Infect. Immun. 78:2620–2630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Heumann D, Barras C, Severin A, Glauser MP, Tomasz A. 1994. Gram-positive cell walls stimulate synthesis of tumor necrosis factor alpha and interleukin-6 by human monocytes. Infect. Immun. 62:2715–2721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hippenstiel S, Opitz B, Schmeck B, Suttorp N. 2006. Lung epithelium as a sentinel and effector system in pneumonia—molecular mechanisms of pathogen recognition and signal transduction. Resp. Res. 7:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hmama Z, et al. 1999. 1α,25-Dihydroxyvitamin D(3)-induced myeloid cell differentiation is regulated by a vitamin D receptor-phosphatidylinositol 3-kinase signaling complex. J. Exp. Med. 190:1583–1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jones MR, Simms BT, Lupa MM, Kogan MS, Mizgerd JP. 2005. Lung NF-kappaB activation and neutrophil recruitment require IL-1 and TNF receptor signaling during pneumococcal pneumonia. J. Immunology 175:7530–7535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kadioglu A, Weiser JN, Paton JC, Andrew PW. 2008. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat. Rev. Microbiol. 6:288–301 [DOI] [PubMed] [Google Scholar]
- 29. Khor CC, et al. 2007. A Mal functional variant is associated with protection against invasive pneumococcal disease, bacteremia, malaria, and tuberculosis. Nat. Genet. 39:523–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Krakauer T. 2002. Stimulant-dependent modulation of cytokines and chemokines by airway epithelial cells: cross-talk between pulmonary epithelial and peripheral blood mononuclear cells. Clin. Diagn. Lab. Immunol. 9:126–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ku CL, et al. 2007. Selective predisposition to bacterial infections in IRAK-4-deficient children: IRAK-4-dependent TLRs are otherwise redundant in protective immunity. J. Exp. Med. 204:2407–2422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Magee AD, Yother J. 2001. Requirement for capsule in colonization by Streptococcus pneumoniae. Infect. Immun. 69:3755–3761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Marks M, et al. 2007. Influence of neutropenia on the course of serotype 8 pneumococcal pneumonia in mice. Infect. Immun. 75:1586–1597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Marriott HM, et al. 2008. Reactive oxygen species regulate neutrophil recruitment and survival in pneumococcal pneumonia. Am. J. Respir. Crit. Care Med. 177:887–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mayer AK, et al. 2007. Differential recognition of TLR-dependent microbial ligands in human bronchial epithelial cells. J. Immunol. 178:3134–3142 [DOI] [PubMed] [Google Scholar]
- 36. Miles AA, Misra SS, Irwin JO. 1938. The estimation of the bactericidal power of the blood. J. Hyg. 38:732–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Morris GE, et al. 2006. Cooperative molecular and cellular networks regulate Toll-like receptor-dependent inflammatory responses. FASEB J. 20:2153–2155 [DOI] [PubMed] [Google Scholar]
- 38. Morris GE, et al. 2005. Agonists of Toll-like receptors 2 and 4 activate airway smooth muscle via mononuclear leukocytes. Am. J. Resp. Crit. Care Med. 171:814–822 [DOI] [PubMed] [Google Scholar]
- 39. Musher DM. 1992. Infections caused by Streptococcus pneumoniae: clinical spectrum, pathogenesis, immunity, and treatment. Clin. Infect. Dis. 14:801–807 [DOI] [PubMed] [Google Scholar]
- 40. Obaro S, Adegbola R. 2002. The pneumococcus: carriage, disease and conjugate vaccines. J. Med. Microbiol. 51:98–104 [DOI] [PubMed] [Google Scholar]
- 41. Opitz B, et al. 2004. Nucleotide-binding oligomerization domain proteins are innate immune receptors for internalized Streptococcus pneumoniae. J. Biol. Chem. 279:36426–36432 [DOI] [PubMed] [Google Scholar]
- 42. Parker LC, et al. 2008. A phosphatidylserine species inhibits a range of TLR- but not IL-1beta-induced inflammatory responses by disruption of membrane microdomains. J. Immunol. 181:5606–5617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pearce BJ, Iannelli F, Pozzi G. 2002. Construction of new unencapsulated (rough) strains of Streptococcus pneumoniae. Res. Microbiol. 153:243–247 [DOI] [PubMed] [Google Scholar]
- 44. Quinton LJ, et al. 2007. Functions and regulation of NF-kappaB RelA during pneumococcal pneumonia. J. Immunol. 178:1896–1903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Riesenfeld-Orn I, Wolpe S, Garcia-Bustos JF, Hoffmann MK, Tuomanen E. 1989. Production of interleukin-1 but not tumor necrosis factor by human monocytes stimulated with pneumococcal cell surface components. Infect. Immun. 57:1890–1893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rijneveld AW, et al. 2001. TNF-alpha compensates for the impaired host defense of IL-1 type I receptor-deficient mice during pneumococcal pneumonia. J. Immunol. 167:5240–5246 [DOI] [PubMed] [Google Scholar]
