

Abstract
Targeting an antigen to Fc receptors (FcR) can enhance the immune response to the antigen in the absence of adjuvant. Furthermore, we recently demonstrated that intranasal immunization with an FcγR-targeted antigen enhances protection against a category A intracellular mucosal pathogen, Francisella tularensis. To determine if a similar strategy could be applied to the important pathogen Streptococcus pneumoniae, we used an improved mucosal FcR-targeting strategy that specifically targets human FcγR type I (hFcγRI). A humanized single-chain antibody component in which the variable domain binds to hFcγRI [anti-hFcγRI (H22)] was linked in a fusion protein with the pneumococcal surface protein A (PspA). PspA is known to elicit protection against pneumococcal sepsis, carriage, and pneumonia in mouse models when administered with adjuvants. Anti-hFcγRI-PspA or recombinant PspA (rPspA) alone was used to intranasally immunize wild-type (WT) and hFcγRI transgenic (Tg) mice in the absence of adjuvant. The hFcγRI Tg mice receiving anti-hFcγRI-PspA exhibited elevated S. pneumoniae-specific IgA, IgG2c, and IgG1 antibodies in serum and bronchoalveolar lavage fluid. Neither immunogen was effective in protecting WT mice in the absence of adjuvant, but when PspA was targeted to hFcγRI as the anti-hFcγRI-PspA fusion, enhanced protection against lethal S. pneumoniae challenge was observed in the hFcγRI Tg mice compared to mice given nontargeted rPspA alone. Immune sera from the anti-hFcγRI-PspA-immunized Tg mice showed enhanced complement C3 deposition on bacterial surfaces, and protection was dependent upon an active complement system. Immune serum also showed an enhanced bactericidal activity directed against S. pneumoniae that appears to be lactoferrin mediated.
INTRODUCTION
Numerous studies have demonstrated that targeting antigens to Fc receptors (FcRs) both in vitro and in vivo can enhance cellular and humoral immune responses (1, 4, 26–28, 53, 69). FcγRs are classified based on their molecular weights, IgG-Fc binding affinities, IgG subclass binding specificities, and cellular distributions. Three subtypes of FcγRs have been described in mice and humans: FcγRI (CD64), FcγRII (CD32), and FcγRIII (CD16). Both human and mouse FcγRI are high-affinity FcγR, which means that they can bind the Fc region of IgG when in monomeric form (25). Unlike the more ubiquitously expressed FcγRII and FcγRIII, FcγRI receptors are constitutively expressed primarily on professional antigen-presenting cells (APCs) (dendritic cells [DC] and macrophages [Mϕ]) (25, 52), and their expression can be induced on polymorphonuclear leukocytes (PMN) (42, 60). FcγRI receptors are activating receptors, providing solely stimulatory signals to the antigen-presenting cell (25). Thus, its distribution on DC and its stimulatory-only nature make FcγRI a particularly good target for enhancing an immune response.
The targeting of antigens to FcγR on DC can enhance presentation of such antigens (a key component of an effective immune response [48]) and potentiate immune responses by increasing the expression of major histocompatibility complex class II (MHC-II) and accelerating the maturation of DC (4). For example, when a tetanus toxin C (TTC) fragment in a γFc fusion protein was targeted to FcγRs, it was found to be superior to the commercial vaccine (TT plus alum) in inducing TT-specific antibodies in vivo (15). Mice immunized with the TTC-γFc fusion protein were fully protected from a lethal challenge of tetanus toxin. However, despite the potential benefit of targeting antigens to FcγR as a vaccine strategy, there have been few examinations of the use of FcγR targeting in generating immune protection against infectious agents, particularly mucosal pathogens. Consequently, our group was the first to show that targeting inactivated Francisella tularensis live vaccine strain to FcγR intranasally (i.n.) in the form of monoclonal antibody-inactivated F. tularensis complexes provided enhanced protection against subsequent mucosal challenge with the live pathogen compared to inactivated F. tularensis alone (53). The fact that immunizations with monoclonal antibody-inactivated F. tularensis were i.n. and that no adjuvant was required to achieve full protection emphasize the significance of this approach. Here, we build upon the approach of using FcγR-targeted immunogens as mucosal vaccines by utilizing a more finely defined targeting approach with recombinant techniques and testing its potential to elicit protection for a universal common mucosal pathogen, Streptococcus pneumoniae.
S. pneumoniae (pneumococcus) is an extracellular, Gram-positive bacterium, and unlike F. tularensis, it is a ubiquitous human pathogen which is responsible for significant morbidity and mortality worldwide. S. pneumoniae is generally regarded as the most common bacterial etiology of community-acquired pneumonia and meningitis and is a prominent cause of otitis media, sinusitis, and bronchitis (21, 37). It is responsible for well over 1 million deaths in children under the age of 5, mostly in developing countries (49, 70). While effective pneumococcal conjugate vaccines are available, their substantial cost is beyond the reach of the people most in need. In addition, the limitations against full coverage of all pneumococci by conjugate vaccines have led to increases in the incidence of serotypes not covered by the vaccines. Effective protein-based vaccines have the potential to be more cost-effective, to provide better coverage of all strains, and to fit with new improved strategies for vaccinations, via mucosal routes without needles, without need for refrigeration and, possibly, without adjuvants.
One of the more significant protein targets for vaccine design and development against S. pneumoniae infection is the pneumococcal surface protein A (PspA). It is present in virtually all pneumococci (30) while not present in closely related streptococcal species. The N-terminal half is a coiled-coil protein that is antigenically varied, being mostly found in one of five clades representative of two major antigenic families (31). Despite its variability, PspA appears to be among the most effective protection-eliciting immunogens for prospective pneumococcal protein-based vaccines as it has been shown to elicit protection against colonization, pneumonia, bacteremia, sepsis, and otitis media in various model systems (9, 14). In all cases, protection with PspA has required the use of appropriate adjuvants. However, while PspA plus adjuvant has proven promising as a protein-based vaccine, given its potential to protect against multiple strains of S. pneumoniae, there is still a need for a more efficient mucosal vaccine delivery strategy, in particular, one that does not require the use of traditional adjuvants, which can be expensive, toxic, and/or limited in their ability to generate appropriate immune responses.
Previous studies by this laboratory have shown that targeting antigens to human FcγR type I (hFcγRI) in vivo in the form of an anti-hFcγRI-antigen fusion protein administered parenterally (intradermally [i.d.]) increased production of antigen-specific antibodies as well as Th1 and Th2 cytokines (1). In addition, studies targeting a “weak antigen” to hFcγRI demonstrated that antigen-specific antibody responses were enhanced when hFcγRI transgenic (Tg) mice were used (36). Thus, we sought to combine hFcγRI targeting with the use of PspA as a strategy to induce protective anti-PspA responses in the absence of adjuvant.
In this study we used a recombinant DNA approach to produce a PspA fusion protein that was targeted to hFcγRI (anti-hFcγRI-PspA) in the hFcγRI Tg mouse pneumococcal sepsis model. Targeting PspA to hFcγRI is shown to be effective at enhancing protection against S. pneumoniae challenge in the absence of adjuvant but only in Tg mice in which the specific hFcγRI target resides on DC and Mϕ. The enhanced protection was associated with enhanced antibody production characteristic of a mixed Th1/Th2 response. Immune sera from anti-FcγRI-PspA-immunized Tg mice were also found to have increased bactericidal activity against pneumococci. The increased bactericidal activity correlated with PspA-specific antibodies in the serum and was consistent with the concept that PspA-specific antibodies may override PspA's normal blockage of bacterial killing by lactoferrin.
MATERIALS AND METHODS
Cells.
U937 is a human myeloid cell line which expresses human FcγRI (ATCC, Manassas, VA). U937 cells were grown in RPMI 1640 medium (CellGro, Manassas, VA) containing 10% fetal bovine serum ([FBS] HyClone, Logan, UT), 100 U/ml penicillin, 100 μg/ml streptomycin (Gibco, Carlsbad, CA), and 0.02 mg/ml gentamicin (Sigma-Aldrich, St. Louis, MO). Mouse peritoneal exudate cells/macrophages (PEC/Mϕ) were obtained from either C57BL/6 or hFcγRI Tg mice at 8 to 12 weeks of age. The mouse MHC-IIb restricted PspA-specific CD4+ T cell hybridoma (B6D2), obtained from C57BL/6 mice, was provided by Clifford Snapper (Uniformed Services University, Bethesda, MD). These cells were grown in Dulbecco's modified Eagle's medium ([DMEM] CellGro) supplemented with 10% FBS (HyClone), minimal essential medium (MEM) nonessential amino acids (CellGro), 1 mM sodium pyruvate (CellGro), 50 μM 2-mercaptoethanol ([2-ME] Bio-Rad, Hercules, CA), 0.01 M HEPES (Gibco), and 100 U/ml penicillin and 100 μg/ml streptomycin (Gibco). The B6D2 hybridoma produces interleukin-2 (IL-2) in response to PspA (18). DC were generated from bone marrow cells isolated from either C57BL/6 wild-type (WT) or hFcγRI Tg mice 8 to 12 weeks of age. Specifically, bone marrow cells were cultured for a week in RPMI 1640 (CellGro) containing 10% FBS (HyClone), 2 mM l-glutamine (CellGro), MEM nonessential amino acids (CellGro), 1 mM sodium pyruvate (CellGro), 50 μM 2-ME (Bio-Rad), and 100 U/ml penicillin and 100 μg/ml streptomycin (Gibco, Carlsbad, CA) supplemented with 50 ng/ml of mouse recombinant FLT3 (R&D Systems, Minneapolis, MN).
The A66.1 S. pneumoniae challenge strain, which expresses pneumococcal polysaccharide serotype 3, was provided by David E. Briles (University of Alabama at Birmingham, Birmingham, AL) and was cultured at 37°C in Todd-Hewitt broth (Becton Dickinson, San Jose, CA) until mid-log phase; the culture was washed three times with phosphate-buffered saline (PBS), resuspended in fresh broth containing 15% glycerol, and stored in liquid nitrogen until use. Importantly, S. pneumoniae strains are differentiated by the capsular polysaccharide coat, which stimulates the production of serotype-specific protective antibody. There are approximately 92 different serotypes of S. pneumoniae (29, 40). For this model, serotype 3 was used due to its virulence in the experimental mouse model and its cognate PspA clade type (56, 57).
