

Abstract
Rap proteins in Bacillus subtilis regulate the phosphorylation level or the DNA-binding activity of response regulators such as Spo0F, involved in sporulation initiation, or ComA, regulating competence development. Rap proteins can be inhibited by specific peptides generated by the export-import processing pathway of the Phr proteins. Rap proteins have a modular organization comprising an amino-terminal alpha-helical domain connected to a domain formed by six tetratricopeptide repeats (TPR). In this study, the molecular basis for the specificity of the RapA phosphatase for its substrate, phosphorylated Spo0F (Spo0F∼P), and its inhibitor pentapeptide, PhrA, was analyzed in part by generating chimeric proteins with RapC, which targets the DNA-binding domain of ComA, rather than Spo0F∼P, and is inhibited by the PhrC pentapeptide. In vivo analysis of sporulation efficiency or competence-induced gene expression, as well as in vitro biochemical assays, allowed the identification of the amino-terminal 60 amino acids as sufficient to determine Rap specificity for its substrate and the central TPR3 to TPR5 (TPR3-5) repeats as providing binding specificity toward the Phr peptide inhibitor. The results allowed the prediction and testing of key residues in RapA that are essential for PhrA binding and specificity, thus demonstrating how the widespread structural fold of the TPR is highly versatile, using a common interaction mechanism for a variety of functions in eukaryotic and prokaryotic organisms.
INTRODUCTION
The initiation of sporulation in the Gram-positive organism Bacillus subtilis is regulated by the complex phosphorelay signal transduction system. In this system, sporulation-activating signals are sensed by multiple sensor histidine kinases whose activation results in autophosphorylation and phosphoryl transfer to an intermediate component, the Spo0F response regulator. From phosphorylated Spo0F (Spo0F∼P), the phosphoryl group is then transferred to the Spo0B phosphotransferase, which then relays it to the Spo0A response regulator and transcription factor. The phosphorylation level of Spo0A in the cell is the determining factor of whether sporulation will initiate or not in response to the activating signals sensed by the kinases (7, 15, 22, 23).
In order to counteract the kinase activities and prevent untimely initiation of sporulation, a number of phosphatases exist to respond to physiological states antithetical to sporulation, such as growth and competence for DNA transformation. There are two families of phosphatases, classified by structure and substrate specificity: the Spo0E-like family and the Rap family (30, 32).
The members of the Spo0E family of phosphatases (Spo0E, YisI, and YnzD) are small proteins (56 to 85 amino acids) identified by a conserved sequence motif, SQ/RE/DLD, in which the aspartate in the fifth position is the essential catalytic residue. Spo0E-like phosphatases specifically dephosphorylate the Spo0A∼P protein via a mechanism involving the conserved aspartate residue protruding from the α2 helix of the double helix fold that characterizes the structure of these proteins (14, 26).
The Rap family comprises 11 members in B. subtilis. RapA, -B, -E, and -H act by dephosphorylating the Spo0F∼P response regulator upon induction by the competence pathway (RapA, -E, and -H) or upon induction by conditions promoting cell growth (RapB) (21, 33, 40). RapJ also dephosphorylates Spo0F∼P in vitro and inhibits sporulation when overexpressed in vivo (27; our unpublished data). The RapC and RapF proteins, together with the dual-specificity RapH protein, inhibit the DNA binding activity of the ComA response regulator for competence development (4, 8, 40, 41), while RapG inhibits the DNA-binding activity of the DegU regulator for degradative enzyme production, competence, and motility (25). The RapI protein serves a role in the movement of the ICEBs1 transposon (2). The function of RapD and RapK is still unknown.
Rap proteins are approximately 380 amino acids long and are characterized by a central domain composed of five tetratricopeptide repeats (TPR) that are predicted to form a groove-like structure potentially critical for protein-protein interaction (11, 31). N-terminal to the first TPR is a stretch of approximately 100 amino acids that is highly conserved among Rap proteins. The central TPR-containing domain is separated from a C-terminal domain carrying a sixth TPR by an approximately 40-amino-acid-long connector region. While the manuscript was in preparation, the structure of the RapH protein in complex with Spo0F was reported. The structure showed that the amino-terminal domain of RapH consists of a 3-helix bundle followed by a 6-unit TPR domain as predicted (27).
Rap proteins are commonly paired with specific Phr penta- or hexapeptides that serve to inhibit their phosphatase activity or their binding to the response regulator carboxy-terminal domain. Phr peptides originate by posttranslational processing of the product of the phr gene, which is genetically linked and often cotranscribed with the upstream rap gene (24, 29, 35).
Our previous studies identified two missense mutations in RapA that abolished the PhrA pentapeptide inhibitory effect on the phosphatase (D192N and P259L). Positioning of these residues on a model structure of RapA and inferences from available structures of TPR-containing proteins in complex with their target peptides led us to hypothesize that Phr peptides bind to their partner Rap proteins in an extended conformation along the concave surface formed by the TPR3 to TPR5 (TPR3-5) motifs of the TPR domain (8, 16, 31, 37).
In this study, we used biochemical and genetic approaches to understand the molecular determinants that allow the RapA protein to identify its specific substrate and inhibitor, as well as the mechanism underlying the inhibition of RapA activity by the PhrA pentapeptide.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Escherichia coli DH5α was used for plasmid construction and propagation. Strains were grown in lysogeny broth (LB) supplemented with ampicillin (100 μg/ml) or kanamycin (30 μg/ml).
