

Abstract
Streptomyces coelicolor exhibits a major secondary metabolism, deriving important amounts of glucose to synthesize pigmented antibiotics. Understanding the pathways occurring in the bacterium with respect to synthesis of oligo- and polysaccharides is of relevance to determine a plausible scenario for the partitioning of glucose-1-phosphate into different metabolic fates. We report the molecular cloning of the genes coding for UDP- and ADP-glucose pyrophosphorylases as well as for glycogen synthase from genomic DNA of S. coelicolor A3(2). Each gene was heterologously expressed in Escherichia coli cells to produce and purify to electrophoretic homogeneity the respective enzymes. UDP-glucose pyrophosphorylase (UDP-Glc PPase) was characterized as a dimer exhibiting a relatively high Vmax in catalyzing UDP-glucose synthesis (270 units/mg) and with respect to dTDP-glucose (94 units/mg). ADP-glucose pyrophosphorylase (ADP-Glc PPase) was found to be tetrameric in structure and specific in utilizing ATP as a substrate, reaching similar activities in the directions of ADP-glucose synthesis or pyrophosphorolysis (Vmax of 0.15 and 0.27 units/mg, respectively). Glycogen synthase was arranged as a dimer and exhibited specificity in the use of ADP-glucose to elongate α-1,4-glucan chains in the polysaccharide. ADP-Glc PPase was the only of the three enzymes exhibiting sensitivity to allosteric regulation by different metabolites. Mannose-6-phosphate, phosphoenolpyruvate, fructose-6-phosphate, and glucose-6-phosphate behaved as major activators, whereas NADPH was a main inhibitor of ADP-Glc PPase. The results support a metabolic picture where glycogen synthesis occurs via ADP-glucose in S. coelicolor, with the pathway being strictly regulated in connection with other routes involved with oligo- and polysaccharides, as well as with antibiotic synthesis in the bacterium.
INTRODUCTION
Gram-positive bacteria of the genus Streptomyces exhibit a particularly complex morphological differentiation, with the formation of a highly branched substrate and aerial mycelium that generates spores from septation of the aerial hyphae (23, 33). Also characteristic of Streptomyces spp. is the occurrence of a flush secondary metabolism, with these bacteria producing almost two-thirds of all known natural antibiotics (9). Streptomyces coelicolor A3(2) is the producer of three pigmented antibiotics, and it has a well-characterized genetics; these characteristics make it one of the preferred strains for research within this genus of bacteria. Although the complete genome of this S. coelicolor strain was elucidated about 10 years ago (5), advanced knowledge of the microorganism at the biochemical level is far from being reached. Most of the studies carried out with Streptomyces spp. have been biased toward the understanding of its morphological and physiological differentiation, while very little has been done on the biochemistry of the enzymes involved in the metabolic pathways.
Monosaccharide interconversion and synthesis of oligo- and polysaccharides are key processes for bacterial metabolism. Specifically, in actinomycetes, different saccharides were found to be decisive for cellular differentiation and to provide carbon skeletons necessary for antibiotic synthesis. It has been established that trehalose and glycogen metabolism have a complex interplay with differentiation (43) and that phosphorylated hexoses are critical metabolites for the occurrence of secondary metabolism in S. coelicolor. In this context, the study of enzymes determining the use of Glc-1P for the production of different carbohydrates is critical for a better understanding of the developmental biology, physiology, and metabolism of this microorganism. The fate of Glc-1P in saccharide anabolism involves a first step where the Glc moiety is “activated” by formation of nucleoside-diphospho-Glc (NDP-Glc), which is catalyzed by different NDP-Glc pyrophosphorylases. Later, different enzymes (i.e., glycosyl transferases) having specificity toward one NDP-Glc lead the monosaccharide to the multifaceted routes for carbohydrates.
Here we report the molecular cloning of three genes from S. coelicolor encoding key enzymes of carbohydrate metabolism: UDP-glucose pyrophosphorylase (UDP-Glc PPase), ADP-Glc PPase, and glycogen synthase (GSase). The recombinant enzymes were purified and characterized in their kinetic, regulatory, and structural properties. The results are analyzed in the framework of the involvement of the three enzymes in hexose-P metabolic partitioning in actinomycetes. Interestingly, the properties of regulation that we found for the ADP-Glc PPase from S. coelicolor are distinct from those known for the enzyme from other bacteria. Our studies lead to a better understanding of the occurrence and regulation of glycogen biosynthesis in a specific genus of actinobacteria, in association with particular metabolic characteristics related to Gram-positive bacteria.
MATERIALS AND METHODS
Chemicals.
Inorganic pyrophosphatase, Glc-1P, ATP, UTP, Fru-6P, Man-6P, PEP, pyruvate, rabbit liver glycogen, ADP-Glc, UDP-Glc, and oligonucleotides were acquired from Sigma-Aldrich (St. Louis, MO). All other reagents were of the highest quality available.
Bacterial strains and growth conditions.
Escherichia coli TOP10 F′ and E. coli BL21(DE3) (Invitrogen) were used in this study. Luria-Bertani (LB) medium was used for growth of E. coli strains. The following antibiotics were added to the media when necessary: kanamycin (50 μg ml−1), ampicillin (100 μg ml−1), and chloramphenicol (20 μg ml−1).
DNA manipulation.
DNA restriction enzymes were used as recommended by the manufacturer (New England BioLabs). DNA manipulations were performed using standard protocols (39). DNA fragments were purified from agarose gels with the GFX PCR DNA and gel band purification kit (GE Healthcare). Plasmids were prepared using a QIAprep spin miniprep kit (Qiagen). Deep Vent DNA polymerase was used in all PCRs according to the supplier's instructions (New England BioLabs).
Cloning of glgC, glgA, and gtaB.
Three genes from S. coelicolor, glgC (GeneID, 1096384), glgA (GeneID, 1096385), and gtaB (GeneID, 1098616), coding for ADP-Glc PPase, GSase, and UDP-Glc PPase, respectively, were amplified by PCR using genomic DNA as a template. The 5′ primers used were designed to have an NdeI site overlapping the translational initiation codon, changing the GTG start codon to ATG when required. The 3′ primers contained an EcoRI site downstream of the stop codon. Table 1 shows oligonucleotide pairs (forward and reverse) used for cloning the different genes. PCRs were performed using a DNA Thermal Cycler 480 (Perkin-Elmer) with the following cycling parameters: 30 cycles of 30 s at 94°C, 30 s at annealing temperature (TA), and 80 s at 72°C. TA was 63°C for glgC, 65°C for glgA, and 68°C for gtaB. Each PCR product was cloned into pCR-BluntII-TOPO (Invitrogen) and sequenced to confirm that it was free of errors. These plasmids were digested with NdeI and EcoRI and the inserts cloned into identical sites of pET28a vector in order to express proteins as His tag fusions. The resulting pET28a-derived plasmids containing glgC, glgA, and gtaB were named pAGP, pGSA, and pUGP, respectively.
Table 1.