Mice.
WT C57BL/6 mice were obtained from Taconic Laboratories (Germantown, NY). Heterozygote hFcγRI Tg mice, on a C57BL/6 background, were generated as previously described (28) and were provided by Medarex Inc. (Bloomsbury, NJ). It is important to emphasize that in this study WT mice will serve as negative controls since the vaccine will be targeted to hFcγRI, which is present only in the hFcγRI Tg mice and not in the WT mice. All mice were housed in the Animal Resources Facility at Albany Medical College under pathogen-free conditions. Mice were provided with water and food ad lib during the course of each experiment. All animal studies were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC).
Construction, screening, isolation, and testing of recombinant anti-hFcγRI-PspA fusion protein.
Medarex Inc. generously provided the humanized bivalent anti-hFcγRI (H22) construct (pJG582) required to generate the construct encoding the anti-hFcγRI-PspA fusion protein. The pUAB055 plasmid encoding 303 amino acids of PspA_Rx1 has been previously described (5). The anti-hFcγRI-PspA construct was made by fusing the PspA-His tag DNA 3′ to the heavy-chain (VH) and light-chain (VL) variable regions within the pJG582 construct (Fig. 1A to C). Specifically, in order to generate the anti-hFcγRI-PspA construct, a 918-bp gene fragment encoding the N-terminal region of PspA and containing a His tag (PspA-303-His) was amplified using PCR. During this procedure, unique SalI and NotI restriction sites were generated at the 5′ and 3′ ends, respectively, and the PCR product was ligated into the pJG582 vector between XhoI and NotI restriction sites (Fig. 1A). Ampicillin-resistant transformants were screened for insertion by PCR, followed by restriction endonuclease analysis and sequencing of miniprep DNA to confirm the in-frame fusion construct. Plasmid DNA was used for liposome-mediated transfection of 1 × 104 NSO cells (ATCC), a non-Ig-synthesizing murine myeloma cell line (68). Supernatants from transfected hybridomas were then screened by flow cytometry for the presence of the fusion protein. For this purpose, hFcγRI-expressing U937 cells (ATCC), hFcγRI Tg PEC/Mϕ, or hFcγRI Tg DC were incubated for 2 h at 4°C with either supernatants or purified anti-hFcγRI-PspA fusion protein from transfected hybridomas in the presence of human IgG block (12 mg/ml; Sigma-Aldrich) followed by a 1-h incubation with rabbit anti-PspA polyclonal antibody (4ABO55) and a subsequent 30-min incubation with goat anti-rabbit IgG-fluorescein isothiocyanate (FITC) (Caltag Laboratories, Burlingame, CA). Anti-PspA and anti-His tag enzyme-linked immunosorbent assays (ELISAs) were also used to confirm the presence of anti-hFcγRI-PspA in the supernatants of cultured transfected hybridomas. Briefly, wells of a 96-well plate were coated with protein L, which binds the anti-hFcγRI variable region of the fusion protein; they were washed three times, and supernatants from the anti-FcγRI-PspA transfected hybridoma cells (clones) were added for 1 h at room temperature. Following three additional washes with PBS, rabbit anti-PspA or mouse anti-His tag monoclonal antibody was added for 1 h, followed by three washes. Finally, anti-rabbit-alkaline phosphatase (AP) or anti-mouse-AP was added for 1 h, respectively; the wells were washed again, and AP substrate was added. The anti-hFcγRI-PspA fusion protein was purified using a nickel affinity column or protein L, also as previously described (51, 68). Purified anti-hFcγRI-PspA fusion protein was run on an 8% polyacrylamide SDS-PAGE gel (Pierce, Rockford, IL). The gel was then stained for 1 h with Imperial Protein Stain (Pierce). The endotoxin level for the purified anti-hFcγRI-PspA was determined utilizing the toxin sensor Chromogenic LAL Endotoxin Assay Kit (GenScript, Piscataway, NJ).
Fig 1.

Generation of a recombinant anti-hFcγRI-PspA fusion protein. (A) As depicted, the variable regions of an hFcγRI-specific monoclonal antibody (heavy and light chains) were linked together. At the 3′ end is the DNA sequence for the FcγRI-specific targeting component. PspA DNA was inserted between the restriction sites Xhol and Notl. (B) A representation of the folded protein structure of the recombinant anti-hFcγRI-PspA fusion protein. (C) The specificity of the anti-hFcγRI-PspA is directed outside the binding domain for antibody Fc on human FcγRI. The detection of anti-hFcγRI-PspA in the supernatants of transfected NSO cells was confirmed by Anti-PspA and anti-His Tag ELISAs of hybridoma supernatants (D). Each bar represents the average of three replicates ± standard deviation. (E) SDS-PAGE gel of the purified anti-hFcγRI-PspA fusion protein (FP). Std, molecular mass standard. (F) Anti-hFcγRI-PspA binding to hFcγRI-expressing U937 cells. (G) Anti-hFcγRI-PspA binding to hFcγRI-expressing PEC/Mϕ from Tg mice. (H) Anti-hFcγRI-PspA binding to hFcγRI-expressing DC from Tg mice. (I) PspA presentation by PEC/Mϕ from Tg mice cultured with a PspA-specific mouse (C57BL/6) T cell line (B6D2) and rPspA or anti-hFcγRI-PspA. After 30 h of incubation at 37°C, supernatants were collected, and the levels of IL-2 were measured by CBA (*, P < 0.1; **, P < 0.05). The experiments shown in panels D to G and I are representative of a minimum of two independent experiments. MFI, mean fluorescence intensity.
Antigen presentation assays.
The PspA-specific T cell hybridoma (B6D2) (1 × 105 cells/well) was cocultured with either WT or Tg PEC/Mϕ (2 × 105 cells/well) with titrating amounts of recombinant PspA (rPspA) or anti-hFcγRI-PspA, ranging in concentration from 0 to 5 μg/ml PspA. Cells were incubated for 30 h at 37°C in 5% CO2. Subsequently, the supernatants were collected, and the content of IL-2 was measured using cytometric bead array (CBA) Flex kits (BD Biosciences-BD Pharmingen, San Diego, CA). The data were acquired on a FACSArray instrument (BD Immunocytometry Systems) and analyzed using FCAP software, version 1.0.1 (BD Immunocytometry Systems).
Detection of hFcγRI expressed on lung DC and Mϕ from Tg mice.
Lungs from WT and Tg mice were isolated, homogenized, and digested with collagenase IV (Sigma-Aldrich) for 1 h at 37°C. Red blood cells were lysed utilizing ammonium chloride, and then the cell suspension was washed three times with PBS containing 2 mg/ml bovine serum albumin ([BSA] Sigma-Aldrich) and 0.1% azide (Sigma-Aldrich). A total of 1 × 106 cells were then incubated for 30 min with 80 μg/ml of hFcγRI-specific mouse monoclonal antibody 22-FITC (Medarex Inc.), allophycocyanin (APC)-conjugated anti-CD11c (eBioscience, San Diego, CA), phycoerythrin (PE)-conjugated anti-CD11b (BD Immunocytometry Systems), and peridinin chlorophyll protein (PerCP)-Cy5.5 conjugated anti-Gr-1 antibodies (eBioscience). Following the incubation, cells were washed again, and fluorescence was detected by flow cytometry using a BD LSRII flow cytometer (BD Immunocytometry Systems). Data analysis was performed using FlowJo, version 8.8.6 (Tree Star, Inc., Ashland, OR).
Immunization and challenge experiments.
Mice were generally divided into three groups consisting of 4 to 6 mice/group, 8 to 12 weeks of age. WT mice (negative controls) or Tg mice were immunized i.n. with 20 μl of PBS, 5 μg or 25 μg of anti-hFcγRI-PspA, or an amount of rPspA equivalent to that present in the anti-hFcγRI-PspA for each condition tested on day 0 and boosted either once (day 21) or twice (days 14 and 28). Prior to the immunization, mice were bled, and serum was isolated and tested for the presence of S. pneumoniae-specific antibodies. Immunized mice were challenged i.n. 2 weeks following the last boost with 1 × 106 CFU of live S. pneumoniae bacteria and monitored for at least 21 days. This challenge dose was chosen based on its ability to cause 50% of WT mice to die in the absence of immunization. Use of this lower challenge dose was necessary to optimize detection of differences in survival between PBS-treated and anti-hFcγRI-PspA-immunized groups. The Tg mice were slightly more susceptible than WT mice, with this same challenge dose representing approximately two times the 50% lethal dose (LD50). The latter challenge scenario resulted in between 80 to 100% of unimmunized Tg mice dying at the LD50 for WT mice. Infectious challenge of animals was performed with strain A66.1, which is a mouse-virulent capsule type 3 pneumococcus (6) and expresses family 1, clade 2, PspA serologically similar to the family 1, clade 2, PspA of strain Rx1 (31). Immunization with Rx1 PspA has been shown previously to protect mice, in the presence of adjuvant, from in vitro grown A66.1 (8). The exact numbers of CFU administered were verified by culturing and counting the inoculum subsequent to the infectious challenge.
Quantification of bacterial burden.
Following immunization and challenge, mice were euthanized at various time intervals, and tissues such as lung, liver, and spleen were collected aseptically in PBS containing a protease inhibitor mixture (1 tablet in 10 ml of sterile PBS) (Roche Diagnostics, Indianapolis, IN) and subjected to mechanical homogenization using a Mini-BeadBeater-8 (BioSpec Products Inc., Bartlesville, OK). Supernatants were then diluted 10-fold in sterile saline, and 10 μl of each dilution was spotted onto blood agar plates in duplicate and incubated at 37°C for 24 h. The numbers of colonies on the plates were counted, and results are expressed as log10 CFU/ml for the respective tissue. The remaining tissue homogenate was spun at 14,000 × g for 20 min, and the clarified supernatant was stored at −20°C for cytokine analysis.
In vitro cytokine production.
