

Abstract
Interleukin-17 (IL-17) is a key factor in T helper type 17 (Th17) lineage host responses and plays critical roles in immunological control of a variety of infectious diseases. Although Legionella pneumophila, an intracellular bacterium found widely in the environment, often causes a serious and life-threatening pneumonia in humans, the contribution of IL-17 to immune function during Legionella pneumonia is unknown. In the present study, we used an experimental Legionella pneumonia infection to clarify the role of IL-17 in the resulting immune response. We observed robust production of pulmonary IL-17A and IL-17F (IL-17A/F), peaking on day 1 and declining thereafter. Upregulated production of tumor necrosis factor alpha (TNF-α), IL-6, and IL-1β, but not monocyte chemotactic protein 1 (MCP-1), was observed in Legionella-infected bone marrow-derived macrophages from BALB/c mice that had been stimulated with IL-17A or IL-17F. A significant decrease in the production of proinflammatory cytokines IL-6 and TNF-α was observed in IL-17A/F-deficient mice (BALB/c background) infected with L. pneumophila. Moreover, we found impaired neutrophil migration and lower numbers of chemokines (KC, LIX, and MIP-2) in IL-17A/F-deficient mice. IL-17A/F-deficient mice also eliminated L. pneumophila more slowly and were less likely to survive a lethal challenge. These results demonstrate that IL-17A/F plays a critical role in L. pneumophila pneumonia, probably through induction of proinflammatory cytokines and accumulation of neutrophils at the infection site.
INTRODUCTION
Interleukin-17 (IL-17) has recently received considerable attention because of its important role in cross talk between the innate and adaptive immune systems (9). It contributes to host responses to a variety of immunological and inflammatory disorders, such as collagen disease (4), allograft rejection (45), tumor (2), allergy (21), and infectious diseases resulting from infection with Klebsiella pneumoniae, Bordetella pertussis, Streptococcus pneumoniae, and Candida albicans (29). IL-17 is secreted by a variety of cells, including T helper type 17 (Th17) cells, natural killer cells, and γδ T cells, thereby inducing Th17-type host responses characterized by production of proinflammatory cytokines and neutrophil recruitment (46). Specifically, IL-17 upregulates several chemokines, such as macrophage inflammatory protein 1 (MIP-1), MIP-2, and monocyte chemoattractant protein 1 (MCP-1), which, in turn, attract inflammatory cells to the site of infection. IL-17 also promotes granulopoiesis via the induction of granulocyte colony-stimulating factor (G-CSF) and stem cell factor (SCF) and potentiates neutrophilic cytotoxicity and phagocytosis (35). It is reported to enhance the function and survival of recruited macrophages in the airway (36).
Because IL-17 is a strong inducer of neutrophil-chemotactic factors, it has been suggested to play an important role in the pathogenesis, severity, and outcome in pneumonia caused by infection with Legionella pneumophila. L. pneumophila is a ubiquitous, Gram-negative facultative intracellular bacillus that frequently causes a life-threatening pneumonia, especially in immunocompromised individuals (23, 32). Legionella infections usually result from inhalation of contaminated aerosols from environmental sources. Once the bacteria are in the lungs, they predominantly infect and multiply within monocytes and macrophages (13, 24, 28). Mortality rates of up to 50% have been reported, illustrating the fact that Legionella pneumonia remains a challenging infectious disease (8, 30, 41).
Cellular immunity and Th1-type cytokine responses are believed to be integral to defense mechanisms against Legionella species, as in cases of other intracellular bacterial infections. Th1-type cytokines such as gamma interferon (IFN-γ), tumor necrosis factor alpha (TNF-α), and IL-12, play a crucial role in defense against Legionella infection (7, 10, 39). However, the roles of IL-17 in pathogenesis of, and host defenses against, Legionella pneumonia still require elucidation.
In the present study, we examined the contribution of IL-17 to pathogenesis and lethal sensitivity of L. pneumophila pneumonia in mice. We focused our efforts on IL-17A and IL-17F (IL-17A/F), two main factors in the IL-17 protein family that are known to play an important role in antimicrobial host defenses and immunological responses to a variety of infections, including fungi, bacteria, viruses, and parasites (6, 15, 16, 26, 29). IL-17A and IL-17F appear to bind the same receptor complexes, comprising IL-17 receptor A and IL-17 receptor C (42, 54), and therefore appear to have similar biological functions (16). We measured quantities of these chemokines in the lungs of mice that had been infected with Legionella pneumonia and examined stimulation of proinflammatory cytokines in response to various levels of IL-17A and IL-17F. We also compared proinflammatory cytokine induction levels, leukocyte numbers, gene expression of CXC chemokines, and total numbers of bacteria in the lungs of wild-type and IL-17A/F-deficient mice (BALB/c background). Finally, we investigated whether survival was lower in IL-17A/F-deficient mice with Legionella pneumonia. Cumulatively, our data strongly indicate that IL-17 is a vital part of the host immune response against Legionella infections.