Oligonucleotide sequences of the specific primers used to amplify glgC, glgA, and gtaB genes from S. coelicolor A3(2)
| Primer | Sequencea | Restriction site |
|---|---|---|
| glgC-fow | 5′-TCATATGCTCGGCATCGTGCTGGCGGGCGG-3′ | NdeI |
| glgC-rev | 5′-AGAATTCCACGGCACCCGCTGTCCCTTGCCC-3′ | EcoRI |
| glgA-fow | 5′-TCATATGCGCGTGGGACTGCTGAGCCGAG-3′ | NdeI |
| glgA-rev | 5′-AGAATTCCAAGCCTGCTTGAGGATCTCCTCG-3′ | EcoRI |
| gtaB-fow | 5′-TCATATGACTCAGTCCCACCCCAGGATCAGC-3′ | NdeI |
| gtaB-rev | 5′-AGAATTCCAACGTCCCTGCATCTCCTCGGC-3′ | EcoRI |
Restriction sites are indicated in bold.
Expression of ADP-Glc PPase, GSase, and UDP-Glc PPase.
As a general procedure, E. coli strains harboring pGro7 (Takara) and the different expression plasmids were cultured overnight at 37°C in LB with appropriate antibiotics and then subcultured by 1:100 dilutions in fresh medium and grown to an optical density at 600 nm (OD600) of 0.6. Chaperones and recombinant gene expression were induced by the addition of 2 mg/ml l-arabinose and 0.4 mM IPTG, respectively, and cultures were incubated at 20°C for 20 h with shaking at 150 rpm. Cells were harvested by centrifugation, washed once with ice-cold 50 mM Tris-HCl buffer, pH 8.0, and suspended in an extraction buffer containing 50 mM Tris-HCl, pH 8.0, 500 mM NaCl, and 1 mM phenylmethylsulfonyl fluoride (PMSF). In the case of GSase, the extraction buffer was supplemented with 2 mM β-mercaptoethanol and 0.1 mM EDTA. Cells were disrupted using a VibraCell VCX 130 (Sonics) device with alternating pulses (3 s on, 2 s off) for 5 min in an ice-cold bath. After disruption, crude extracts were clarified by centrifugation for 30 min at 15,000 × g.
The coexpression of chaperones GroES and GroEL encoded by plasmid pGro7 was found to be essential to obtain ADP-Glc PPase and GSase as soluble proteins. UDP-Glc PPase, however, did not require pGro7 coexpression, and it was obtained in its soluble form by inducing its expression with 0.25 mM IPTG, followed by incubation at 25°C for 20 h at 150 rpm.
Purification of His-tagged enzymes.
ADP-Glc PPase, GSase, and UDP-Glc PPase from S. coelicolor A3(2) were expressed as N-terminal His tag fusions, in order to facilitate their subsequent purification. Enzymes were purified by affinity chromatography, using Ni-nitrilotriacedic acid (NTA) agarose resin (Invitrogen) according to the protocol supplied by the manufacturer. Briefly, crude extract fractions prepared in buffer H (20 mM Tris-HCl, pH 8.0, 500 mM NaCl, 10 mM imidazole) were loaded onto columns previously equilibrated with the same buffer. After extensive washing with buffer H, samples were eluted by means of a linear gradient to buffer I (20 mM Tris-HCl, pH 8.0, 500 mM NaCl, 300 mM imidazole). Elution fractions containing the corresponding recombinant enzyme were analyzed by SDS-PAGE (30) to check for purity.
For each recombinant enzyme, active fractions eluted from the Ni-NTA agarose column were pooled, dialyzed to remove imidazole, and supplemented with 10% (vol/vol) glycerol. For GSase, the enzyme was dialyzed against a buffer containing 20 mM triethanolamine-HCl, pH 8.0, and 20% (vol/vol) glycerol. The three recombinant enzymes were stable for at least 6 months when stored at −80°C under the conditions specified above.
Protein measurement.
Protein concentration was determined by the method of Bradford (11), using bovine serum albumin (BSA) as a standard. Coomassie brilliant blue staining was utilized in SDS-PAGE.
Enzyme activity assays.
ADP-Glc PPase and UDP-Glc PPase activities were determined at 37°C in both the NDP-Glc synthesis (assay A) and pyrophosphorolysis (assay B) directions. Unless otherwise specified, conditions for the different assays were as follows.
(i) Assay A.
Synthesis of ADP-Glc or UDP-Glc was assayed by following the formation of Pi (after hydrolysis of PPi by inorganic pyrophosphatase) by the colorimetric method previously described (18). Reaction mixtures contained 50 mM MOPS (morpholinepropanesulfonic acid), pH 8.0, 10 mM MgCl2, 2 mM ATP or UTP, 0.2 mg/ml BSA, 0.0005 unit/μl yeast inorganic pyrophosphatase, and the appropriate enzyme dilution. Assays were initiated by the addition of 1 mM Glc-1P in a total volume of 50 μl. Reaction mixtures were incubated for 10 min at 37°C and terminated by adding reactive malachite green (18). The complex formed with the released Pi was measured at 630 nm with an ELISA (enzyme-linked immunosorbent assay) EMax detector (Molecular Devices).
Alternatively, mainly in experiments performed to analyze the effect of Pi on ADP-Glc PPase, assay A was replaced by the radiometric coupled assay method of Yep et al. (45), measuring the synthesis of ADP-[14C]Glc from [14C]Glc-1P and ATP. The standard reaction mixture contained 100 mM MOPS buffer (pH 8.0), 10 mM MgCl2, 1 mM [14C]Glc-1P (100 to 1,000 cpm/nmol), 1 mM ATP, 0.0005 unit/μl inorganic pyrophosphatase, and 0.2 mg/ml BSA plus enzyme in a total volume of 0.2 ml. The reaction mixtures were incubated for 10 min at 37°C, and the reactions were terminated by heating in a boiling-water bath for 1 min. The ADP-[14C]Glc was then converted to [14C]glycogen by the addition of glycogen synthase (from E. coli) and nonradioactive glycogen as a primer. Glycogen formed was precipitated and washed, and the radioactivity was measured in a scintillation counter.
(ii) Assay B.
Pyrophosphorolysis of ADP-Glc or UDP-Glc was followed by the formation of [32P]ATP or [32P]UTP, respectively, from 32PPi, as previously described (19). The reaction mixtures contained 50 mM MOPS buffer, pH 8.0, 5 mM MgCl2, 1 mM ADP-Glc or UDP-Glc, 1 mM 32PPi (3,000 cpm/nmol), 10 mM NaF, 0.2 mg/ml BSA, and enzyme in a final volume of 150 μl. The reactions were started with a 32PPi addition and after 10 min of incubation at 37°C were stopped with 1 ml of cold 10% (vol/vol) trichloroacetic acid. The [32P]NTP formed was measured following a modified protocol, as described elsewhere (19).
(iii) Glycogen synthase.
The assay was conducted as described in reference 21, using a medium that contained (unless otherwise specified) 1 mM ADP-[14C]Glc (500 to 1,500 cpm/nmol), 10 mM MgCl2, 2.5 mg/ml rabbit liver glycogen, 50 mM bicine-NaOH (pH 8.0), and 0.5 mg/ml BSA in a total volume of 100 μl. The assays were started by adding 20 μl of GSase from S. coelicolor dissolved in 20 mM triethanolamine-HCl, pH 8.0.
In all of the assay procedures, one unit of activity (U) is defined as the amount of enzyme catalyzing the formation of 1 μmol of product per min, under conditions described above in each case.
Kinetic studies.