Spleen cells were harvested from immunized mice and cultured as previously described, with some modification (1). Briefly, to the individual wells of a 24-well plate, 1 ml of RPMI 1640 medium plus 10% FBS containing 4 × 106 spleen cells was added. Then, to appropriate wells, 100 μl of 4 × 106 inactivated S. pneumoniae organisms was added in the above culture medium. To inactivate these organisms (to produce inactivated S. pneumoniae), 1 × 108 CFU of live S. pneumoniae bacteria were resuspended in 1 ml of sterile PBS (Cellgro), which then was added to 10 ml of 2% paraformaldehyde (Sigma-Aldrich) and incubated for 2 h at room temperature. Fixed bacteria (inactivated S. pneumoniae) were then washed with sterile PBS three times. Inactivation was verified by culturing a 100-μl sample (1 × 107 organisms) of the inactivated S. pneumoniae on sheep blood agar plates (Fisher Scientific, Pittsburgh, PA). Although 24 h is normally sufficient to observe growth of this organism, plates were incubated at 37°C for up to 10 days to make absolutely certain that all bacteria were inactivated. As a negative control, 100 μl of RPMI 1640 medium plus 10% FBS without inactivated S. pneumoniae was added. Spleen cell cultures were then placed in a humidity chamber at 37°C and 5% CO2, and supernatants were harvested after 3 days of incubation. CBA Flex kits (BD Biosciences) were then used for the simultaneous measurement of multiple cytokines in tissue homogenates. Data were acquired on a FACSArray instrument (BD Immunocytometry Systems) and analyzed using FCAP software, version 1.0.1 (BD Immunocytometry Systems).
Measurement of S. pneumoniae-specific antibody production.
ELISA plates were coated with 1×107 CFU/well of live S. pneumoniae bacteria in carbonate buffer (4.3 g/liter sodium bicarbonate [Sigma-Aldrich]) plus 5.3 g/liter sodium carbonate [Sigma-Aldrich], pH 9.4) and incubated for either 2 h at 37°C or overnight at 4°C. Plates were then washed three times with PBS–0.05% Tween and blocked for 2 h with PBS–10% BSA, and then samples were added in 2-fold (BAL fluid) or 10-fold (serum) serial dilutions. Following a 2-h incubation at 37°C, plates were washed, and secondary horseradish peroxidase (HRP)-conjugated anti-mouse antibody was added (anti-IgG, anti-IgA, anti-IgG2c, anti-IgG1, anti-IgG2b, and anti-IgG3 [Caltag Laboratories]). After a 1-h incubation at 37°C, plates were washed, and 3,3′,5,5′-tetramethylbenzidine (TMB) substrate was added (Zymed-Invitrogen, Carlsbad, CA). The sample optical densities (ODs) were read at 650 nm using a kinetic microplate reader (Molecular Devices, Sunnyvale, CA). Antibody titers were determined as the reciprocal dilution that provided an OD reading twice that of the background generated in the presence of PBS.
C3 deposition assay.
To exclude differences in complement levels within individual samples or groups, the endogenous complement in sera from previously immunized WT and Tg mice was first inactivated by incubation at 56°C for 10 min. One hundred microliters of complement-inactivated sera (pooled from each group) was then added to 1 × 107 CFU of S. pneumoniae and incubated for 30 min at 37°C. Samples were then washed with PBS (Na+/Mg2+ [Cellgro]), and 100 ml of mouse complement (Innovative Research, Southfield, MI) was added to each sample. The complement was obtained by collecting serum from the clotted blood of healthy fasted non-Swiss albino mice of mixed sex. Following a 30-min incubation at 37°C, samples were washed and incubated with 100 μl of FITC-rabbit anti-mouse C3 monoclonal antibody (1:1,000 dilution; Immunology Consultants Laboratories, Newberg, OR) for 30 min at 37°C. Samples were then washed again, and C3 deposition was assessed by flow cytometry utilizing an LSRII flow cytometer (BD Immunocytometry Systems).
Complement depletion in vivo.
WT and Tg mice received injections of cobra venom factor (CVF) (Sigma-Aldrich) intraperitoneally (i.p.) at 200 mg/kg of mouse weight on day 0 and every 3 days thereafter. To confirm depletion of complement, sera were obtained from CVF-treated mice on days 0, 1, 2, 3, and 4 after CVF administration and used to coat ELISA plates (50 μl/well). Plates were then washed, and 100 μl of mouse anti-C3 (Invitrogen) was added per well and incubated for 1 h at 37°C. Following incubation, plates were washed, and an AP-conjugated anti-mouse polyclonal antibody (Caltag laboratories) was added. Phosphate substrate (Sigma-Aldrich) was then added to detect complement C3.
Measurement of bactericidal activity in sera from immunized mice.
To determine if sera from immunized mice exhibited bactericidal activity, S. pneumoniae organisms (1 × 107 CFU) were resuspended and incubated for 2 to 5 h at 37°C in various dilutions of pooled sera from immunized mice. Importantly, results obtained were similar at 2 and 5 h. To assess bactericidal activity, following incubation at 37°C in the presence of sera from immunized mice, two strategies were utilized. First, samples were diluted 10-fold in sterile saline, and 10 μl of each dilution was spotted onto blood agar plates in duplicate and incubated at 37°C for 24 h. The numbers of colonies on the plates were counted, and results are expressed as log10 CFU/ml for the respective tissue. Second, bacterial samples were stained with Sytox green, a nucleic acid stain (Invitrogen), which can stain only cells with a punctured/permeabilized membrane, at a concentration of 5 μM for 10 min at room temperature. Bacterial samples were then washed with PBS-BSA-azide and fixed with 2% paraformaldehyde. Bacterial permeability, as indicated by Sytox green-positive cells, was then measured by flow cytometry and indicated by increased mean fluorescence intensity.
To determine if lactoferrin, a molecule known to be bactericidal for S. pneumoniae (39, 58), is responsible for the bactericidal activity we observed with S. pneumoniae, we first determined if the strain we were working with was susceptible to lactoferrin-mediated bactericidal activity. Specifically, 1 × 106 organisms were resuspended and incubated in Hanks balanced salt solution (HBSS; Gibco) containing 2% FBS, alone or with 10 μg/ml polyclonal rabbit anti-PspA antibody for 1 h at 37°C. Following incubation, samples were washed, and either medium or human lactoferrin protein (Abcam, Cambridge, MA) was added to each sample at concentrations ranging from 1 to 50 μg/ml. After an additional 2 h of incubation at 37°C, each sample was serially diluted and plated out on blood agar plates in order to obtain a final CFU count. To determine if lactoferrin specifically was responsible for the bactericidal activity observed when sera from mice immunized with anti-hFcγRI-PspA were used, sera from immunized mice were incubated for 1 h at 37°C alone or with polyclonal (1 to 50 μg/ml) rabbit anti-lactoferrin antibody (Thermo-Fisher Scientific, Waltham, MA). These mixtures were then added to 1 × 106 CFU of S. pneumoniae organisms, which were resuspended and incubated for 2 h at 37°C. After incubation, each S. pneumoniae sample was serially diluted and plated on blood agar plates in order to determine a final CFU count as described above.
Statistical analysis.
In order to compare survival curves, Kaplan-Meier double log rank analyses were used. Statistical data for bacterial burden and cytokine analysis were generated using one-way analysis of variances (ANOVA) on day 7, which is the peak of infection. Antibody titers were assessed using an unpaired two-tailed t test. GraphPad Prism 4 provided the software for the statistical analysis (San Diego, CA).
RESULTS
Generation and testing of the anti-hFcγRI-PspA fusion protein.
In order to target the protective S. pneumoniae immunogen PspA to hFcγRI, we constructed a fusion protein consisting of PspA linked to a bivalent variable region fragment specific for hFcγRI. The presence of anti-hFcγRI-PspA (Fig. 1A to C) in the NSO cell supernatants was confirmed by an anti-PspA and an anti-His tag ELISA (Fig. 1D), and the fusion protein was subsequently purified. The purified anti-hFcγRI-PspA fusion protein produced a band at approximately 145 kDa (Fig. 1E). Endotoxin levels averaged 3.3 ± 2.3 endotoxin units (EU)/ml, which is above the level normally present in sterile water (0.25 to 0.5 EU/ml) but below levels generally acceptable for vaccination (5 EU/ml). To assess effective binding of anti-hFcγRI-PspA to hFcγRI, we incubated cells that expressed hFcγRI (U937, Tg PEC/Mϕ, and Tg DC) with an anti-hFcγRI-PspA fusion protein, followed by the addition of rabbit anti-PspA polyclonal antibody and FITC-conjugated anti-rabbit IgG polyclonal antibody. A concentration-dependent binding of anti-hFcγRI-PspA to the cell surface of U937 cells, PEC/Mϕ, and DC was observed via flow cytometry (Fig. 1F to H, respectively). We then determined whether targeting PspA to hFcγRI increased antigen presentation following binding in the presence of hFcγRI-expressing APCs. For this purpose, a PspA-specific T cell hybridoma (B6D2) was cultured with PEC/Mϕ from mice with either nontargeted rPspA or anti-hFcγRI-PspA. Increased production of IL-2 by PspA-specific B6D2 cells was observed in response to increased concentrations of anti-hFcγRI-PspA compared to nontargeted rPspA in the presence of Tg PEC/Mϕ (Fig. 1I). In the case where WT PEC/Mϕ were used in place of Tg PEC/Mϕ, the production of IL-2 measured in the presence of anti-hFcγRI-PspA was the same as that in the presence of nontargeted rPspA (data not shown).
Targeting PspA to hFcγRI in vivo enhances protection against lethal S. pneumoniae challenge.