MATERIALS AND METHODS
Animals.
Specific-pathogen-free 5- to 8-week-old male BALB/c (Charles River Laboratories, Kanagawa, Japan), C57BL/6J (Charles River Laboratories), and A/J (Sankyo Laboratory, Tokyo, Japan) mice were quarantined for 1 week after reception. IL-17A/IL-17F double knockout mice (Il17a−/− Il17f−/−) on a BALB/c genetic background were previously established at the Institute of Medical Science, University of Tokyo (12, 27). All the studies were performed in mice of the BALB/c background. All mice were housed under specific-pathogen-free conditions within the animal care facility at Toho University School of Medicine (Tokyo, Japan) until the day of sacrifice. Experiments were conducted according to our institution's ethical guidelines for animal experiments and safety guidelines for gene manipulation experiments. Animal protocols were approved by the institutional animal care and use committee (approval number 10-52-54).
L. pneumophila inoculation.
Clinical isolates of L. pneumophila Suzuki, a serogroup 1 strain stocked at Toho University Hospital (38), were prepared as previously reported (37). Animals were anesthetized intramuscularly with ketamine at 7 mg/kg of body weight and xylazine at 15 mg/kg. Their tracheas were exposed, and 30 μl of bacterial suspension was administered via a sterile 26-gauge needle. Skin incisions were closed with surgical staples.
Preparation of BM-Mϕ and their stimulation with L. pneumophila and recombinant IL-17.
Bone marrow-derived macrophages (BM-Mϕ) were isolated from the femurs of male BALB/c, C57BL/6J, and A/J mice as previously described (3). The cells were cultured in 96-well tissue culture plates (105 cells per well) and used for BM-Mϕ experiments. After the medium had been changed, BM-Mϕ were infected with L. pneumophila at the indicated multiplicity of infection (MOI) for 2 h. At the end of the infection period (time zero), nonphagocytosed and nonadherent bacteria were removed by two washes with fresh medium. The cells were subsequently incubated at 37°C in a humidified atmosphere containing 5% CO2. At the indicated time points, culture supernatants were collected, and the infected macrophages were lysed as previously described (51).
So that we could investigate the effects of elevated IL-17A and IL-17F levels during Legionella pneumonia on other inflammatory cytokines, we incubated murine BM-Mϕ for 24 h with increasing doses of IL-17A or IL-17F (1 pg/ml to 10 ng/ml) in the presence or absence of L. pneumophila infection (MOI of 1.0). Culture supernatants of five different samples were then collected and centrifuged at 10,000 × g for 5 min. Samples were stored at −40°C until used for enzyme-linked immunosorbent assays (ELISAs) (see below). Recombinant, endotoxin-free mouse IL-17A and IL-17F were obtained from R&D Systems (Minneapolis, MN).
BAL and collection of BALF.
At 1, 2, 4, and 7 days after infection, mice were sacrificed by CO2 asphyxia. Their tracheas were exposed and intubated using a polyethylene catheter with a 1.7-mm outer diameter. Bronchoalveolar lavage (BAL) was performed with 3 ml of phosphate-buffered saline (PBS) containing a complete protease inhibitor cocktail tablet (Roche, Indianapolis, IN). BAL fluid (BALF) samples were pooled for each animal. Leukocyte numbers were counted with a hemocytometer. Differential counts were performed on cytospin preparations (Cytospin 3 centrifuge; Shandon Scientific, Ltd., Astmoor, United Kingdom) stained with May-Giemsa stain. The remaining BALF was stored at −20°C until subsequent use.
Lung harvests for analysis.
After the mice had been sacrificed, their chests were opened, and the pulmonary vasculature was perfused with 1 ml of saline via the right ventricle. Whole lungs were removed, and left lungs were homogenized under a vented hood with a homogenizer (IKA Japan K.K., Osaka, Japan) in 1 ml of water containing a complete protease inhibitor cocktail tablet and 0.2 mM phenylmethanesulfonyl fluoride. Portions of homogenates (10 μl) were inoculated onto buffered charcoal-yeast extract agar supplemented with α-ketoglutaric acid (BCYEα) after serial 1:10 dilution in saline. The remaining homogenates (900 μl) were exposed to cell lysis buffer (0.5% Triton X-100, 150 mM NaCl, and 15 mM Tris base) for 30 min at 4°C and then centrifuged at 10,000 × g for 20 min. Supernatants were collected and stored at −20°C until use. Right lungs were treated with RNAlater (Ambion, Austin, TX) and stored at 4°C until use in evaluation of mRNA expression (see below).