Saturation curves were performed by assaying the respective enzyme activities at saturating levels of a fixed substrate and different concentrations of the variable substrate. The experimental data were plotted as enzyme activity (U/mg) versus substrate (or effector) concentration (mM), and the kinetic constants were determined by fitting the data to the Hill equation as described elsewhere (2). Fitting was performed with the Levenberg-Marquardt nonlinear least-squares algorithm provided by the computer program Origin7.0. Hill plots were used to calculate the Hill coefficient (nH), the maximal velocity (Vmax), and the kinetic constants that correspond to the activator, substrate, or inhibitor concentrations giving 50% of the maximal activation (A0.5), velocity (S0.5), or inhibition (I0.5). All kinetic constants are the means of at least three sets of data, which were reproducible within ±10%.
Determination of native molecular mass.
The native molecular masses of ADP-Glc PPase, GSase, and UDP-Glc PPase from S. coelicolor A3(2) were determined by gel filtration with a Tricorn 5/200 column (GE Healthcare). We used a gel filtration calibration kit-high molecular weight (GE Healthcare) with protein standards including thyroglobulin (669 kDa), ferritin (440 kDa), aldolase (158 kDa), conalbumin (75 kDa), and ovalbumin (44 kDa). The column void volume was determined using a dextran blue loading solution (Promega). The mass of each protein was extrapolated from the standard semilog curve Kav versus log MM (molecular mass). Kav is the parameter defined by the following equation: Kav = (Ve−Vo)/(Vc−Vo), where Ve is the elution volume, Vo is the void volume, and Vc is the column volume.
RESULTS AND DISCUSSION
Cloning and heterologous expression of the glgC, glgA, and gtaB genes.
The identification of the nucleotide sequences encoding ADP-Glc PPase (GeneID, 1096384; glgC gene) and UDP-Glc PPase (GeneID, 1098616; gtaB gene), as well as a putative glycosyl transferase (GeneID, 1096385; glgA gene), in the database of the S. coelicolor A3(2) genome project (NCBI accession number, NC_003888.3) encouraged us to perform the molecular cloning of full-length genes. Thus, glgC (1,200 bp in length), glgA (1,064 bp), and gtaB (912 bp) genes were amplified from genomic DNA in individual single-step PCR procedures and their identities were confirmed by DNA sequencing in both directions. It is predicted that glgC, gtaB, and glgA encode proteins of 399, 304, and 387 amino acids with respective molecular masses of 42.7, 33.0, and 42.0 kDa. Plasmid pUGP (see details under Materials and Methods) was used to transform competent E. coli BL21(DE3) cells, whereas pAGP and pGSA were used to transform competent E. coli BL21(DE3)/pGro7 cells. The strategy of coexpression with chaperonins was critical to conveniently obtain both enzymes, ADP-Glc PPase and GSase, in corresponding soluble fractions; attempts to express them in the absence of the pGro7 plasmid resulted in insoluble recombinant proteins (data not shown). In order to optimize the expression of the desired enzymes, we analyzed different culture conditions (time and temperature of induction as well as IPTG concentration) in each case. After inducing expression and obtaining crude extracts from the transformed cells, the His-tagged recombinant proteins were purified by immobilized metal (Ni2+) affinity chromatography.
Figure 1 illustrates the purity and quaternary structure exhibited by recombinant ADP-Glc PPase, UDP-Glc PPase, and GSase after purification. SDS-PAGE (Fig. 1A) analysis revealed that the recombinant enzymes were purified to a high degree. The apparent molecular masses of the recombinant proteins were slightly higher than the theoretical values predicted for the three enzymes (Fig. 1A, lanes I, II, and III), after the His tag attached to the pET expression vectors. Notably, UDP-Glc PPase was expressed at high levels, resulting in nearly 30 mg/ml of pure enzyme after a single purification step (Fig. 1A, lane II). When the coexpression strategy was employed, such as in the cases of ADP-Glc PPase and GSase, chaperonins were almost completely removed in the 30 mM imidazole washing step, after which the recombinant enzymes were obtained with a high degree of purity (Fig. 1A, lanes I and III, respectively). On the other hand, native molecular masses of the S. coelicolor A3(2) recombinant enzymes were determined by gel filtration chromatography, as shown in Fig. 1B. The molecular mass determined for ADP-Glc PPase fits well with a tetrameric protein, which is in agreement with the quaternary structure of the enzyme from other sources characterized so far (3, 4). In addition, the active form of the GSase exhibited a dimeric structure which is also in accordance with previous reports (13, 40). The UDP-Glc PPase became active as a dimer, and no higher oligomerization states were observed for the S. coelicolor enzyme; these results differ from the report of a multioligomeric structure for the M. tuberculosis H37Rv UDP-Glc PPase (31).
Fig 1.
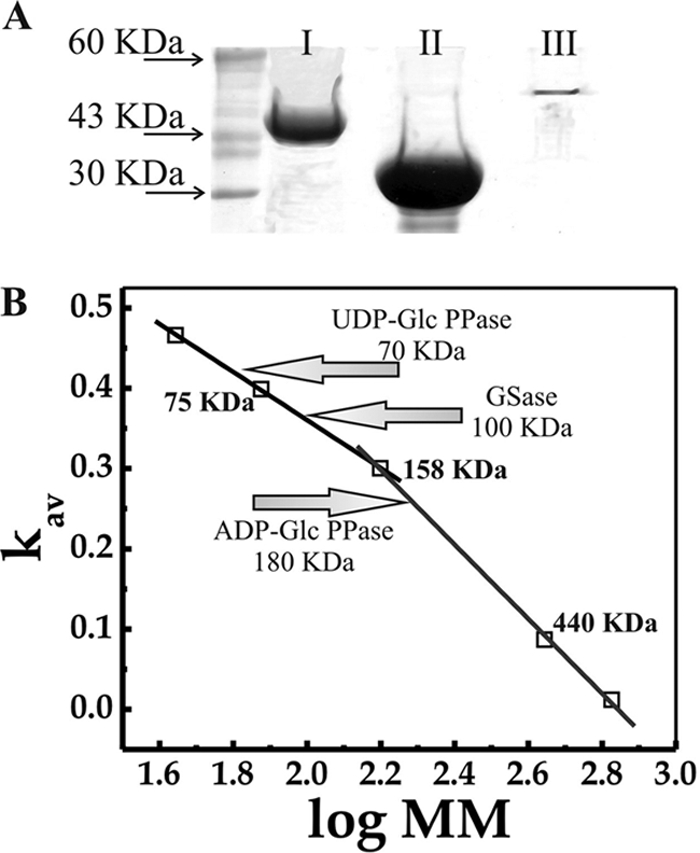
(A) SDS-PAGE analysis of purified ADP-Glc PPase (I), UDP-Glc PPase (II), and GSase (III) from S coelicolor A3(2) expressed in E. coli BL21(DE3) (UDP-Glc PPase) or E. coli BL21(DE3)/[pGro7] (GSase and ADP-Glc PPase). Left lane corresponds to molecular mass standards. (B) Molecular mass determination, performed from size exclusion chromatography, as detailed in Materials and Methods.
Kinetic characterization of ADP-Glc PPase, UDP-Glc PPase, and GSase.