The increased binding and antigen presentation of PspA following targeting to hFcγRI in vitro led us to hypothesize that targeting PspA to the same receptor in vivo would enhance protection against lethal challenge with S. pneumoniae. However, before conducting in vivo experiments to test this hypothesis, we first verified the presence of hFcγRI on mucosal Mϕ and DC from the Tg mice (Fig. 2A and B, respectively). In addition, studies titrating the dose of S. pneumoniae in WT versus Tg mice indicated that the infectious dose of S. pneumoniae that produced the LD50 for the WT mice (1 × 106 CFU) was approximately 2× LD50 for Tg mice and produced between 80 to 100% lethality in these mice. Thus, to establish a successful and robust immunization strategy and detect differences in protection between WT and Tg mice, we initially varied the dose of anti-hFcγRI-PspA, as well as the number of booster immunizations (Fig. 2C to H). Immunization i.n. with 5 μg of anti-hFcγRI-PspA followed by a single boost did not provide enhanced protection for the Tg mice (Fig. 2D). However, when the dose was increased to 25 μg of anti-hFcγRI-PspA with the single boost approach, we observed improved survival in Tg versus WT mice when we compared relative differences between unimmunized and immunized mice (Fig. 2E and F). Furthermore, Tg mice were fully protected against i.n. infection with S. pneumoniae at a dose of 25 μg of anti-hFcγRI-PspA following two boosts (days 14 and 28 postimmunization) (Fig. 2G and H). Importantly, in WT mice, which lack the hFcγRI target, this nonadjuvanted immunization with anti-hFcγRI-PspA could not protect against S. pneumoniae challenge any better than the PBS control at any dose (Fig. 2C, E, and G), indicating that the humanized hFcγRI-specific component itself, when linked to PspA and utilized in the absence of hFcγRI-expressing APC, cannot generate the enhanced protection against S. pneumoniae challenge observed in the presence of hFcγRI-expressing APCs. A slight decrease in survival of unimmunized Tg versus WT mice was also observed, demonstrating a differential susceptibility to S. pneumoniae challenge between Tg and WT mice (Fig. 2C to H).
Fig 2.

Anti-hFcγRI-PspA induces protection without adjuvant but only in hFcγRI Tg mice. (A) Expression of hFcγRI by lung Mϕ from Tg (gray peak) versus WT (black peak) mice. (B) Expression of hFcγRI by lung DC from Tg (gray peak) versus WT (black peak) mice. FITC-A, FITC-antibody fluorescence. (C to H) C57BL/6 WT and Tg mice were immunized i.n. with 5 μg (C and D) or 25 μg (E, F, G, and H) of anti-hFcγRI-PspA or PBS (day 0) and then boosted once, on day 21, (C, D, E, and F), or twice, on days 14 and 28 (G and H) postimmunization. Two weeks following the final boost, mice were challenged i.n. with S. pneumoniae bacteria (1 × 106 CFU), and their survival was monitored for 21 days. Five to eight mice were used per group. Representative survival curves from a minimum of three separate experiments are presented. In the experiment shown in panel F, P = 0.138 for the PBS- versus anti-hFcγRI-PspA-immunized group of Tg mice. In the experiment shown in panel H, P = 0.004 for the PBS- versus anti-hFcγRI-PspA-immunized group of Tg mice.
The full protection that could be induced in Tg mice following anti-hFcγRI-PspA immunization was also consistent with a reduced bacterial burden observed on days 4 and 7 post-S. pneumoniae infection in the lung, liver, and spleen of anti-hFcγRI-PspA-immunized Tg animals compared to PBS-immunized mice (Fig. 3B, D, and F, respectively).
Fig 3.

Anti-hFcγRI-PspA reduces bacterial burden (CFU) in hFcγRI Tg mice. C57BL/6 WT and Tg mice were immunized i.n. with 25 μg of anti-hFcγRI-PspA or PBS (day 0) and then boosted twice on days 14 and 28. On day 42 mice were challenged i.n. with S. pneumoniae bacteria (1 × 106 CFU). On days 2, 4, and 7 postinfection the lung, liver, and spleen of WT and Tg mice were harvested, and bacterial burden was measured by plating on blood agar plates, as described in Materials and Methods (*, P < 0.1; **, P < 0.05).
Targeting PspA to hFcγRI enhances S. pneumoniae-specific antibody and cytokine responses and produces a mixed Th1/Th2-like profile.
The importance of antibody in protective immune responses against extracellular pathogens such as S. pneumoniae has been widely established (10, 13, 20, 47, 74). Hence, we measured the levels of S. pneumoniae-specific antibody in the sera and BAL fluids of immunized mice. Tg mice immunized with anti-hFcγRI-PspA versus PBS had significantly higher levels of total IgG and IgA antibodies in their sera (Fig. 4A and B) and BAL fluids (Fig. 4C and D) than WT mice immunized with anti-hFcγRI-PspA. To determine the Th-type profile generated, we also analyzed antibody isotypes produced following immunization with anti-hFcγRI-PspA. For this purpose, we assessed the levels of S. pneumoniae-specific IgG2c (Th1 type) and IgG1 (Th2 type) antibodies in the sera and BAL fluids of immunized mice. The levels of these two isotypes were significantly enhanced in the Tg mice versus the WT animals immunized with anti-hFcγRI-PspA (Fig. 5A to D), suggesting that targeting PspA to hFcγRI induces strong S. pneumoniae-specific humoral responses of a mixed Th1/Th2 type. Other IgG isotypes tested (IgG2b and IgG3) were also found to be elevated in anti-hFcγRI-PspA-immunized mice (data not shown). To confirm the mixed nature of the Th response, we examined cytokine production profiles in vitro, secreted by spleen cells, following immunization with anti-hFcγRI-PspA. For this purpose, splenocytes from Tg and WT mice immunized with anti-hFcγRI-PspA were obtained and cultured for 3 days with fixed (inactivated) S. pneumoniae organisms. Supernatants were then collected, and the cytokine levels were determined by CBA analysis. Both Th1-like (IL-2, IFN-γ, and TNF-α) and Th2-like (IL-4 and IL-5) cytokines were elevated in splenocyte cultures from Tg mice immunized with anti-hFcγRI-PspA versus similarly immunized WT counterparts, confirming the generation of a mixed Th1/Th2 recall response upon targeting PspA to hFcγRI (Fig. 5E).
Fig 4.

Targeting PspA to hFcγRI enhances S. pneumoniae-specific antibody responses in hFcγRI Tg mice. C57BL/6 WT and Tg mice were immunized i.n. with PBS or with 25 μg of anti-hFcγRI-PspA (day 0) and boosted twice (days 14 and 28). On day 42 the serum and BAL fluid were collected, and the S. pneumoniae-specific IgG and IgA antibodies were measured by ELISA, as described in Materials and Methods. Antibody data for this experiment represent the mean of 4 to 6 mice ± standard deviation. The results presented are representative of two independent experiments, each experiment using 4 to 6 mice per group (*, P < 0.1; **, P < 0.05).
Fig 5.

Targeting PspA to hFcγRI induces a mixed Th1/Th2-like cytokine response. (A to D) C57BL/6 WT and Tg mice were immunized i.n. with anti-hFcγRI-PspA or PBS (day 0) and then boosted twice on days 14 and 28. On day 42, mouse sera, and BAL fluids were collected, and the S. pneumoniae-specific IgG2c and IgG1 antibody levels were measured by ELISA, as described in Materials and Methods. Antibody data represent the mean of 4 to 6 mice ± standard deviation. (E) WT and Tg mice were immunized i.n. as described above. Splenocytes were harvested on day 10 after the second boost (day 38) and cultured with fixed bacteria (inactivated S. pneumoniae) for 3 days. Supernatants were obtained, and Th1 or Th2 cytokine concentrations were measured by CBA, as described in Materials and Methods. The results are representative of two independent experiments, with each experiment using 4 to 6 mice per group (*, P < 0.1; **, P < 0.05).
In order to exclude any potential effects of differential susceptibility between Tg and WT mice on the enhanced protection we observed using anti-hFcγRI-PspA immunization, we then compared protection generated by immunizing WT versus Tg mice with anti-hFcγRI-PspA versus the nontargeted rPspA (Fig. 6A and B). Consistent with our studies comparing WT to Tg mice, protection was significantly enhanced when Tg mice were immunized with anti-hFcγRI-PspA versus rPspA alone at equivalent doses of PspA (Fig. 6B). For WT mice, we observed no significant difference between anti-hFcγRI-PspA and rPspA immunizations (Fig. 6A). In addition, S. pneumoniae-specific antibody responses were similarly of a mixed Th1/Th2-like profile in the sera of Tg mice immunized with either rPspA or anti-hFcγRI-PspA (Fig. 6C to F), suggesting that targeting PspA to hFcγRI, while enhancing the overall response, does not significantly alter the Th1 versus Th2 response to PspA.
Fig 6.

Targeting PspA to hFcγRI enhances protection against S. pneumoniae infection compared to the nontargeted rPspA and generates a mixed Th1/Th2-like antibody response. (A and B) C57BL/6 WT and Tg mice were immunized i.n. with 25 μg of anti-hFcγRI-PspA, the rPspA equivalent to the anti-hFcγRI-PspA, or PBS (day 0) and then boosted twice on days 14 and 28. On day 42 mice were challenged with S. pneumoniae (1 × 106 CFU), and their survival was monitored for 21 days. Six to 8 mice were used per group. Survival curves representative of three independent experiments are shown. In the experiment shown in panel B, P = 0.045 for the rPspA versus anti-hFcγRI-PspA-immunized Tg group. (C to F) WT and Tg mice were immunized as described above for panels A and B. On day 42, mouse serum was collected, and the S. pneumoniae-specific IgG, IgA, IgG2c, and IgG1 antibody levels were measured by ELISA. Antibody data represent the mean of 4 to 6 mice ± standard deviation. The results are representative of two independent experiments (*, P < 0.1; **, P < 0.05).
Enhanced protection against S. pneumoniae infection generated by anti-hFcγRI-PspA is complement dependent.
It has been previously demonstrated that PspA-specific antibody can enhance complement activation in the presence of S. pneumoniae, further enhancing complement-mediated opsonophagocytosis (20, 44, 45, 46, 50, 54, 55, 62, 63, 71). Thus, we examined whether the enhanced antibody production to S. pneumoniae following immunization with anti-hFcγRI-PspA enhanced complement C3 deposition on S. pneumoniae. We measured the deposition of C3 on the surface of S. pneumoniae cells by flow cytometry using sera from WT and Tg mice immunized with PBS, rPspA, and anti-hFcγRI-PspA. The highest deposition of C3 was observed with immune sera obtained from anti-hFcγRI-PspA-immunized Tg mice (Fig. 7A). In order to eliminate the possibility of higher initial concentrations of C3 in the serum samples of anti-hFcγRI-PspA-immunized Tg mice, the experiment was repeated by heat inactivating the samples prior to incubation with S. pneumoniae, followed by the addition of 100 μl of mouse complement to every sample, thereby equalizing the amount of complement in all samples. The results obtained were similar to those of nontreated serum (data not shown).