Cytokine/chemokine measurements.
Cytokines and chemokines were measured with IL-17A, IL-17F, IL-6, TNF-α, MCP-1, and MIP-2 mouse ELISA kits (R&D Systems), according to the manufacturer's protocols.
Gene expression analysis.
Total RNA was isolated from lungs using RiboPure kits (Ambion), according to the manufacturer's instructions. Turbo DNA-free kits (Ambion) were used to treat RNA with DNase, after which it was reverse transcribed using a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). Data analysis utilizing the SYBR green real-time reverse transcription-PCR (RT-PCR) technique was performed on an ABI Prism 7000 sequence detector system (Applied Biosystems). We used the following PCR primers: keratinocyte-derived chemokine (KC), 5′-GCTGGGATTCACCTCAAGAA-3′ (forward) and 5′-TCTCCGTTACTTGGGGACAC-3′ (reverse); MIP-2, 5′-CGCCCAGACAGAAGTCATAG-3′ (forward) and 5′-TCCTCCTTTCCAGGTCAGTTA-3′ (reverse); LIX, 5′-GGTCCACAGTGCCCTACG-3′ (forward) and 5′-GCGAGTGCATTCCGCTTA-3′ (reverse); IL-12p35, 5′-CACCCTTGCCCTCCTAAACC-3′ (forward) and 5′-CACCTGGCAGGTCCAGAGA-3′ (reverse); IFN-γ, 5′-GGATATCTGGAGGAACTGGCAA-3′ (forward) and 5′-TGATGGCCTGATTGTCTTTCAA-3′ (reverse); IL-4, 5′-CTCATGGAGCTGCAGAGACTCTT-3′ (forward) and 5′-CATTCATGGTGCAGCTTATCGA-3′ (reverse); β-actin, 5′-AGAGGGAAATCGTGCGTGAC (forward) and 5′-CAATAGTGATGACCTGGCCGT (reverse) (31, 33, 49). Relative fold changes in transcript levels were calculated using the 2−ΔΔCT (where CT is threshold cycle) method (22) using the housekeeping gene β-actin as a reference standard for the amount loaded and the quality of the cDNA.
Statistical analysis.
All values are expressed as means ± standard error (SE). Data were analyzed with one-way analyses of variance (ANOVAs), t tests, and chi-square tests. Survival curves were constructed using the Kaplan-Meier method and were analyzed by log rank tests. Significance was defined as a P value of <0.05.
RESULTS
Production of IL-17A and IL-17F during Legionella pneumonia.
Levels of IL-17A and IL-17F in lung tissue of BALB/c mice peaked 1 day after inoculation of L. pneumophila and then rapidly declined to baseline values over the next 6 days (Fig. 1).
Fig 1.

Production of IL-17A and IL-17F in the lung during L. pneumophila pneumonia in a mouse model. IL-17A (A) and IL-17F (B) levels are shown as a function of time after the inoculation of approximately 105 CFU of L. pneumophila. All bars indicate mean ± SE (n = 6/group). *, P < 0.05 in comparison with a negative control. Similar results were obtained across three repeat experiments.
Effects of IL-17A and IL-17F on production of proinflammatory cytokines in macrophages.
As shown in Fig. 2, the addition of IL-17A or IL-17F greatly increased Legionella-induced production of IL-6, TNF-α, and IL-1β, from 3- to over 10-fold by cultured macrophages from BALB/c mice. These cytokines were maximally induced by different optimal concentrations of IL-17A and IL-17F though increases were significantly different from baseline levels at all doses (1 pg to 10 ng). Similar results in IL-6 and TNF-α production were observed in C57BL/6J and A/J mice (see Fig. S1 in the supplemental material). On the other hand, the proinflammatory mediator response in the uninfected cells was only slightly augmented by stimulation with either IL-17A or IL-17F.
Fig 2.

Stimulation of macrophage (BALB/c) proinflammatory cytokines, as indicated on the figure, by IL-17A and IL-17F at 24 h after infection with L. pneumophila (106 CFU/ml; MOI of 1.0). All bars indicate mean ± SE (n = 5/group). *, P < 0.01 in a comparison with the infection-only group; **, P < 0.05 in a comparison between IL-17A and IL-17F. The results are representative of three independent experiments.
Proinflammatory cytokine production in the lungs of IL-17A/F-deficient mice infected with Legionella.
In the lungs of wild-type BALB/c mice infected with Legionella, there was a sharp increase in IL-6, TNF-α, and IL-1β levels on day 2 (Fig. 3A to C). In contrast, IL-17A/F-deficient mice (BALB/c background) either had significantly lower peaks (TNF-α) (Fig. 3B) or no peak at all (IL-6) (Fig. 3A) on day 2. Corresponding to the data generated by our BM-Mϕ experiments, no significant differences in MCP-1 production were observed between wild-type and IL-17A/F-deficient mice (Fig. 3D).