- 47. Rijneveld AW, et al. 2003. Interleukin-1 receptor antagonist transiently impairs antibacterial defense but not survival in murine pneumococcal pneumonia. Eur. Cytokine Netw. 14:242–245 [PubMed] [Google Scholar]
- 48. Robson RL, Reed NA, Horvat RT. 2006. Differential activation of inflammatory pathways in A549 type II pneumocytes by Streptococcus pneumoniae strains with different adherence properties. BMC Infect. Dis. 6:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schmeck B, et al. 2006. Pneumococci induced TLR- and Rac1-dependent NF-kappaB-recruitment to the IL-8 promoter in lung epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 290:L730–L737 [DOI] [PubMed] [Google Scholar]
- 50. Schwende H, Fitzke E, Ambs P, Dieter P. 1996. Differences in the state of differentiation of THP-1 cells induced by phorbol ester and 1,25-dihydroxyvitamin D3. J. Leukocyte Biol. 59:555–561 [PubMed] [Google Scholar]
- 51. Selinger DS, Reed WP. 1979. Pneumococcal adherence to human epithelial cells. Infect. Immun. 23:545–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Standiford TJ, et al. 1990. Interleukin-8 gene expression by a pulmonary epithelial cell line. A model for cytokine networks in the lung. J. Clin. Invest. 86:1945–1953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Standiford TJ, Kunkel SL, Phan SH, Rollins BJ, Strieter RM. 1991. Alveolar macrophage-derived cytokines induce monocyte chemoattractant protein-1 expression from human pulmonary type II-like epithelial cells. J. Biol. Chem. 266:9912–9918 [PubMed] [Google Scholar]
- 54. Strieter RM, Kunkel SL, Keane MP, Standiford TJ. 1999. Chemokines in lung injury: Thomas A. Neff lecture. Chest 116:103S–110S [DOI] [PubMed] [Google Scholar]
- 55. Sun K, Salmon SL, Lotz SA, Metzger DW. 2007. Interleukin-12 promotes gamma interferon-dependent neutrophil recruitment in the lung and improves protection against respiratory Streptococcus pneumoniae infection. Infect. Immun. 75:1196–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sutherland AM, et al. 2011. A nonsynonymous polymorphism of IRAK4 associated with increased prevalence of Gram-positive infection and decreased response to Toll-like receptor ligands. J. Innate Immun. 3:447–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Talbot UM, Paton AW, Paton JC. 1996. Uptake of Streptococcus pneumoniae by respiratory epithelial cells. Infect. Immun. 64:3772–3777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Thomas LH, Wickremasinghe MI, Sharland M, Friedland JS. 2000. Synergistic upregulation of interleukin-8 secretion from pulmonary epithelial cells by direct and monocyte-dependent effects of respiratory syncytial virus infection. J. Virol. 74:8425–8433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tong HH, Blue LE, James MA, DeMaria TF. 2000. Evaluation of the virulence of a Streptococcus pneumoniae neuraminidase-deficient mutant in nasopharyngeal colonization and development of otitis media in the chinchilla model. Infect. Immun. 68:921–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ward JR, et al. 2009. A central role for monocytes in Toll-like receptor-mediated activation of the vasculature. Immunology 128:58–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ware LB, Matthay MA. 2000. The acute respiratory distress syndrome. New Engl. J. Med. 342:1334–1349 [DOI] [PubMed] [Google Scholar]
- 62. Weiser JN, et al. 2003. Antibody-enhanced pneumococcal adherence requires IgA1 protease. Proc. Natl. Acad. Sci. U. S. A. 100:4215–4220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wickremasinghe MI, Thomas LH, Friedland JS. 1999. Pulmonary epithelial cells are a source of IL-8 in the response to Mycobacterium tuberculosis: essential role of IL-1 from infected monocytes in a NF-kappa B-dependent network. J. Immunol. 163:3936–3947 [PubMed] [Google Scholar]
- 64. Winder AA, Wohlford-Lenane C, Scheetz TE. 2009. Differential effects of cytokines and corticosteroids on Toll-like receptor 2 expression and activity in human airway epithelia. Resp. Res. 10:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Witzenrath M, et al. 2011. The NLRP3 inflammasome is differentially activated by pneumolysin variants and contributes to host defense in pneumococcal pneumonia. J. Immunol. 187:434–440 [DOI] [PubMed] [Google Scholar]
- 66. Xu F, et al. 2008. Modulation of the inflammatory response to Streptococcus pneumoniae in a model of acute lung tissue infection. Am. J. Resp. Cell. Mol. Biol. 39:522–529 [DOI] [PubMed] [Google Scholar]
- 67. Zwijnenburg PJ, van der Poll T, Florquin S, Roord JJ, Van Furth AM. 2003. IL-1 receptor type 1 gene-deficient mice demonstrate an impaired host defense against pneumococcal meningitis. J. Immunol. 170:4724–4730 [DOI] [PubMed] [Google Scholar]