Recombinant ADP-Glc PPase and UDP-Glc PPase were assayed for enzymatic activity in both the forward (NDP-Glc synthesis) and the reverse (NDP-Glc pyrophosphorolysis) directions of catalysis, as described in Materials and Methods. The respective kinetic parameters are shown in Table 2. It is worth noting that, to the best of our knowledge, this is the first report on the kinetic characterization of an ADP-Glc PPase from a Gram-positive, G+C-rich organism. The S. coelicolor ADP-Glc PPase exhibited affinity for substrates that, as a whole, are on the order of those previously reported for ADP-Glc PPases from other sources (Table 2), including Gram-negative bacteria and higher plants (3, 4). On the other hand, UDP-Glc PPase activity (Table 2) is three orders of magnitude higher than activity of the analogous enzyme from Mycobacterium tuberculosis H37Rv (31), a closely related organism, which has been produced with a similar expression system. Also, a particular behavior was observed in S. coelicolor UDP-Glc PPase concerning UTP consumption, since it was not possible to reach a clear plateau for the substrate saturation curves under the assay conditions (even when using up to 20 mM UTP and 50 mM Mg2+), from which we estimate a rough Vmax value for the physiological activity of the enzyme (Table 2). This odd characteristic disagrees with respect to the function of UTP and Mg2+ in catalysis, as it is known for other NDP-Glc PPases that the nucleotide binds first to the enzyme and that the major substrate would be the complex it forms with the divalent cation (6, 17, 27). However, results reported for the UDP-Glc PPase from Xanthomonas spp. could solve the conflict, as it was found that UTP may play a role in favoring interaction between subunits and enhancing the enzyme stability and activity (10).
Table 2.
Kinetic parameters for ADP-Glc PPase, UDP-Glc PPase, and GSase from S. coelicolor A3(2)a
| Enzyme | Substrate | S0.5 (mM) | nH | Vmax(U/mg) |
|---|---|---|---|---|
| ADP-Glc PPase | ||||
| Assay A | ATP | 0.39 ± 0.03 | 2.4 | 0.15 ± 0.01 |
| Glc-1P | 0.37 ± 0.04 | 0.7 | ||
| Mg2+ | 1.74 ± 0.14 | 1.6 | ||
| Assay B | ADP-Glc | 2.10 ± 0.06 | 3.0 | 0.27 ± 0.02 |
| PPi | 0.35 ± 0.05 | 0.7 | ||
| Mg2+ | 1.81 ± 0.16 | 1.7 | ||
| UDP-Glc PPase | ||||
| Assay A | UTP | >10 | 270.00 ± 4.00b | |
| Glc-1P | 0.06 ± 0.01 | 2.4 | ||
| Mg2+ | 0.46 ± 0.03 | 2.9 | ||
| Assay B | UDP-Glc | 0.38 ± 0.03 | 1.1 | 1.00 ± 0.05 |
| PPi | 0.80 ± 0.04 | 1.7 | ||
| Mg2+ | 0.36 ± 0.02 | 1.9 | ||
| GSase | ||||
| ADP-Glc | 0.13 ± 0.01 | 1.4 | 0.10 ± 0.01 | |
| Glycogen [mg/ml] | 0.31 ± 0.03 | 0.9 |
Assays were carried out as described in Materials and Methods. Hill plots were used to calculate the Hill coefficient (nH), the maximal velocity (Vmax), and S0.5, the respective substrate concentrations giving 50% of Vmax. Values for Vmax, S0.5, and nH are average numbers from three independent experiments, using regression analysis.
Because of the nonsaturating behavior observed for UTP (see the text), this value is apparent and calculated from saturation curves for Glc-1P using 10 mM UTP and 5 mM Mg2+.
Another remarkable result in Table 2 is the unusually high ratio of activities of sugar-nucleotide synthesis/pyrophosphorolysis exhibited by S. coelicolor UDP-Glc PPase. The Vmax shown in Table 2 in the direction of UDP-Glc synthesis is even higher than that corresponding to the enzyme from Xanthomonas spp., previously reported as significantly elevated between UDP-Glc PPases from prokaryotic sources (10). Interestingly, kinetic properties of the S. coelicolor UDP-Glc PPase agree with the occurrence of high activity in the physiological direction of the reaction, which could be relevant for the metabolism in this microorganism. Supporting this view is the recent report by Zhou and coworkers (46) demonstrating that levels of UDP-Glc synthesis affects production of antibiotics and modifies carbon fluxes in Streptomyces hygroscopicus.
GSase activity was analyzed with respect to its capacity for elongating a preformed molecule of glycogen; the results of this characterization are also summarized in Table 2. Although the parameters for S. coelicolor GSase differ from those obtained for the E. coli homolog (44), they are very close to the parameters obtained in the characterization of the starch synthase III from Arabidopsis thaliana (14). It is well established that starch synthesis in plants and bacterial glycogen synthesis share similar properties in relation to their mechanisms and regulation steps (3, 4). The presence of Mg2+ was essential for the activity of both pyrophosphorylases but not for GSase, though the activity of the latter enzyme was slightly increased (1.5-fold) in the presence of 10 mM Mg2+. Also, in the presence of Mn2+, GSase retained 47% of the activity reached with Mg2+; other divalent cations, such as Co2+, Ni2+, Zn2+, and Cu2+, inhibited the enzyme by more than 90% (data not shown).
In general, pyrophosphorylases are relatively specific for their substrates, although some exceptions have been reported. For example, dTTP is a poor substrate for UDP-Glc PPase from E. coli, whereas GDP-mannose (GDP-Man) PPases from M. tuberculosis and from Leptospira interrogans exhibit promiscuity in the use of nucleotide triphosphates (NTPs) as substrates (1, 36). Different NTPs (ATP, UTP, ITP, GTP, CTP, dTTP) at an up to 5 mM final concentration were assayed as alternative substrates for both ADP-Glc PPase and UDP-Glc PPase in the NDP-Glc synthesis direction of catalysis. It was observed that ADP-Glc PPase was strictly specific for ATP whereas UDP-Glc PPase was active with dTTP as an alternative substrate to UTP. Regarding nucleotide utilization, the S. coelicolor UDP-Glc PPase depicted saturation curves for dTTP allowing determination of an unambiguous S0.5 value for this substrate (Table 3); the latter represented a remarkable difference with respect to UTP utilization, where no saturation was observed, as described above. Analysis of the data presented in Tables 2 and 3 shows that when UDP-Glc PPase is assayed in the presence of UTP as a substrate, it exhibits a Vmax about 3-fold higher and an S0.5 for the cosubstrate Glc-1P almost one order of magnitude lower than the corresponding values obtained in the presence of dTTP. The property of the S. coelicolor UDP-Glc PPase of using dTTP is similar to that of the UDP-Glc PPase from Xanthomonas spp. (10). In addition, neither ADP-Glc PPase nor UDP-Glc PPase utilized mannose-1P (Man-1P) as a substrate, and when both enzymes were evaluated as GDP-Man PPases (assayed with GTP and Man-1P), no detectable activity was observed.
Table 3.
Kinetic parameters for S. coelicolor A3(2) UDP-Glc PPase in a synthetic reactiona
| Substrate | S0.5(mM) | nH | Vmax(U/mg) |
|---|---|---|---|
| TTP | 0.79 ± 0.06 | 1.1 | 94 ± 2 |
| Glc-1P | 0.45 ± 0.02 | 2.4 | 94 ± 2 |
dTTP and Glc-1P were used as substrates. Vmax, S0.5, and nHwere calculated, as detailed in Materials and Methods, from averaged data from three independent experiments, using regression analysis.