Fig 3.

Proinflammatory cytokine production in the lungs of wild-type and IL-17A/F-deficient mice (BALB/c background) during the week following infection with L. pneumophila. The production of IL-6, TNF-α, IL-1β, and MCP-1 proteins was examined at 1, 2, 4, and 7 days after infection with L. pneumophila (105 CFU). All bars indicate mean ± SE (n = 6/group). *, P < 0.05 in comparisons between Il17a−/− Il17f−/− (A/F*) and wild-type (WT) mice. Results are representative of two independent experiments.
Leukocyte accumulation and gene expression of chemokines and cytokines.
In the week following infection with Legionella, wild-type and IL-17A/F-deficient mice (BALB/c background) had similar BALF total cell numbers (Fig. 4A). However, there were significant differences in proportions of specific inflammatory cells. Specifically, on day 2, neutrophils, macrophages, and lymphocytes constituted 71.8, 25.9, and 2.3% of the cells in wild-type BALF and 47.5, 48.3, and 4.2% of cells in BALF of IL-17A/F-deficient mice, respectively.
Fig 4.

Leukocyte accumulation and gene expression of CXC chemokines in lungs of BALB/c mice infected with Legionella. (A) Total cell numbers and proportions of different inflammatory cells in the BALF of wild-type and IL-17A/F-deficient (A/F*) mice (BALB/c background) at 1, 2, 4, and 7 days after infection with 105 CFU of L. pneumophila. *, P < 0.05 for a comparison between the neutrophil numbers of Il17a−/− Il17f−/− (A/F*) and wild-type (WT) mice. (B) Expression of the chemokines CXCL-1 (KC), CXCL-5 (LIX), CXCL-2 (MIP-2), IL-12p35, IFN-γ, and IL-4 in whole-lung tissue of mice at 48 h after infection with L. pneumophila. Data are expressed as fold increase. *, P < 0.05 for a comparison between Il17a−/− Il17f−/− (A/F*) and wild-type (WT) mice. Bars indicate mean ± SE (n = 5/group). Results are representative of three independent experiments.
Correspondingly, gene expression of the pulmonary CXC chemokines KC, LIX, and MIP-2 were lower in IL-17A/F-deficient mice than in wild-type mice (Fig. 4B). In addition, significantly lower MIP-2 levels were observed in the lungs of IL-17A/F-deficient mice (Fig. 4C). Wild-type and IL-17A/F-deficient mice did not have significantly different levels of IL-12, IFN-γ, or IL-4 gene expression (Fig. 4B).
Bacterial numbers in mice challenged with sublethal doses of L. pneumophila.
Pulmonary bacterial burdens were similar in wild-type and IL-17A/F-deficient mice (BALB/c background) on days 1, 2, and 4 after infection with L. pneumophila (105 CFU) (Fig. 5). However, on day 7, IL-17A/F-deficient mice had approximately quadruple the number of bacteria found in wild-type mice.
Fig 5.
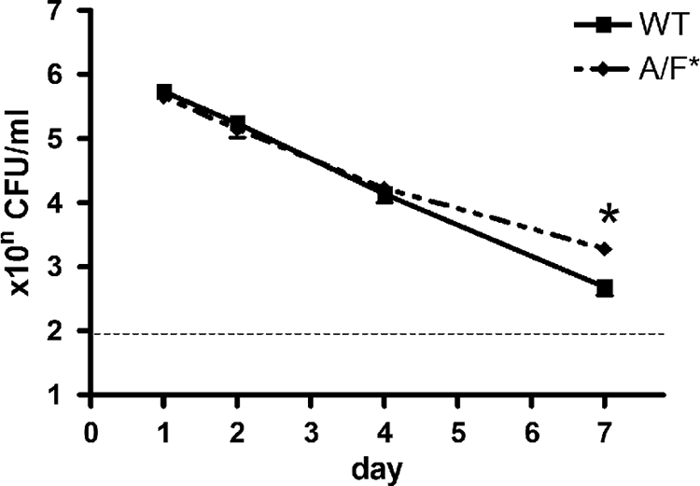
Numbers of bacteria in the lungs of wild-type and IL-17A/F-deficient mice (BALB/c background) infected with L. pneumophila. Bacteria were counted at 1, 2, 4, and 7 days after inoculation of 105 CFU of L. pneumophila. *, P < 0.01 for a comparison between Il17a−/− Il17f−/− (A/F*) and wild-type (WT) mice. Bars indicate mean ± SE (n = 6/group). Results are representative of two independent experiments.
Survival of mice challenged with lethal doses of L. pneumophila.