Fig 7.
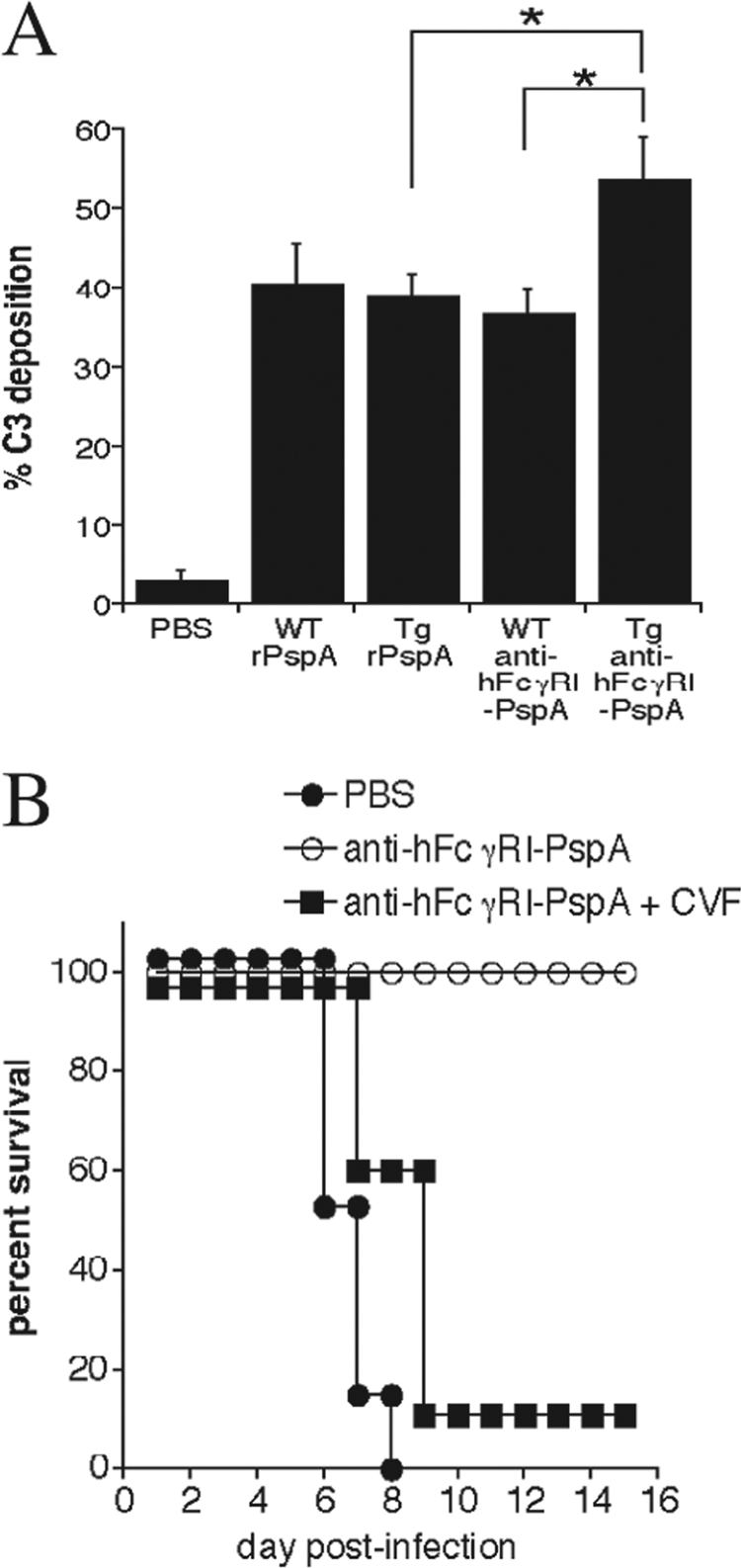
Targeting PspA to hFcγRI generates protection against S. pneumoniae, which is complement dependent. (A) Heat-inactivated sera from C57BL/6 WT and Tg mice, immunized with 25 μg of anti-hFcγRI-PspA, the rPspA equivalent to the anti-hFcγRI-PspA, or PBS, were incubated with 1 × 107 CFU of S. pneumoniae for 20 min at 4°C at a 1:2 dilution. Each bacterial sample was then incubated with 100 μl of mouse serum containing complement for 30 min. Deposition of C3 on S. pneumoniae was assessed by the addition of anti-C3 FITC monoclonal antibody followed by flow cytometry and analysis of the percentage of C3-positive bacteria (% C3) (*, P < 0.1). (B) Tg mice were immunized i.n. with PBS or 25 μg of anti-hFcγRI-PspA (day 0) followed by two boosts (days 14 and 28). One day prior to infection with S. pneumoniae (day 42), a group of hFcγRI-PspA-immunized mice received an i.p. injection of CVF (+CVF), which was repeated every 3 days thereafter. Survival of mice was monitored for at least 21 days. Five to 6 mice were used per group. The data are representative of a minimum of two independent experiments. For the anti-hFcγRI-PspA versus anti-hFcγRI-PspA +CVF group, P = 0.005.
In order to further demonstrate that complement does play a critical role in the protection generated by immunization with anti-hFcγRI-PspA in vivo, anti-hFcγRI-PspA-immunized Tg mice were treated with cobra venom factor (CVF) to deplete C3 in vivo. Upon a single administration (i.p.) of CVF, the levels of C3 in the sera of mice were reduced by 95% and remained at these levels for at least 3 days (data not shown). Consistent with a role for C3 deposition in vivo, complement (C3) depletion in anti-hFcγRI-PspA-immunized Tg mice abrogated the protection previously established against lethal S. pneumoniae challenge (Fig. 7B). Thus, these results highlight the importance of complement in clearing S. pneumoniae infection following immunization with anti-hFcγRI-PspA.
Increased bactericidal activity present in sera from anti-hFcγRI-PspA-immunized Tg mice is mediated by lactoferrin.
Published studies indicate that complement functions to protect against S. pneumoniae infection primarily through the mediation of opsonophagocytosis and not complement-mediated lysis (13, 33). Nevertheless, we examined whether increased C3 deposition enhances bactericidal activity against S. pneumoniae. To accomplish this, we incubated serum samples from anti-hFcγRI-PspA Tg mice with S. pneumoniae for 2 to 5 h at 37°C and subsequently plated bacteria on sheep blood agar plates, as noted in Materials and Methods. A reduction in bacterial viability of up to 90% was evident when sera from anti-hFcγRI-PspA-immunized Tg mice were used compared to sera obtained from PBS-immunized Tg mice (Fig. 8A). In addition, sera from Tg mice immunized with rPspA exhibited significantly less bactericidal activity than sera from anti-hFcγRI-PspA-immunized Tg mice (data not shown). The stimulation of bactericidal activity in immune serum was dependent on PspA-specific antibodies in that preincubation with rPspA abrogated the increased bactericidal activity. In addition, use of heat-inactivated (HI) immune serum also eliminated the bactericidal effect (Fig. 8A and B). This was particularly notable since complement is heat labile and since the classical complement pathway is antibody dependent. To further confirm the observed impact on bacterial viability observed above, S. pneumoniae bacteria incubated with immune serum for 4 h at 37°C were stained with Sytox green, a DNA-specific stain that permeates damaged cell membranes but not intact bacteria. These results supported the conclusion that immunization of Tg mice with anti-hFcγRI-PspA results in the production of antibody that mediates enhanced bactericidal activity. Once again, the bactericidal activity was neutralized following preincubation of the immune serum with rPspA (Fig. 8C), again indicating that PspA-specific antibody was involved.
Fig 8.

Increased bactericidal activity generated by anti-hFcγRI-PspA immunization is mediated by lactoferrin. (A) Bacterial viability was assessed by culturing 1 × 107 CFU of S. pneumoniae with immune serum as well as heat-inactivated (H.I.) sera from WT and Tg immunized mice for 2 to 5 h at 37°C at different dilutions as depicted on the x axis. Bacterial samples were then serially diluted and spotted on blood agar plates as described in Materials and Methods. (B) The effect of rPspA on neutralizing complement activity was assessed by preincubating serum with rPspA at different concentrations for 30 min before a the killing assay was performed with a 1:2 dilution of serum. (C) A second measure of bactericidal activity was also assessed using a Sytox green assay for bacterial permeability. A total of 1 × 106 CFU of S. pneumoniae was incubated with a 1:2 dilution of sera from PBS-, rPspA-, or anti-hFcγRI-PspA-immunized mice for 2 to 5 h at 37°C in the presence or absence of rPspA. Sytox green was added for a further 10-min incubation, and fluorescence/permeability was measured by flow cytometry. (D) To determine if lactoferrin could mediate bactericidal activity against the S. pneumoniae strain used in these studies in a PspA-dependent manner, organisms were incubated with medium alone or with medium containing 10 μg/ml polyclonal rabbit anti-PspA antibody for 1 h at 37°C. Following incubation, samples were washed, and either medium or human lactoferrin protein was added to each sample at concentrations indicated on the x axis. After an additional 2 h of incubation at 37°C, each sample was serially diluted and plated out on blood agar plates in order to obtain a final CFU count. (E) To determine if lactoferrin is the source of bactericidal activity observed when sera from mice immunized with the anti-hFcγRI-PspA were used, sera from unimmunized mice or anti-hFcγRI-PspA-immunized mice were incubated for 1 h at 37°C with rabbit antilactoferrin antibody (Thermo-Fisher Scientific) at concentrations indicated on the x axis. The serum-antilactoferrin mixtures were then added to 1 × 106 CFU of S. pneumoniae and incubated for 2 h at 37°C. After incubation, each S. pneumoniae sample was serially diluted and plated on blood agar plates in order to determine a final CFU count. Statistical analysis was performed where depicted (*, P < 0.1; **, P < 0.05).