On the other hand, GSase was analyzed with respect to its NDP-Glc specificity. We found that the S. coelicolor GSase strictly used ADP-Glc as the glucosyl donor for significant elongation of a preformed glycogen molecule. UDP-Glc rendered negligible enzyme activity even at concentrations as high as 10 mM (namely, 40-fold the GSase S0.5 for ADP-Glc). In addition, UDP-Glc was not an inhibitor of ADP-Glc transferring activity. We also analyzed soluble starch and pectin as S. coelicolor GSase substrates, but neither of them was employed for the elongation reaction (data not shown).
Analysis of possible allosteric effectors.
Activation inhibition assays were performed for both ADP-Glc PPase and UDP-Glc PPase with intermediates of central metabolic pathways for carbon and energy of the cell, which are known to be important effectors of characterized ADP-Glc PPases from different sources (3, 4). The metabolites utilized were phosphoenolpyruvate (PEP), fructose-1,6-bisphosphate (Fru-1,6-bisP), pyruvate (Pyr), 3-phosphoglyceric acid (3-PGA), Glc-6P, ribose-5P (Rib-5P), fructose-6P (Fru-6P), Man-1P, Man-6P, Pi, AMP, ADP, NAD+, NADH, NADP+, and NADPH. The concentrations of the effectors analyzed went up to 5 mM while the substrates of the reaction mixtures were maintained at saturating concentrations. As shown in Fig. 2, under these conditions Pyr, Fru-6P, Rib-5P, Man-6P, PEP, and Glc-6P worked as activators of ADP-Glc PPase from S. coelicolor A3(2). Saturation curves (Fig. 3) showed a distinctive behavior for each allosteric positive modulator, increasing the enzyme activity between 3- and 33-fold, as shown in Fig. 2 (note the logarithmic scale utilized for enzyme activity) and Fig. 3. Conversely, Pi and NADPH were found to be important as inhibitors of ADP-Glc PPase activity (Fig. 2), with the two metabolites exhibiting similar concentration-dependent effects (Fig. 4). It is noteworthy that neither Glc-6P nor Man-6P was ever reported as the main effector of any bacterial ADP-Glc PPase characterized previously; moreover, this is the first time that an ADP-Glc PPase shows inhibition by NADPH (3, 4). Recently, Boehlein et al. (6, 7) showed that Fru-6P and Glc-6P are effectors of the maize ADP-Glc PPase, both hexose-P activating the enzyme to extents similar to that for (although with higher A0.5 than) 3-PGA, the primary activator of the plant enzyme. For the maize enzyme, the authors determined an A0.5 of 4 mM for Glc-6P, which is about 18-fold higher than that corresponding to 3-PGA (6, 7).
Fig 2.

Relative ADP-Glc PPase activity (synthetic reaction) assayed in the absence or in the presence of different metabolites (at 2 mM).
Fig 3.
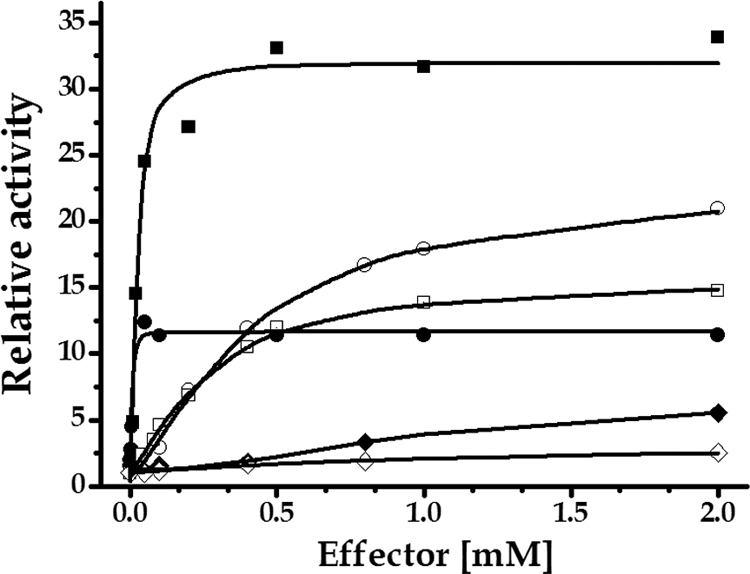
Saturation plots for S. coelicolor A3(2) ADP-Glc PPase effectors. Assays were conducted as synthetic reactions in which Glc-1P and ATP concentrations were 1 mM. Symbols: filled square, Glc-6P; empty circle, Fru-6P; empty square, PEP; filled circle, Man-6P; filled rhombus, Rib-5P; empty rhombus, Pyr. A0.5 values were calculated, as the respective activator concentrations giving 50% of the maximal activation.
Fig 4.
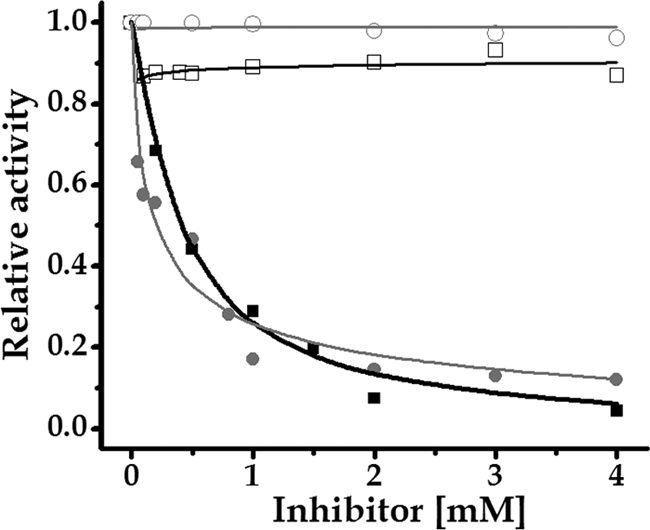
Inhibitory effect of NADPH and Pi on the activity of ADP-Glc PPase from S. coelicolor. The enzyme activity was assayed in the ADP-Glc synthesis direction as described in Materials and Methods in the absence of allosteric positive modulator (filled squares and black curves, NADPH; filled circles and gray curves, Pi) or in the presence of 0.5 mM Glc-6P (empty squares, NADPH; empty circles, Pi).
In the analysis of UDP-Glc PPase, none of the metabolites assayed affected the enzyme activity. These results are in agreement with data reported for bacterial UDP-Glc PPases, which in general are not influenced by allosteric effectors. Also, metabolites from the tricarboxylic acid cycle were evaluated for their activation-inhibition effects. None of them affected the activity of either ADP-Glc PPase or UDP-Glc PPase (data not shown). On the other hand, GSase enzyme activity was not modified when the different metabolites, including those found to be effectors for S. coelicolor ADP-Glc PPase, were analyzed. This is in accordance with the fact that in bacteria the reaction performed by ADP-Glc PPase is the key step in the pathway leading to glycogen biosynthesis, which is under allosteric regulation by intermediary metabolites corresponding to the principal pathway of carbon utilization in the organism (3, 4).
Kinetic characterization of recombinant ADP-Glc PPase effectors.