When challenged with Legionella (∼107 CFU), wild-type mice had significantly higher survival rates than IL-17A/F-deficient mice (BALB/c background) (69% versus 38% at day 21; P < 0.05) (Fig. 6). These results indicate that IL-17A/F activity is critical in preventing lethality of infection with Legionella pneumonia.
Fig 6.
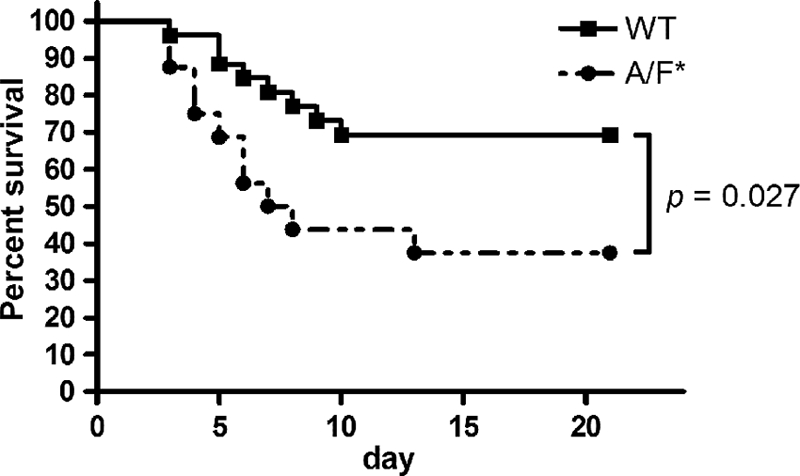
Survival curves for wild-type (WT; n = 26) and IL-17A/F-deficient (A/F*; n = 16) mice (BALB/c background) infected with L. pneumophila. Results are representative of two independent experiments.
DISCUSSION
An increasing number of studies have documented the importance of IL-17 in immune responses to a variety of infectious diseases, especially those caused by extracellular pathogens. Our current results are the first to demonstrate the protective functions of IL-17A/F against L. pneumophila, an intracellular pneumonia pathogen. Further, our findings suggest that the beneficial effects of IL-17A/F may be associated with robust induction of inflammatory cytokines and recruitment of neutrophils, leading to clearance of L. pneumophila from the lungs and decreases in pneumonia-associated mortality.
The addition of IL-17A and IL-17F greatly increased Legionella-induced production of IL-6, TNF-α, and IL-1β but not MCP-1. Maximum cytokine inductions were produced by different concentrations of IL-17A and IL-17F, with higher levels of IL-17 tending to lead to suppression. Although the mechanisms behind, and implications of, these results are unknown, significantly stronger induction of IL-6 was observed in the presence of IL-17A than with IL-17F. These data suggest that IL-17-dependent cytokine/chemokine production has fine-tuning mechanisms, including optimal concentrations, IL-17 receptor specificity, intracellular signaling, and a positive/negative feedback loop (48). It is likely that these cytokines positively regulate the Th1 axis (47) and simultaneously induce both Th17-type cellular responses and IL-17 production (44). In contrast, however, our results demonstrate that there is no difference in MCP-1, IFN-γ, IL-12, and IL-4 levels in control and IL-17-deficient mice. Thus, we conclude that IL-17 is likely not associated with induction of these cytokines/chemokines in our model of Legionella pneumonia.
Few previous studies have employed IL-17-deficient or IL-17 receptor-deficient mice in order to investigate the cytokine's role in immune responses to pathogenesis. Among IL-17 receptor-deficient mice intranasally challenged with Klebsiella pneumoniae, neutrophil recruitment and bacterial clearance were impaired, and lethal sensitivity was higher (50). Likewise, IL-17 receptor-deficient mice with systemic Candida albicans infections had higher fungal burdens and impaired influx of neutrophils into the kidneys, leading to higher mortality rates (14). Recent data from a Listeria monocytogenes infection model demonstrated that IL-17 produced by γδ T cells plays a critical role in neutrophil recruitment and eradication of bacteria (11, 25). IL-17 has also been shown to have a crucial role in the host defense against Salmonella enterica (34), Toxoplasma gondii (18), and Chlamydia trachomatis (52) as IL-17 blockades or deficiencies during infection led to impairment of neutrophil recruitment into the infection site. This is the same pattern observed in the Legionella pneumonia model studied here.
We found a significant reduction of neutrophil numbers in the BALF of IL-17A/F-deficient mice, a pattern that was consistent with our real-time PCR and ELISA results indicating a suppression of CXC chemokines (KC, LIX, and MIP-2). These chemokines, as well as CXCR-2-mediated neutrophil accumulation, have previously been shown to exert their protective effect during Legionella infections (40).