Despite the above observations being consistent with a role for complement in the bactericidal activity against S. pneumoniae following immunization of Tg mice with anti-hFcγRI-PspA, other evidence suggested an alternative explanation. Unlike complement, lactoferrin can be bactericidal for S. pneumoniae (34, 59). Thus, we sought to first establish/confirm that lactoferrin was bactericidal for the S. pneumoniae strain being utilized in our studies and that this bactericidal activity was regulated by PspA. In fact, this was the case. While up to 50 μg/ml lactoferrin had no effect on bacterial numbers in the absence of anti-PspA blocking antibody, in the presence of 1 μg/ml lactoferrin and 10 μg/ml anti-PspA antibody, bacterial numbers were reduced by 73% (Fig. 8D). This number increased to 95% when the concentration of lactoferrin reached 50 μg/ml (Fig. 8D). Furthermore, the reduction in bacterial numbers observed in the presence of sera from anti-hFcγRI-PspA-immunized mice was eliminated in a dose-dependent manner by titrating in anti-lactoferrin antibody (Fig. 8E).
DISCUSSION
In these studies, we utilized an hFcγRI-targeting recombinant immunogen (anti-hFcγRI-PspA) administered i.n. as an adjuvant-independent vaccine. The vaccine elicited full protection against lethal challenge with S. pneumoniae, a common extracellular mucosal pathogen, in Tg mice capable of binding this immunogen via hFcγRI. WT mice lacking hFcγRI were not protected, and the response was similar and weak as in Tg mice given nonadjuvanted nontargeted antigen. Importantly, the hFcγRI target is expressed on Mϕ and DC in our hFcγRI Tg mouse model, as it is in humans (Fig. 2A and B). This paper represents only the second study, the first also being by this group (53), to demonstrate the generation of enhanced protective immune responses against a bacterial mucosal pathogen upon targeting a protective immunogen to FcγR after intranasal instillation. This study is also the first to use recombinant DNA technology to produce a fusion protein capable of targeting the protective immunogen specifically to hFcγRI, thereby avoiding the inhibitory Fc receptor (FcγRIIB) and enhancing protection against mucosal challenge with a clinically relevant bacterial pathogen. In addition, we accomplish this in the absence of adjuvant. We also demonstrate that the observed protection is complement dependent and that utilization of anti-hFcγRI-PspA as a mucosal immunogen enhances not only C3 deposition on S. pneumoniae but also PspA-specific antibody-dependent lactoferrin-mediated bactericidal activity against S. pneumoniae.
Previously, we established that formation of immune complexes (monoclonal antibody-inactivated F. tularensis complex) between inactivated F. tularensis plus antilipopolysaccharide (LPS) IgG2a monoclonal antibody and subsequent immunization of WT mice i.n. enhanced protection against the live vaccine strain (LVS) and, more significantly, the highly virulent strain SchuS4 of F. tularensis (53). However, the utilization of monoclonal antibody-inactivated F. tularensis targets multiple FcγR types, including FcγRIIB, an inhibitory FcγR which negatively regulates the immune response when engaged (23), including by limiting production of antibody and protection against S. pneumoniae infection (12, 17, 24). Antibody plays a critical role in the prevention of pneumococcal disease through neutralization of virulence factors such as PspA and clearance of infecting pneumococcal organisms through opsonophagocytosis (16). Therefore, we sought to generate and test a vaccine that targets the protective immunogen, PspA, to a specific human activating FcγR, hFcγRI, thereby bypassing interactions with FcγRIIB, while at the same time targeting a human FcγR in a unique hFcγRI Tg mouse model.
We also selected a uniquely beneficial antigen, PspA. Early studies on the use of PspA as a vaccine have focused on its now established ability to protect against infection with pneumococci expressing various capsular serotypes. Thus, this provides significant impetus for focusing on this particular antigen as a vaccine immunogen. In a sepsis model, subcutaneous or i.p. immunization with full-length family 1 PspA significantly increased the survival rate of mice intravenously challenged with multiple pneumococcal strains of the same PspA family (43). The protective effect of PspA immunization against otitis media has also been investigated. Rodents passively immunized with antibody for family 1 PspA and challenged with homologous pneumococcal cells through middle ear inoculation exhibited reduced inflammation and damage of the tympanic membrane (5, 7). However, although PspA is a promising vaccine candidate against pneumococcal disease, it still has limitations when used in its native form due to its limited ability to induce strong immune responses, particularly when it is used without adjuvant. Therefore, a number of studies have been conducted in which PspA has been administered with various adjuvants, such as cholera toxin and IL-12. Both adjuvants augmented IgA and systemic IgG levels, minimizing bacteremia in immunized mice (3, 73). Unfortunately, the potential toxic side effects of these adjuvants, accompanied by the lack of FDA approval, negatively impact the clinical significance of these studies. A strategy that eliminates the need for adjuvant would therefore be a significant advancement in mucosal vaccine technology, particularly when PspA is used as the protective immunogen. By targeting PspA to hFcγRI, the limited stimulatory capacity of PspA can potentially be overcome as previously shown, despite the absence of adjuvant (1, 27, 53).
Consistent with the latter statement, these studies, which do not utilize adjuvant and target PspA to hFcγRI in a hFcγRI Tg mouse model, demonstrate enhanced protection against infection with S. pneumoniae (Fig. 2 and 6). Although differential sensitivity of WT versus Tg mice to S. pneumoniae challenge was a complicating factor, the two approaches we chose to compare FcR-targeted to nontargeted immunogen clearly show the increased effectiveness of the hFcγRI-targeted immunogen. Specifically, when PBS or hFcγRI-targeted immunogen is administered i.n. to WT versus Tg mice and the mice are challenged with an equal number of bacteria, there is clearly no significant difference in survival rates between groups of WT mice immunized with PBS or anti-hFcγRI-PspA. However, there is a very clear difference between survival of these same groups of mice when the mice express hFcγRI (Tg mice) (Fig. 2). Despite this, we also verified the above observation by excluding differential susceptibility as an issue. Specifically, only Tg mice were used and immunized with PBS, rPspA, or anti-FcγRI-PspA. Also in this case, it was clearly evident that hFcγRI-targeted immunogen was superior in terms of protecting the Tg mice from subsequent challenge (Fig. 6A and B).
Protection also correlates with increased levels of S. pneumoniae-specific IgG and IgA (Fig. 4 and 6) as well as increased T cell cytokine responses in Tg mice immunized with the hFcγRI-targeted PspA (Fig. 5E). Importantly, while IgG mediates opsonophagocytosis of S. pneumoniae by phagocytic cells (2), IgA has also been shown to play a protective role against S. pneumoniae infection (22, 61). In the case of antibody responses, it is also important that we measured antibody responses to live intact S. pneumoniae bacteria, as apposed to purified PspA. This was done to avoid any potential misinterpretations resulting from PspA not being in its native conformation or not being localized within its native environment. Specifically, conformational differences between purified PspA and cell wall-bound PspA, as well as the presence of carbohydrates surrounding PspA on the bacterial surface, could have a significant impact on antibody binding and thus titers. In addition, since these studies are primarily focused on development of a vaccine against intact S. pneumoniae bacteria and, specifically, PspA localized on the bacterial surface (not purified PspA), measurement of antibody binding to live intact S. pneumoniae expressing PspA seemed most appropriate. However, it is also important that the antibody-mediated bactericidal activity generated by our anti-hFcγRI-PspA fusion protein was eliminated by adsorption with purified rPspA (Fig. 8B and C). Thus, the antibody response generated by immunization of Tg mice with anti-hFcγRI-PspA, while detected using live S. pneumoniae, is PspA specific. In addition, our antibody and cytokine data indicate the generation of a mixed Th1/Th2 like response, highlighting a potential role for enhanced CD4+ T lymphocyte responses in protection (Fig. 5). In fact, an effective adaptive immune response, including the generation of both humoral and cellular (Th) immune responses, is often necessary for resolving mucosal infections (19, 38). More specifically, in a number of studies in which murine knockout models were utilized, not only were antibodies against S. pneumoniae key for resolving infection, but also the presence of both CD4+ and CD8+ T lymphocytes was necessary for survival, even following passive immunization of mice (35, 41, 64). Lastly, the Th1 versus Th2 response plays a significant role in determining the antibody isotype response, which in turn also dictates complement and FcγR involvement in bacterial clearance (52).
Another particularly unique advantage of utilizing PspA as an immunogen involves its ability, while on the bacterial surface, to negatively regulate the activity of immune mediators and the potential to block that activity with the generation of anti-PspA antibodies as a result of effective immunization. Although the biological function of PspA is not entirely understood, S. pneumoniae virulence is believed to be related, in part, to the ability of PspA to inhibit complement activation on the bacterial cell surface (65, 72). Indeed, the importance of the complement system in host defense against S. pneumoniae infection has been extensively studied in animal models (11, 32, 55). Activation of the complement system leads to deposition of complement component C3 fragments on the surface of S. pneumoniae (66, 67). Specifically, this effect was inferred by observing, after in vitro opsonization, an increase in C3b deposition on the surface of an S. pneumoniae mutant that does not express surface-anchored PspA (56). Furthermore, sera from mice infected with S. pneumoniae capsular type 3 strain expressing family 1, clade 2, PspA protein had higher levels of circulating C3 than that from mice infected with a PspA-deficient S. pneumoniae strain (65). Consistent with the above, our study indicates that utilization of anti-hFcγRI-PspA as an immunogen serves to neutralize PspA via enhanced production of anti-PspA antibodies, thereby leading to increased complement deposition on S. pneumoniae (Fig. 7A). Similarly, PspA negatively regulates bactericidal activity against S. pneumoniae mediated by lactoferrin, which can be reversed via anti-hFcγRI-PspA immunization (Fig. 8) through the generation of high anti-S. pneumoniae (PspA) antibody titers. Thus, the immune mechanisms by which FcR-targeted immunogens provide enhanced protection against S. pneumoniae challenge likely involve increased opsonophagocytosis mediated by the binding of anti-PspA antibody to S. pneumoniae, as well as increased C3 binding and bactericidal activity mediated by lactoferrin. Importantly, the latter two mechanisms represent unique functions and a clear advantage of using PspA as a vaccine antigen against S. pneumoniae.