Characterization of the effectors of the ADP-Glc PPase enzyme is particularly important in order to have a better comprehension of the glycogen synthesis and regulation in S. coelicolor A3(2). ADP-Glc PPases are allosterically regulated by compounds belonging to the main carbon assimilation route in the organism (3, 4). We found that S. coelicolor ADP-Glc PPase exhibits high promiscuity regarding allosteric effectors compared to other bacterial systems (3, 4). This remarkable feature may indicate the relevance in connecting the polysaccharide synthesis with different metabolic processes. Thus, we conducted effector curves in the presence of a saturating concentration of the substrates, to obtain the kinetic parameters shown in Table 4. From these results, as well as from those shown in Fig. 3, it can be deduced that a major effector molecule for ADP-Glc PPase is Glc-6P, since it increases Vmax 33-fold with an A0.5 value of 0.03 mM. Although Man-6P has the lowest A0.5 value (0.01 mM) among the activators found, it increased ADP-Glc PPase activity 12-fold, which is a lower activation level than those produced by Fru-6P (21-fold) and PEP (14-fold) (Table 4 and Fig. 3). On the other hand, Pyr proved to be the less effective activator, increasing by only 3-fold the ADP-Glc PPase Vmax. Rib-5P, Fru-6P, and PEP showed an appreciable activation effect (Fig. 3), with higher A0.5 values than Glc-6P. In the ADP-Glc pyrophosphorolysis process of reaction, all effectors showed comparable activations, with A0.5 values in the same order of magnitude, compared with those for the synthetic (physiological) direction of catalysis (data not shown).
Table 4.
Kinetic parameters for allosteric effectors of S. coelicolor A3(2) ADP-Glc PPase in a physiological reaction (ADP-Glc synthesis)a
| Effector | A0.5 (mM) | nH | Activation (fold) |
|---|---|---|---|
| Pyr | 0.62 ± 0.06 | 1.2 | 3 |
| Rib-5P | 1.20 ± 0.06 | 2.1 | 7 |
| Man-6P | 0.011 ± 0.001 | 2.0 | 12 |
| PEP | 0.25 ± 0.05 | 1.0 | 14 |
| Fru-6P | 0.34 ± 0.04 | 1.4 | 21 |
| Glc-6P | 0.03 ± 0.01 | 1.3 | 33 |
Hill plots were used to calculate the nH (Hill coefficient) and the A0.5 (activator concentration giving 50% of the maximal activation).
ADP-Glc PPase kinetics in the presence of a saturating concentration of effectors.
To further characterize the specific action of an allosteric effector, it is relevant to establish, in detail, how its binding to a site other than the active site modifies the kinetic properties of one enzyme, including the specific activity as well as the affinity toward substrates. The kinetic parameters for S. coelicolor ADP-Glc PPase were determined in the ADP-Glc synthetic direction of reaction in the presence of each of the different effectors, and they were compared with those obtained in the absence of any of them (Table 5). It can be observed that PEP and Man-6P are the metabolites producing significant modifications of S0.5 for substrates in the enzyme, increasing 5-fold the affinity for both Glc-1P and ATP. In a complete analysis of positive modulators, it is remarkable that the strong effect detected for Glc-6P, which exerted the highest increase in Vmax and also changed the affinity for both substrates employed in the physiological reaction, catalyzed the enzyme, with a 3- to 4-fold increase in affinity (Table 5). S. coelicolor A3(2) ADP-Glc PPase seems to have characteristics similar to those of characterized orthologous enzymes from Gram-negative organisms in respect to their activation by signals of high carbon and energy in the intracellular environment, although it differs from them in specificity for the regulatory metabolites, being activated by Pyr, Rib-5P, Fru-6P, Man-6P, PEP, and particularly Glc-6P.
Table 5.
Kinetic parameters for S. coelicolor A3(2) ADP-Glc PPasea
| Substrate |
S0.5 (mM) |
|
|---|---|---|
| Glc-1P | ATP | |
| No effector | 0.37 ± 0.03 | 0.39 ± 0.04 |
| Pyr | 0.17 ± 0.02 | 0.38 ± 0.03 |
| PEP | 0.07 ± 0.01 | 0.10 ± 0.01 |
| Man-6P | 0.09 ± 0.01 | 0.07 ± 0.01 |
| Rib-5P | 0.13 ± 0.01 | 0.31 ± 0.02 |
| Fru-6P | 0.34 ± 0.03 | 0.18 ± 0.02 |
| Glc-6P | 0.19 ± 0.03 | 0.13 ± 0.01 |
Kinetic parameters were determined in the presence or absence of saturating concentrations of each activator. S0.5, the substrate concentration giving 50% of Vmax in the absence or in the presence of 2 mM each specified activator.
The ADP-Glc PPase from S. coelicolor was also characterized for those metabolites that resulted in negative modulators. We determined I0.5 values of 0.41 mM and 0.19 mM for NADPH and Pi, respectively, in a synthetic reaction (Fig. 4). The inhibition of S. coelicolor ADP-Glc PPase by NADPH is a particularly notable case of regulation, since this metabolite was never reported as an inhibitor of any ADP-Glc PPase (3, 4, 37), although it was reported as an activator of the ADP-Glc PPase from E. coli (20). In addition, Pi was largely characterized as an inhibitor of many ADP-Glc PPases from photosynthetic organisms (both prokaryotes and eukaryotes) as well as in the bacterium Agrobacterium tumefaciens (3, 4). We also observed cross talk between positive and negative modulators of the enzyme activity. Thus, increasing amounts of Glc-6P avoided the inhibitory effect of NADPH and Pi, becoming almost impervious to inhibition in the presence of 0.5 mM Glc-6P (Fig. 4). This situation is similar to that occurring with ADP-Glc PPase in cyanobacteria, where the presence of 3-PGA (an activator of the cyanobacterial ADP-Glc PPase) produces an increment in the I0.5 for Pi (an inhibitor) (3, 4).
Concluding remarks.
Figure 5 depicts metabolic routes occurring in S. coelicolor for the utilization of Glc-6P, as inferred from the genome information available and from previous studies (5, 8, 9, 25, 34, 38, 42). In this context we highlight the relevance of each of the enzymes studied in this work and how they integrate with the other major pathways involved in glucose utilization. The kinetic characterization of UDP-Glc PPase, ADP-Glc PPase, and GSase and, particularly, the regulatory properties found for ADP-Glc PPase contribute to a better understanding of how the flux of Glc-6P can be directed to different metabolic fates. The hexose-P is a node of a key metabolic enclave in the bacterium, from which carbohydrates flow toward oxidative or conservative pathways. Oxidation of Glc-6P occurs mainly via the Embden-Meyerhof-Parnas pathway (EMPP) and the oxidative pentose-P pathway (OPPP), fueling the cell with either ATP or NADPH as well as with useful intermediates for secondary metabolite biosynthesis (8, 25, 38). On the other hand, Glc-1P is the initial metabolite of routes allowing sugar/NDP-sugar interconversion, synthesis of oligo- and polysaccharide structural components, or production of reserve glucans (glycogen) (12, 32). A critical point determining metabolic fates for Glc-1P/Glc-6P within a cell is the presence or absence of UDP-sugar PPase, an enzyme that so far has been found in plants and protozoa (15, 16, 22, 26, 28, 29, 41). We omitted this enzyme in the metabolic map detailed in Fig. 5 after finding no putative gene encoding it on the S. coelicolor genome in an exhaustive search performed using as templates known sequences of UDP-sugar PPases from protozoa (Leishmania major, Trypanosoma cruzi) and plants (Arabidopsis thaliana, Glycine max). The closer relative we found in such a search was the UDP-Glc PPase characterized herein, sharing only 5 to 6% identity with templates. The latter reinforces the apparent absence of UDP-sugar PPase in S. coelicolor in the work detailed by Kleczkowski et al. (26), where the authors conclude that based on comparison of sequences derived from cDNA and/or genomic clones, in most bacteria it is not possible to distinguish that enzyme from UDP-Glc (or UDP-glucosamine) PPases.