Chemokine gene expression in whole-lung tissue could reflect either altered expression of genes or altered cell composition. Modest levels of the latter were observed in the BALF of IL-17-deficient mice. In the future, it will be important to analyze cell composition in whole-lung tissue in addition to examining chemokine gene expression in each cell type (e.g., epithelial cells, neutrophils, and macrophages). The role of epithelial cells, a major source of chemokines in the lung, may be of particular interest.
Previously (37, 39, 40), we have administered the study animals one of two types of bacterial dosage: sublethal (105 CFU) or lethal (107 CFU). It may be difficult to clearly define our model (true infection, inflammation, or acute damage) because there is no growth of bacteria in the lungs. Thus, in order to appropriately evaluate host responses, it may be necessary to employ an A/J mouse or guinea pig model, in which multiplication of Legionella organisms has been observed. Unfortunately, these are not currently available.
There was a 5-day delay between the maximum neutrophil count (day 2) and the decreased pulmonary bacterial burden (day 7). Thus, we cannot definitively conclude that IL-17 is responsible for neutrophil-mediated direct killing of L. pneumophila. We have previously reported that neutrophils play a crucial role during primary L. pneumophila infection, not via direct killing but by having more immunomodulatory effects (40). In particular, we found that, during the acute phase of infection, neutrophils may be a source of IL-12 in the lungs. This, in turn, drives Th1-type host responses, including IFN-γ induction and the resultant eradication of L. pneumophila at the later phase. However, among the IL-17A/F-deficient mice in our current study, we did not observe any variations in the expression of IL-12 and IFN-γ. IL-17 may have been involved in induction of antimicrobial peptides, such as β-defensin-2, lipocalin 2, and members of the S100 family, from colonic, skin, and airway epithelium (1, 5, 17, 20, 53, 54) although the effects of these antibacterial factors on L. pneumophila have been less characterized in in vitro and in vivo pneumonia models.
Th17-driven host defense systems can be positively and/or negatively affected by a variety of factors and cells. In studying the activity of Th17 host systems against infectious diseases, the timing of analysis, infection site, species of the infecting organism, and host specificity will all influence the measured response. Cross talk and synergistic interplay between the Th17 and Th1 domains is plausible as these axes are an integral part of the immune response against several different species of intracellular microorganisms (19, 43).
Cumulatively, our results indicate that IL-17 is an important part of the host immune response to Legionella pneumonia as it is upregulated soon after infection, stimulates the production of several important factors in the immune response, and is associated with decreases in intrapulmonary bacteria and with increases in survival rates. Additional research into IL-17 and the Th17 network as a whole is likely to provide new insights into the pathogenesis of the bacteria and novel therapeutic approaches in the treatment of Legionella pneumonia.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to K. Kawakami for providing the anti-γδ TCR antibody-producing hybridoma. We thank all of the staff and A. Ishizaka for very helpful suggestions and advice during this project.
This work was supported by JSPS KAKENHI (20790577, 22790958).
Footnotes
Published ahead of print 5 December 2011
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1. Aujla SJ, et al. 2008. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat. Med. 14:275–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Benchetrit F, et al. 2002. Interleukin-17 inhibits tumor cell growth by means of a T-cell-dependent mechanism. Blood 99:2114–2121 [DOI] [PubMed] [Google Scholar]
- 3. Byrne B, Swanson MS. 1998. Expression of Legionella pneumophila virulence traits in response to growth conditions. Infect. Immun. 66:3029–3034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chabaud M, Fossiez F, Taupin JL, Miossec P. 1998. Enhancing effect of IL-17 on IL-1-induced IL-6 and leukemia inhibitory factor production by rheumatoid arthritis synoviocytes and its regulation by Th2 cytokines. J. Immunol. 161:409–414 [PubMed] [Google Scholar]