The specific cellular mechanisms by which FcR targeting of antigen at mucosal sites induces an enhanced immune response are under investigation in our laboratory. Two such mechanisms are, however, demonstrated in this report and include enhanced antigen (PspA) binding to APCs (Fig. 1G and H) and enhanced presentation of antigen (PspA) (Fig. 1I). Additional data from our laboratory (data not shown) also suggest that several other mechanisms are involved. These include induction of DC maturation, more rapid antigen internalization by APCs, antigen persistence within APCs, and enhanced antigen trafficking to APCs within the lungs and nasal-associated lymphoid tissue (NALT) of mice immunized with FcR-targeted immunogen.
Finally, it should be noted that the anti-hFcγRI component of the anti-hFcγRI-PspA fusion protein is humanized. This was done in anticipation of eventually utilizing this same anti-hFcγRI-targeting component in human vaccines. As would be expected when a humanized protein is injected into mice, an antihuman response to the humanized portion of the anti-hFcγRI-PspA fusion protein was observed in anti-hFcγRI-PspA-immunized mice (data not shown). Importantly, such a response does have the potential to limit the function of the anti-hFcγRI-PspA fusion protein upon repeated administration in the mouse model. Despite this potential limitation, however, enhanced protection was observed. Furthermore, any potential impact of the antihuman response on the anti-hFcγRI-PspA function in mice would likely produce an underestimate of the effectiveness of this vaccine in humans. Specifically, if humans were immunized with this humanized anti-hFcγRI-PspA fusion protein, humanization would eliminate or minimize such deleterious responses.
In conclusion, by utilizing recombinant DNA technology, we have generated a fusion protein that targets the protective S. pneumoniae immunogen PspA to hFcγRI in a hFcγRI Tg mouse model. Intranasal immunization of Tg mice expressing this receptor with anti-hFcγRI-PspA generates robust cellular and humoral immune responses and enhances protection against lethal S. pneumoniae challenge compared to immunization with nontargeted rPspA. This protection is complement dependent. Furthermore, enhanced C3 deposition as well as lactoferrin-mediated bactericidal activity is observed. These results, combined with our previous study demonstrating enhanced protection against the intracellular bacterium F. tularensis when an FcR-targeted immunogen was administered i.n., provide strong evidence that targeting protective immunogens to FcγR i.n. has significant potential to eliminate the need for mucosal adjuvants while also providing an extremely versatile mucosal vaccine strategy against a wide range of pathogens. In addition, these studies provide additional evidence that utilizing PspA as an immunogen against S. pneumoniae provides unique advantages in that it does not just generate antibody that can serve to bind to and opsonize S. pneumoniae organisms but can also generate antibody that essentially neutralizes the inhibitory activities of PspA, thereby allowing increased C3 deposition and lactoferrin-mediated killing of S. pneumoniae organisms.
ACKNOWLEDGMENTS
These studies were funded by grants from the National Institutes of Health (1R01AI076408 and 1R21AI065476).
The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
We thank Yili Lin for her flow cytometry expertise as well as Kristen A. Porter and Carlos de Noronha for their assistance in figure design.
Footnotes
Published ahead of print 12 December 2011
REFERENCES
- 1. Adamova E, et al. 2005. Enhanced antigen-specific antibody and cytokine responses when targeting antigen to human FcGAMMA receptor type I using an anti-human FcGAMMA receptor type I-streptavidin fusion protein in an adjuvant-free system. Immunol. Invest. 34:417–429 [DOI] [PubMed] [Google Scholar]
- 2. Alonso De Velasco E, et al. 1995. Anti-polysaccharide immunoglobulin isotype levels and opsonic activity of antisera: relationships with protection against Streptococcus pneumoniae infection in mice. J. Infect. Dis. 172:562–565 [DOI] [PubMed] [Google Scholar]
- 3. Arulanandam BP, Lynch JM, Briles DE, Hollingshead S, Metzger DW. 2001. Intranasal vaccination with pneumococcal surface protein A and interleukin-12 augments antibody-mediated opsonization and protective immunity against Streptococcus pneumoniae infection. Infect. Immun. 69:6718–6724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boruchov AM, et al. 2005. Activating and inhibitory IgG Fc receptors on human DCs mediate opposing functions. J. Clin. Invest. 115:2914–2923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Briles DE, et al. 2000. Intranasal immunization of mice with a mixture of the pneumococcal proteins PsaA and PspA is highly protective against nasopharyngeal carriage of Streptococcus pneumoniae. Infect. Immun. 68:796–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Briles DE, Crain MJ, Gray BM, Forman C, Yother J. 1992. Strong association between capsular type and virulence for mice among human isolates of Streptococcus pneumoniae. Infect. Immun. 60:111–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Briles DE, et al. 2000. The potential to use PspA and other pneumococcal proteins to elicit protection against pneumococcal infection. Vaccine 18:1707–1711 [DOI] [PubMed] [Google Scholar]
- 8. Briles DE, et al. 2000. Immunization of humans with recombinant pneumococcal surface protein A (rPspA) elicits antibodies that passively protect mice from fatal infection with Streptococcus pneumoniae bearing heterologous PspA. J. Infect. Dis. 182:1694–1701 [DOI] [PubMed] [Google Scholar]
- 9. Briles DE, Jonsdottir I, Hollingshead SK. 2008. Animal models in invasive pneumococcal disease. In Siber GR, Klugman KP, Makela PH. (ed), Pneumococcal vaccines: impact of the conjugate vaccine. ASM Press, Washington, DC [Google Scholar]
- 10. Briles DE, et al. 1996. Systemic and mucosal protective immunity to pneumococcal surface protein A. Ann. N. Y. Acad. Sci. 797:118–126 [DOI] [PubMed] [Google Scholar]
- 11. Brown JS, et al. 2002. The classical pathway is the dominant complement pathway required for innate immunity to Streptococcus pneumoniae infection in mice. Proc. Natl. Acad. Sci. U. S. A. 99:16969–16974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brownlie RJ, et al. 2008. Distinct cell-specific control of autoimmunity and infection by FcγRIIb. J. Exp. Med. 205:883–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bruyn GA, Zegers BJ, van Furth R. 1992. Mechanisms of host defense against infection with Streptococcus pneumoniae. Clin. Infect. Dis. 14:251–262 [DOI] [PubMed] [Google Scholar]
- 14. Campos IB, et al. 2008. Nasal immunization of mice with Lactobacillus casei expressing the pneumococcal surface protein A: induction of antibodies, complement deposition and partial protection against Streptococcus pneumoniae challenge. Microbes Infect. 10:481–488 [DOI] [PubMed] [Google Scholar]
- 15. Chargelegue D, et al. 2005. Highly immunogenic and protective recombinant vaccine candidate expressed in transgenic plants. Infect. Immun. 73:5915–5922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chiavolini D, Pozzi G, Ricci S. 2008. Animal models of Streptococcus pneumoniae disease. Clin. Microbiol. Rev. 21:666–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Clatworthy MR, Smith KG. 2004. FcγRIIb balances efficient pathogen clearance and the cytokine-mediated consequences of sepsis. J. Exp. Med. 199:717–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Colino J, et al. 2009. Parameters underlying distinct T cell-dependent polysaccharide-specific IgG responses to an intact gram-positive bacterium versus a soluble conjugate vaccine. J. Immunol. 183:1551–1559 [DOI] [PubMed] [Google Scholar]
- 19. Cooper AM, et al. 2002. Mice lacking bioactive IL-12 can generate protective, antigen-specific cellular responses to mycobacterial infection only if the IL-12 p40 subunit is present. J. Immunol. 168:1322–1327 [DOI] [PubMed] [Google Scholar]
- 20. Darrieux M, et al. 2007. Fusion proteins containing family 1 and family 2 PspA fragments elicit protection against Streptococcus pneumoniae that correlates with antibody-mediated enhancement of complement deposition. Infect. Immun. 75:5930–5938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fedson DS. 1999. The clinical effectiveness of pneumococcal vaccination: a brief review. Vaccine 17(Suppl 1):S85–S90 [DOI] [PubMed] [Google Scholar]
- 22. Fukuyama Y, et al. 2010. Secretory-IgA antibodies play an important role in the immunity to Streptococcus pneumoniae. J. Immunol. 185:1755–1762 [DOI] [PubMed] [Google Scholar]
- 23. Gerber JS, Mosser DM. 2001. Stimulatory and inhibitory signals originating from the macrophage Fcγ receptors. Microbes Infect. 3:131–139 [DOI] [PubMed] [Google Scholar]
- 24. Gessner JE, Heiken H, Tamm A, Schmidt RE. 1998. The IgG Fc receptor family. Ann. Hematol. 76:231–248 [DOI] [PubMed] [Google Scholar]
- 25. Gosselin EJ, Bitsaktsis C, Li Y, Iglesias BV. 2009. Fc receptor-targeted mucosal vaccination as a novel strategy for the generation of enhanced immunity against mucosal and non-mucosal pathogens. Arch. Immunol. Ther. Exp. (Warsz.) 57:311–323 [DOI] [PubMed] [Google Scholar]
- 26. Gosselin EJ, et al. 1992. Enhanced antigen presentation using human Fc gamma receptor (monocyte/macrophage)-specific immunogens. J. Immunol. 149:3477–3481 [PubMed] [Google Scholar]
- 27. Guyre PM, et al. 1997. Increased potency of Fc-receptor-targeted antigens. Cancer Immunol. Immunother. 45:146–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Heijnen IA, et al. 1996. Antigen targeting to myeloid-specific human Fc gamma RI/CD64 triggers enhanced antibody responses in transgenic mice. J. Clin. Invest. 97:331–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Henrichsen J. 1995. Six newly recognized types of Streptococcus pneumoniae. J. Clin. Microbiol. 33:2759–2762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hollingshead SK, et al. 2006. Pneumococcal surface protein A (PspA) family distribution among clinical isolates from adults over 50 years of age collected in seven countries. J. Med. Microbiol. 55:215–221 [DOI] [PubMed] [Google Scholar]