Fig 5.

Metabolic scenario for Glc-6P partitioning in S. coelicolor, with emphasis on the involvement of ADP-Glc PPase, UDP-Glc PPase, and GSase activities and the differential regulation of the first enzyme. Green (with color degree directly proportional to stronger effect) and red boxes mark metabolites behaving as allosteric activators and inhibitors, respectively, of ADP-Glc PPase. Wide gray arrows symbolize metabolic fluxes involving several reactions. Abbreviations: EMPP, Embden-Meyerhof-Parnas pathway; OPPP, oxidative pentose-P pathway, ManB, P-mannose mutase; Pgm, P-glucomutase; Gmpp, GDP-Man pyrophosphorylase; Pgi, P-glucose isomerase.
The carbon flux analysis detailed in Fig. 5 indicates that the use of Glc-1P by UDP-Glc PPase, leading to sugar interconversion and structural polyglycans, is highly active, and it would be under poor (if any) regulation, according to our kinetic studies on this enzyme. Conversely, the flux of Glc-1P to storage polysaccharides via ADP-Glc would be limiting and finely regulated in connection with levels of energy and reductive pathway in the cell, considering the differences in Vmax and regulatory properties determined for ADP-Glc PPase in comparison with those of the enzyme synthesizing UDP-Glc. Thus, an observation of activation of ADP-Glc PPase by Glc-6P, Fru-6P, PEP, Pyr, and Rib-5P agrees with the fact that synthesis of glycogen is upheld when high levels of intermediates support the functioning of the oxidative pathways providing ATP and NADPH for anabolism. Reciprocally, the polysaccharide buildup is inhibited under low energy conditions, represented by the increase in Pi levels. This scenario for the regulation of ADP-Glc PPase by levels of energy (coincident with the fact that the enzyme uses ATP to synthesize ADP-Glc) is common to previous reports concerning the enzyme from different prokaryotic or eukaryotic sources (3, 4), but for the S. coelicolor enzyme the response to Glc-6P and Rib-5P as main allosteric activators is distinctive.
The activation by Man-6P found for the S. coelicolor ADP-Glc PPase is also a novel feature of this enzyme. It may be speculated that this regulation leads to Glc storage as glycogen when carbon skeletons exceed the demands of anabolic routes. As illustrated in Fig. 5, Man-6P is converted to Man-1P by ManB, an enzyme that can also catalyze conversion of Glc-6P to Glc-1P (42) (Fig. 5). Man-1P is needed for GDP-mannose (GDP-Man) production, which is an important precursor for several bioactive products in Streptomyces spp., including the antifungal polyenes, the antibacterial antibiotics hygromycin A and mannopeptimycins, and the anticancer agent bleomycin (35). Also, Man-P may be involved in biosynthesis of mannose-containing polysaccharides, which are not clearly identified as components in Streptomyces spp. but which have been extensively characterized in mycobacteria (24). Of particular interest is the observed inhibition of the S. coelicolor enzyme by NADPH (never found for other ADP-Glc PPases), as this could be interpreted as a mechanism to direct carbon skeletons primarily to synthesize antibiotics (rather than to accumulate glycogen) when reductive power is available and supposedly subutilized due to relatively low levels of carbon intermediates. The latter findings, and the fact that there is an interplay between Glc-6P activation and NADPH inhibition of the ADP-Glc PPase, support a mechanism to balance carbon utilization with supporting levels of necessary reducing equivalents for the key secondary metabolism that takes place in S. coelicolor.
ACKNOWLEDGMENTS
This work was supported by grants from CONICET (PIP 2519), ANPCyT (PICT'08 1640 and 1754 to H.G. and A.A.I., respectively) and UNL (CAID Orientados y Redes). M.D.A.D. is a fellow from CONICET, and S.P., H.G., and A.A.I. are investigators from the same institution.
Footnotes
Published ahead of print 30 December 2011
REFERENCES
- 1. Asencion Diez MD, et al. 2010. Functional characterization of GDP-mannose pyrophosphorylase from Leptospira interrogans serovar Copenhageni. Arch. Microbiol. 192:103–114 [DOI] [PubMed] [Google Scholar]
- 2. Ballicora MA, et al. 2007. Identification of regions critically affecting kinetics and allosteric regulation of the Escherichia coli ADP-glucose pyrophosphorylase by modeling and pentapeptide-scanning mutagenesis. J. Bacteriol. 189:5325–5333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ballicora MA, Iglesias AA, Preiss J. 2003. ADP-glucose pyrophosphorylase, a regulatory enzyme for bacterial glycogen synthesis. Microbiol. Mol. Biol. Rev. 67:213–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ballicora MA, Iglesias AA, Preiss J. 2004. ADP-glucose pyrophosphorylase: a regulatory enzyme for plant starch synthesis. Photosynth. Res. 79:1–24 [DOI] [PubMed] [Google Scholar]
- 5. Bentley SD, et al. 2002. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417:141–147 [DOI] [PubMed] [Google Scholar]
- 6. Boehlein SK, Shaw JR, Hannah LC, Stewart JD. 2010. Probing allosteric binding sites of the maize endosperm ADP-glucose pyrophosphorylase. Plant Physiol. 152:85–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Boehlein SK, Shaw JR, Stewart JD, Hannah LC. 2008. Heat stability and allosteric properties of the maize endosperm ADP-glucose pyrophosphorylase are intimately intertwined. Plant Physiol. 146:289–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Borodina I, Krabben P, Nielsen J. 2005. Genome-scale analysis of Streptomyces coelicolor A3(2) metabolism. Genome Res. 15:820–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Borodina I, et al. 2008. Antibiotic overproduction in Streptomyces coelicolor A3 2 mediated by phosphofructokinase deletion. J. Biol. Chem. 283:25186–25199 [DOI] [PubMed] [Google Scholar]
- 10. Bosco MB, Machtey M, Iglesias AA, Aleanzi M. 2009. UDPglucose pyrophosphorylase from Xanthomonas spp. Characterization of the enzyme kinetics, structure and inactivation related to oligomeric dissociation. Biochimie 91:204–213 [DOI] [PubMed] [Google Scholar]
- 11. Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254 [DOI] [PubMed] [Google Scholar]
- 12. Brana AF, Mendez C, Diaz LA, Manzanal MB, Hardisson C. 1986. Glycogen and trehalose accumulation during colony development in Streptomyces antibioticus. J. Gen. Microbiol. 132:1319–1326 [DOI] [PubMed] [Google Scholar]
- 13. Buschiazzo A, et al. 2004. Crystal structure of glycogen synthase: homologous enzymes catalyze glycogen synthesis and degradation. EMBO J. 23:3196–3205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Busi MV, et al. 2008. Functional and structural characterization of the catalytic domain of the starch synthase III from Arabidopsis thaliana. Proteins 70:31–40 [DOI] [PubMed] [Google Scholar]