- 5. Chan YR, et al. 2009. Lipocalin 2 is required for pulmonary host defense against Klebsiella infection. J. Immunol. 182:4947–4956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Crowe CR, et al. 2009. Critical role of IL-17RA in immunopathology of influenza infection. J. Immunol. 183:5301–5310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Deng JC, Tateda K, Zeng X, Standiford TJ. 2001. Transient transgenic expression of γ interferon promotes Legionella pneumophila clearance in immunocompetent hosts. Infect. Immun. 69:6382–6390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. el-Ebiary M, et al. 1997. Prognostic factors of severe Legionella pneumonia requiring admission to ICU. Am. J. Respir. Crit. Care. Med. 156:1467–1472 [DOI] [PubMed] [Google Scholar]
- 9. Flierl MA, et al. 2008. Adverse functions of IL-17A in experimental sepsis. FASEB J. 22:2198–2205 [DOI] [PubMed] [Google Scholar]
- 10. Friedman H, Yamamoto Y, Klein TW. 2002. Legionella pneumophila pathogenesis and immunity. Semin. Pediatr. Infect. Dis. 13:273–279 [DOI] [PubMed] [Google Scholar]
- 11. Henry T, et al. 2010. Type I IFN signaling constrains IL-17A/F secretion by γδ T cells during bacterial infections. J. Immunol. 184:3755–3767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Horai R, et al. 2004. TNF-α is crucial for the development of autoimmune arthritis in IL-1 receptor antagonist-deficient mice. J. Clin. Invest. 114:1603–1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Horwitz MA, Silverstein SC. 1980. Legionnaires' disease bacterium (Legionella pneumophila) multiples intracellularly in human monocytes. J. Clin. Invest. 66:441–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huang W, Na L, Fidel PL, Schwarzenberger P. 2004. Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J. Infect. Dis. 190:624–631 [DOI] [PubMed] [Google Scholar]
- 15. Ishigame H, et al. 2009. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity 30:108–119 [DOI] [PubMed] [Google Scholar]
- 16. Iwakura Y, Nakae S, Saijo S, Ishigame H. 2008. The roles of IL-17A in inflammatory immune responses and host defense against pathogens. Immunol. Rev. 226:57–79 [DOI] [PubMed] [Google Scholar]
- 17. Kao CY, et al. 2004. IL-17 markedly up-regulates β-defensin-2 expression in human airway epithelium via JAK and NF-κB signaling pathways. J. Immunol. 173:3482–3491 [DOI] [PubMed] [Google Scholar]
- 18. Kelly MN, et al. 2005. Interleukin-17/interleukin-17 receptor-mediated signaling is important for generation of an optimal polymorphonuclear response against Toxoplasma gondii infection. Infect. Immun. 73:617–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Khader SA, et al. 2005. IL-23 compensates for the absence of IL-12p70 and is essential for the IL-17 response during tuberculosis but is dispensable for protection and antigen-specific IFN-γ responses if IL-12p70 is available. J. Immunol. 175:788–795 [DOI] [PubMed] [Google Scholar]
- 20. Liang SC, et al. 2006. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 203:2271–2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Linden A. 2001. Role of interleukin-17 and the neutrophil in asthma. Int. Arch. Allergy Immunol. 126:179–184 [DOI] [PubMed] [Google Scholar]
- 22. Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔC(T) method. Methods 25:402–408 [DOI] [PubMed] [Google Scholar]
- 23. Marston BJ, Lipman HB, Breiman RF. 1994. Surveillance for Legionnaires' disease. Risk factors for morbidity and mortality. Arch. Intern. Med. 154:2417–2422 [PubMed] [Google Scholar]
- 24. McDade JE, et al. 1977. Legionnaires' disease: isolation of a bacterium and demonstration of its role in other respiratory disease. N. Engl. J. Med. 297:1197–1203 [DOI] [PubMed] [Google Scholar]
- 25. Meeks KD, Sieve AN, Kolls JK, Ghilardi N, Berg RE. 2009. IL-23 is required for protection against systemic infection with Listeria monocytogenes. J. Immunol. 183:8026–8034 [DOI] [PubMed] [Google Scholar]
- 26. Miyazaki Y, et al. 2010. IL-17 is necessary for host protection against acute-phase Trypanosoma cruzi infection. J. Immunol. 185:1150–1157 [DOI] [PubMed] [Google Scholar]
- 27. Nakae S, et al. 2002. Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity 17:375–387 [DOI] [PubMed] [Google Scholar]
- 28. Nash TW, Libby DM, Horwitz MA. 1984. Interaction between the Legionnaires' disease bacterium (Legionella pneumophila) and human alveolar macrophages. Influence of antibody, lymphokines, and hydrocortisone. J. Clin. Invest. 74:771–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Peck A, Mellins ED. 2010. Precarious balance: Th17 cells in host defense. Infect. Immun. 78:32–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pedro-Botet ML, et al. 1998. Role of immunosuppression in the evolution of Legionnaires' disease. Clin. Infect. Dis. 26:14–19 [DOI] [PubMed] [Google Scholar]
- 31. Ramesh G, Zhang B, Uematsu S, Akira S, Reeves WB. 2007. Endotoxin and cisplatin synergistically induce renal dysfunction and cytokine production in mice. Am. J. Physiol. Renal Physiol. 293:F325–332 [DOI] [PubMed] [Google Scholar]