- 31. Hollingshead SK, Becker R, Briles DE. 2000. Diversity of PspA: mosaic genes and evidence for past recombination in Streptococcus pneumoniae. Infect. Immun. 68:5889–5900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hosea SW, Brown EJ, Frank MM. 1980. The critical role of complement in experimental pneumococcal sepsis. J. Infect. Dis. 142:903–909 [DOI] [PubMed] [Google Scholar]
- 33. Jarva H, Jokiranta TS, Wurzner R, Meri S. 2003. Complement resistance mechanisms of streptococci. Mol. Immunol. 40:95–107 [DOI] [PubMed] [Google Scholar]
- 34. Jedrzejas MJ. 2006. Unveiling molecular mechanisms of pneumococcal surface protein A interactions with antibodies and lactoferrin. Clin. Chim Acta 367:1–10 [DOI] [PubMed] [Google Scholar]
- 35. Kadioglu A, Coward W, Colston MJ, Hewitt CR, Andrew PW. 2004. CD4-T-lymphocyte interactions with pneumolysin and pneumococci suggest a crucial protective role in the host response to pneumococcal infection. Infect. Immun. 72:2689–2697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Keler T, et al. 2000. Targeting weak antigens to CD64 elicits potent humoral responses in human CD64 transgenic mice. J. Immunol. 165:6738–6742 [DOI] [PubMed] [Google Scholar]
- 37. Klein DL. 1999. Pneumococcal disease and the role of conjugate vaccines. Microb. Drug Resist. 5:147–157 [DOI] [PubMed] [Google Scholar]
- 38. Klinman DM, Conover J, Coban C. 1999. Repeated administration of synthetic oligodeoxynucleotides expressing CpG motifs provides long-term protection against bacterial infection. Infect. Immun. 67:5658–5663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kuwata H, Yip TT, Yip CL, Tomita M, Hutchens TW. 1998. Bactericidal domain of lactoferrin: detection, quantitation, and characterization of lactoferricin in serum by SELDI affinity mass spectrometry. Biochem. Biophys. Res. Commun. 245:764–773 [DOI] [PubMed] [Google Scholar]
- 40. Lin J, et al. 2006. Validation of a multiplex pneumococcal serotyping assay with clinical samples. J. Clin. Microbiol. 44:383–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Malley R, et al. 2003. Recognition of pneumolysin by Toll-like receptor 4 confers resistance to pneumococcal infection. Proc. Natl. Acad. Sci. U. S. A. 100:1966–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Matsui T, et al. 2006. CD64 on neutrophils is a sensitive and specific marker for detection of infection in patients with rheumatoid arthritis. J. Rheumatol. 33:2416–2424 [PubMed] [Google Scholar]
- 43. McDaniel LS, McDaniel DO, Hollingshead SK, Briles DE. 1998. Comparison of the PspA sequence from Streptococcus pneumoniae EF5668 to the previously identified PspA sequence from strain Rx1 and ability of PspA from EF5668 to elicit protection against pneumococci of different capsular types. Infect. Immun. 66:4748–4754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mold C, Du Clos TW, Nakayama S, Edwards KM, Gewurz H. 1982. C-reactive protein reactivity with complement and effects on phagocytosis. Ann. N. Y. Acad. Sci. 389:251–262 [DOI] [PubMed] [Google Scholar]
- 45. Mold C, Edwards KM, Gewurz H. 1982. Effect of C-reactive protein on the complement-mediated stimulated of human neutrophils by Streptococcus pneumoniae serotypes 3 and 6. Infect. Immun. 37:987–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Moreno AT, et al. 2010. Immunization of mice with single PspA fragments induces antibodies capable of mediating complement deposition on different pneumococcal strains and cross-protection. Clin. Vaccine Immunol. 17:439–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Musher DM, Watson DA, Baughn RE. 1990. Does naturally acquired IgG antibody to cell wall polysaccharide protect human subjects against pneumococcal infection? J. Infect. Dis. 161:736–740 [DOI] [PubMed] [Google Scholar]
- 48. Neefjes JJ, Momburg F. 1993. Cell biology of antigen presentation. Curr. Opin. Immunol. 5:27–34 [DOI] [PubMed] [Google Scholar]
- 49. O'Brien KL, et al. 2009. Burden of disease caused by Streptococcus pneumoniae in children younger than 5 years: global estimates. Lancet 374:893–902 [DOI] [PubMed] [Google Scholar]
- 50. Ochs MM, et al. 2008. Vaccine-induced human antibodies to PspA augment complement C3 deposition on Streptococcus pneumoniae. Microb. Pathog. 44:204–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Plitnick LM, et al. 2001. Lipoteichoic acid inhibits interleukin-2 (IL-2) function by direct binding to IL-2. Clin. Diagn. Lab Immunol. 8:972–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ravetch JV. 1994. Fc receptors: rubor redux. Cell 78:553–560 [DOI] [PubMed] [Google Scholar]
- 53. Rawool DB, et al. 2008. Utilization of Fc receptors as a mucosal vaccine strategy against an intracellular bacterium, Francisella tularensis. J. Immunol. 180:5548–5557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ren B, et al. 2004. The virulence function of Streptococcus pneumoniae surface protein A involves inhibition of complement activation and impairment of complement receptor-mediated protection. J. Immunol. 173:7506–7512 [DOI] [PubMed] [Google Scholar]
- 55. Ren B, Szalai AJ, Hollingshead SK, Briles DE. 2004. Effects of PspA and antibodies to PspA on activation and deposition of complement on the pneumococcal surface. Infect. Immun. 72:114–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ren B, Szalai AJ, Thomas O, Hollingshead SK, Briles DE. 2003. Both family 1 and family 2 PspA proteins can inhibit complement deposition and confer virulence to a capsular serotype 3 strain of Streptococcus pneumoniae. Infect. Immun. 71:75–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Roche H, Ren B, McDaniel LS, Hakansson A, Briles DE. 2003. Relative roles of genetic background and variation in PspA in the ability of antibodies to PspA to protect against capsular type 3 and 4 strains of Streptococcus pneumoniae. Infect. Immun. 71:4498–4505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Senkovich O, et al. 2007. Structure of a complex of human lactoferrin N-lobe with pneumococcal surface protein a provides insight into microbial defense mechanism. J. Mol. Biol. 370:701–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Shaper M, Hollingshead SK, Benjamin WH, Jr, Briles DE. 2004. PspA protects Streptococcus pneumoniae from killing by apolactoferrin, and antibody to PspA enhances killing of pneumococci by apolactoferrin [corrected]. Infect. Immun. 72:5031–5040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Shen L, Guyre PM, Fanger MW. 1987. Polymorphonuclear leukocyte function triggered through the high affinity Fc receptor for monomeric IgG. J. Immunol. 139:534–538 [PubMed] [Google Scholar]
- 61. Sun K, Johansen FE, Eckmann L, Metzger DW. 2004. An important role for polymeric Ig receptor-mediated transport of IgA in protection against Streptococcus pneumoniae nasopharyngeal carriage. J. Immunol. 173:4576–4581 [DOI] [PubMed] [Google Scholar]
- 62. Suresh MV, Singh SK, Ferguson DA, Jr, Agrawal A. 2006. Role of the property of C-reactive protein to activate the classical pathway of complement in protecting mice from pneumococcal infection. J. Immunol. 176:4369–4374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Szalai AJ, Briles DE, Volanakis JE. 1996. Role of complement in C-reactive-protein-mediated protection of mice from Streptococcus pneumoniae. Infect. Immun. 64:4850–4853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Tian H, Groner A, Boes M, Pirofski LA. 2007. Pneumococcal capsular polysaccharide vaccine-mediated protection against serotype 3 Streptococcus pneumoniae in immunodeficient mice. Infect. Immun. 75:1643–1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tu AH, Fulgham RL, McCrory MA, Briles DE, Szalai AJ. 1999. Pneumococcal surface protein A inhibits complement activation by Streptococcus pneumoniae. Infect. Immun. 67:4720–4724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Walport MJ. 2001. Complement. First of two parts. N. Engl. J. Med. 344:1058–1066 [DOI] [PubMed] [Google Scholar]
- 67. Walport MJ. 2001. Complement. Second of two parts. N. Engl. J. Med. 344:1140–1144 [DOI] [PubMed] [Google Scholar]
- 68. Walsh MC, et al. 2003. A two-component modular approach for enhancing T-cell activation utilizing a unique anti-FcγRI-streptavidin construct and microspheres coated with biotinylated-antigen. Biomol. Eng. 20:21–33 [DOI] [PubMed] [Google Scholar]
- 69. White DM, et al. 2011. Rapid immune responses to a botulinum neurotoxin Hc subunit vaccine through in vivo targeting to antigen-presenting cells. Infect. Immun. 79:3388–3396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Williams BG, Gouws E, Boschi-Pinto C, Bryce J, Dye C. 2002. Estimates of world-wide distribution of child deaths from acute respiratory infections. Lancet Infect. Dis. 2:25–32 [DOI] [PubMed] [Google Scholar]
- 71. Winkelstein JA. 1984. Complement and the host's defense against the pneumococcus. Crit. Rev. Microbiol. 11:187–208 [DOI] [PubMed] [Google Scholar]
- 72. Wu HY, Nahm MH, Guo Y, Russell MW, Briles DE. 1997. Intranasal immunization of mice with PspA (pneumococcal surface protein A) can prevent intranasal carriage, pulmonary infection, and sepsis with Streptococcus pneumoniae. J. Infect. Dis. 175:839–846 [DOI] [PubMed] [Google Scholar]
- 73. Yamamoto S, et al. 1997. A nontoxic mutant of cholera toxin elicits Th2-type responses for enhanced mucosal immunity. Proc. Natl. Acad. Sci. U. S. A. 94:5267–5272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yother J, Forman C, Gray BM, Briles DE. 1982. Protection of mice from infection with Streptococcus pneumoniae by anti-phosphocholine antibody. Infect. Immun. 36:184–188 [DOI] [PMC free article] [PubMed] [Google Scholar]