- 15. Damerow S, et al. 2010. Leishmania UDP-sugar pyrophosphorylase: the missing link in galactose salvage? J. Biol. Chem. 285:878–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dickmanns A, et al. 2011. Structural basis for the broad substrate range of the UDP-sugar pyrophosphorylase from Leishmania major. J. Mol. Biol. 405:461–478 [DOI] [PubMed] [Google Scholar]
- 17. Furlong CE, Preiss J. 1969. Biosynthesis of bacterial glycogen. VII. Purification and properties of the adenosine diphosphoglucose pyrophosphorylase of Rhodospirillium rubrum. J. Biol. Chem. 244:2539–2548 [PubMed] [Google Scholar]
- 18. Fusari C, Demonte AM, Figueroa CM, Aleanzi M, Iglesias AA. 2006. A colorimetric method for the assay of ADP-glucose pyrophosphorylase. Anal. Biochem. 352:145–147 [DOI] [PubMed] [Google Scholar]
- 19. Ghosh HP, Preiss J. 1966. Adenosine diphosphate glucose pyrophosphorylase. A regulatory enzyme in the biosynthesis of starch in spinach leaf chloroplasts. J. Biol. Chem. 241:4491–4504 [PubMed] [Google Scholar]
- 20. Govons S, Gentner N, Greenberg E, Preiss J. 1973. Biosynthesis of bacterial glycogen. XI. Kinetic characterization of an altered adenosine diphosphate-glucose synthase from a “glycogen-excess” mutant of Escherichia coli B. J. Biol. Chem. 248:1731–1740 [PubMed] [Google Scholar]
- 21. Greenberg E, Preiss J. 1964. The occurrence of adenosine diphosphate glucose: glycogen transglucosylase in bacteria. J. Biol. Chem. 239:4314–4315 [PubMed] [Google Scholar]
- 22. Gronwald JW, Miller SS, Vance CP. 2008. Arabidopsis UDP-sugar pyrophosphorylase: evidence for two isoforms. Plant Physiol. Biochem. 46:1101–1105 [DOI] [PubMed] [Google Scholar]
- 23. Hardisson C, Manzanal MB. 1976. Ultrastructural studies of sporulation in Streptomyces. J. Bacteriol. 127:1443–1454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Harris LS, Gray GR. 1977. Acetylated methylmannose polysaccharide of Streptomyces. J. Biol. Chem. 252:2470–2477 [PubMed] [Google Scholar]
- 25. Hodgson DA. 2000. Primary metabolism and its control in streptomycetes: a most unusual group of bacteria. Adv. Microb. Physiol. 42:47–238 [DOI] [PubMed] [Google Scholar]
- 26. Kleczkowski LA, Decker D, Wilczynska M. 2011. UDP-sugar pyrophosphorylase: a new old mechanism for sugar activation. Plant Physiol. 156:3–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kleczkowski LA, Villand P, Preiss J, Olsen OA. 1993. Kinetic mechanism and regulation of ADP-glucose pyrophosphorylase from barley (Hordeum vulgare) leaves. J. Biol. Chem. 268:6228–6233 [PubMed] [Google Scholar]
- 28. Kotake T, et al. 2007. Properties and physiological functions of UDP-sugar pyrophosphorylase in Arabidopsis. Biosci. Biotechnol. Biochem. 71:761–771 [DOI] [PubMed] [Google Scholar]
- 29. Kotake T, et al. 2004. UDP-sugar pyrophosphorylase with broad substrate specificity toward various monosaccharide 1-phosphates from pea sprouts. J. Biol. Chem. 279:45728–45736 [DOI] [PubMed] [Google Scholar]
- 30. Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685 [DOI] [PubMed] [Google Scholar]
- 31. Lai X, Wu J, Chen S, Zhang X, Wang H. 2008. Expression, purification, and characterization of a functionally active Mycobacterium tuberculosis UDP-glucose pyrophosphorylase. Protein Expr. Purif. 61:50–56 [DOI] [PubMed] [Google Scholar]
- 32. Martin MC, Schneider D, Bruton CJ, Chater KF, Hardisson C. 1997. A glgC gene essential only for the first of two spatially distinct phases of glycogen synthesis in Streptomyces coelicolor A3(2). J. Bacteriol. 179:7784–7789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McVittie A. 1974. Ultrastructural studies on sporulation in wild-type and white colony mutants of Streptomyces coelicolor. J. Gen. Microbiol. 81:291–302 [DOI] [PubMed] [Google Scholar]
- 34. Mira de Orduna R, Theobald U. 2000. Intracellular glucose 6-phosphate content in Streptomyces coelicolor upon environmental changes in a defined medium. J. Biotechnol. 77:209–218 [DOI] [PubMed] [Google Scholar]
- 35. Nic Lochlainn L, Caffrey P. 2009. Phosphomannose isomerase and phosphomannomutase gene disruptions in Streptomyces nodosus: impact on amphotericin biosynthesis and implications for glycosylation engineering. Metab. Eng. 11:40–47 [DOI] [PubMed] [Google Scholar]
- 36. Ning B, Elbein AD. 1999. Purification and properties of mycobacterial GDP-mannose pyrophosphorylase. Arch. Biochem. Biophys. 362:339–345 [DOI] [PubMed] [Google Scholar]
- 37. Preiss J. 1984. Bacterial glycogen synthesis and its regulation. Annu. Rev. Microbiol. 38:419–458 [DOI] [PubMed] [Google Scholar]
- 38. Ryu YG, Butler MJ, Chater KF, Lee KJ. 2006. Engineering of primary carbohydrate metabolism for increased production of actinorhodin in Streptomyces coelicolor. Appl. Environ. Microbiol. 72:7132–7139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, third ed., vol 1 Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 40. Sheng F, Jia X, Yep A, Preiss J, Geiger JH. 2009. The crystal structures of the open and catalytically competent closed conformation of Escherichia coli glycogen synthase. J. Biol. Chem. 284:17796–17807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yang T, Bar-Peled M. 2010. Identification of a novel UDP-sugar pyrophosphorylase with a broad substrate specificity in Trypanosoma cruzi. Biochem. J. 429:533–543 [DOI] [PubMed] [Google Scholar]
- 42. Yang YH, et al. 2010. Loss of phosphomannomutase activity enhances actinorhodin production in Streptomyces coelicolor. Appl. Microbiol. Biotechnol. 86:1485–1492 [DOI] [PubMed] [Google Scholar]
- 43. Yeo M, Chater K. 2005. The interplay of glycogen metabolism and differentiation provides an insight into the developmental biology of Streptomyces coelicolor. Microbiology 151:855–861 [DOI] [PubMed] [Google Scholar]
- 44. Yep A, Ballicora MA, Preiss J. 2004. The active site of the Escherichia coli glycogen synthase is similar to the active site of retaining GT-B glycosyltransferases. Biochem. Biophys. Res. Commun. 316:960–966 [DOI] [PubMed] [Google Scholar]
- 45. Yep A, et al. 2004. An assay for adenosine 5′-diphosphate (ADP)-glucose pyrophosphorylase that measures the synthesis of radioactive ADP-glucose with glycogen synthase. Anal. Biochem. 324:52–59 [DOI] [PubMed] [Google Scholar]
- 46. Zhou X, Wu H, Li Z, Zhou X, Bai L, Deng Z. 2011. Overexpression of UDP-glucose pyrophosphorylase increases validamycin A but decreases validoxylamine A production in Streptomyces hygroscopicus var. jinggangensis 5008. Metab. Eng. 13:768–776 [DOI] [PubMed] [Google Scholar]