- 32. Reingold AL. 1988. Role of legionellae in acute infections of the lower respiratory tract. Rev. Infect. Dis. 10:1018–1028 [DOI] [PubMed] [Google Scholar]
- 33. Ruddy MJ, Shen F, Smith JB, Sharma A, Gaffen SL. 2004. Interleukin-17 regulates expression of the CXC chemokine LIX/CXCL5 in osteoblasts: implications for inflammation and neutrophil recruitment. J. Leukoc. Biol. 76:135–144 [DOI] [PubMed] [Google Scholar]
- 34. Schulz SM, Kohler G, Holscher C, Iwakura Y, Alber G. 2008. IL-17A is produced by Th17, γδ T cells and other CD4− lymphocytes during infection with Salmonella enterica serovar Enteritidis and has a mild effect in bacterial clearance. Int. Immunol. 20:1129–1138 [DOI] [PubMed] [Google Scholar]
- 35. Schwarzenberger P, et al. 1998. IL-17 stimulates granulopoiesis in mice: use of an alternate, novel gene therapy-derived method for in vivo evaluation of cytokines. J. Immunol. 161:6383–6389 [PubMed] [Google Scholar]
- 36. Sergejeva S, Ivanov S, Lotvall J, Linden A. 2005. Interleukin-17 as a recruitment and survival factor for airway macrophages in allergic airway inflammation. Am. J. Respir. Cell Mol. Biol. 33:248–253 [DOI] [PubMed] [Google Scholar]
- 37. Tateda K, et al. 2003. Hyperoxia mediates acute lung injury and increased lethality in murine Legionella pneumonia: the role of apoptosis. J. Immunol. 170:4209–4216 [DOI] [PubMed] [Google Scholar]
- 38. Tateda K, et al. 1998. Serum cytokines in patients with Legionella pneumonia: relative predominance of Th1-type cytokines. Clin. Diagn. Lab. Immunol. 5:401–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tateda K, et al. 2001. Early recruitment of neutrophils determines subsequent T1/T2 host responses in a murine model of Legionella pneumophila pneumonia. J. Immunol. 166:3355–3361 [DOI] [PubMed] [Google Scholar]
- 40. Tateda K, et al. 2001. Chemokine-dependent neutrophil recruitment in a murine model of Legionella pneumonia: potential role of neutrophils as immunoregulatory cells. Infect. Immun. 69:2017–2024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tkatch LS, Kusne S, Irish WD, Krystofiak S, Wing E. 1998. Epidemiology of legionella pneumonia and factors associated with legionella-related mortality at a tertiary care center. Clin. Infect. Dis. 27:1479–1486 [DOI] [PubMed] [Google Scholar]
- 42. Toy D, et al. 2006. Cutting edge: interleukin 17 signals through a heteromeric receptor complex. J. Immunol. 177:36–39 [DOI] [PubMed] [Google Scholar]
- 43. Umemura M, et al. 2007. IL-17-mediated regulation of innate and acquired immune response against pulmonary Mycobacterium bovis bacille Calmette-Guerin infection. J. Immunol. 178:3786–3796 [DOI] [PubMed] [Google Scholar]
- 44. Vanaudenaerde BM, et al. 2011. Innate and adaptive interleukin-17-producing lymphocytes in chronic inflammatory lung disorders. Am. J. Respir. Crit. Care Med. 183:977–986 [DOI] [PubMed] [Google Scholar]
- 45. Van Kooten C, et al. 1998. Interleukin-17 activates human renal epithelial cells in vitro and is expressed during renal allograft rejection. J. Am. Soc. Nephrol. 9:1526–1534 [DOI] [PubMed] [Google Scholar]
- 46. Weaver CT, Hatton RD, Mangan PR, Harrington LE. 2007. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu. Rev. Immunol. 25:821–852 [DOI] [PubMed] [Google Scholar]
- 47. Wesa AK, Galy A. 2001. IL-1β induces dendritic cells to produce IL-12. Int. Immunol. 13:1053–1061 [DOI] [PubMed] [Google Scholar]
- 48. Wright JF, et al. 2007. Identification of an interleukin 17F/17A heterodimer in activated human CD4+ T cells. J. Biol. Chem. 282:13447–13455 [DOI] [PubMed] [Google Scholar]
- 49. Yamamoto M, et al. 2008. TSU68 prevents liver metastasis of colon cancer xenografts by modulating the premetastatic niche. Cancer Res. 68:9754–9762 [DOI] [PubMed] [Google Scholar]
- 50. Ye P, et al. 2001. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J. Exp. Med. 194:519–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yoshizawa S, et al. 2005. Legionella pneumophila evades γ interferon-mediated growth suppression through interleukin-10 induction in bone marrow-derived macrophages. Infect. Immun. 73:2709–2717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang F, Meng G, Strober W. 2008. Interactions among the transcription factors Runx1, RORγt and Foxp3 regulate the differentiation of interleukin 17-producing T cells. Nat. Immunol. 9:1297–1306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang Z, Clarke TB, Weiser JN. 2009. Cellular effectors mediating Th17-dependent clearance of pneumococcal colonization in mice. J. Clin. Invest. 119:1899–1909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zheng Y, et al. 2008. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med. 14:282–289 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.