The B. subtilis strains used in this study were derived from the wild-type strain JH642 (trpC2 phe-1), the rapA mutant JH12834 (trpC2 phe-1 rapA::Tn917 erm) (33), and the rapC-lacZ reporter strain JH19278 (spo0A12 abrB::Tn917 rapC::spc amyE::rapC-lacZ aphA-3), which is a derivative of strains JH12923 and JH12963, described by Core and Perego (8).
B. subtilis strains were grown in Schaeffer's sporulation medium (36) supplemented with the appropriate antibiotic at the following concentrations: chloramphenicol, 5 μg/ml; kanamycin, 2 μg/ml; or erythromycin, 1 μg/ml. Competent cells of B. subtilis strains were prepared by the method of Anagnostopoulos and Spizizen (1).
Sporulation assays were carried out in 5 ml of Schaeffer's medium at 37°C for 48 h. Serial dilutions were plated in duplicate before and after treatment with 0.5 ml of CHCl3. Colony counts were averaged, and the percentage of sporulation was calculated as the ratio between the survival and the viable counts.
Plasmid construction.
Plasmid pMP9239 is a derivative of the multicopy vector pBS19 (34) carrying a 1,764-bp fragment spanning from a PstI site located 595 bp upstream of the rapA starting codon to a BsiE site located 26 bp downstream of the rapA stop codon and within the phrA coding sequence. Integration of this plasmid by single crossover in the B. subtilis chromosome results in inactivation of the phrA gene and therefore a sporulation-deficient phenotype.
Plasmid pBS-RapAC1 is a derivative of pMP9239 in which the 3′-end fragment from EcoRI (in the rapA coding sequence) to the KpnI site (in the vector's multiple cloning site) was replaced by a PCR-amplified fragment of the 3′ end of the rapC gene obtained using the oligonucleotide primers OMEcoRISWAP and OM3′Kpn (see Fig. 2).
Fig 2.

Amino acid sequence alignment of RapA and RapC. The alignment was obtained with the ClustalW program. Asterisks indicate identical residues; colons and periods indicate conserved and semiconserved substitutions, respectively. The six TPR domains as defined by amino acid sequence conservation are in the gray boxes (31). The extent of the two α-helices that include each TPR domain, as determined by the crystal structure of RapH, is indicated by the green line (α1) or the yellow line (α2) (27). The position of the RapAC fusions is indicated by the red connectors, while the position of the RapCA fusion is shown by the blue connector. The residues corresponding to the regions in RapH that form four α-helices in the N terminus and the two α-helices in the connecting region between TPR5 and TPR6, identified by the crystal structure of the RapH-Spo0F complex, are shown by the black lines (27). The six residues affected in PhrA binding are shown in red (D192, Y224, N225, N228, H260, and P259).
Plasmid pBS-RapCA1 is a derivative of pBS19 carrying the chimeric rapCA gene expressed from the rapC promoter. This was obtained by PCR amplification of the 5′ end of the rapC gene and its promoter with the oligonucleotide primers OM5′Kpn-RapCBsmA and the amplification of the 3′ end of the rapA gene with the primers RapABsmA-RapA3′Pst. The two fragments were digested with BsmAI and ligated to each other. The product of this ligation was digested with KpnI and PstI and ligated in similarly digested pBS19.
The pBS-RapAC2 chimera was generated with the gene splicing by overlapping extension (SOE) method (17) using the oligonucleotide primer pairs RapA5′Pst-RapASOE-70 and RapCSOE-70-OM3′Kpn. The SOE method was also used for the construction of plasmid pBS-RapAC3 (using the oligonucleotide primer pairs RapA5′Pst-RapASOE-122 and RapCSOE-122-OM3′Kpn) and pBS-RapAC4 (using the oligonucleotide primers RapA5′Pst-RapASOE+105 and RapSOE+105-OM3′Kpn).
The RapAC5 hybrid was generated only in the pET16cat vector by the SOE method using the oligonucleotide primers OL5′Bam-RapAC53′ and RapAC55-OMexp3′. The PCR-amplified fragment was digested with BamHI and cloned in the similarly digested pET16cat vector.
Plasmid pET16cat is a derivative of pET16b (Novagen) carrying, in the NruI site, the chloramphenicol acetyltransferase gene (cat), obtained as a SmaI-HincII fragment from the cat cassette vector pJM105 (28). Plasmid pMP9230 (33) is pET16b (Novagen) carrying the rapA coding sequence obtained by PCR amplification of chromosomal DNA of strain JH642 using the oligonucleotide primers OL5′Bam and OL3′Bam. Cloning this fragment into the BamHI site of pET16b resulted in the fusion of 10 histidine codons to the 5′ end of rapA. Introduction of the cat cassette in the NruI site of pMP9230 resulted in plasmid pMP9230cat, used for site-directed mutagenesis and protein expression.
The rapAC hybrid genes were cloned into the pET16cat vector as BamHI fragments obtained by PCR amplification using the pBS-RapAC plasmid as a template and the OL5′Bam-OMexp3′ pair of oligonucleotide primers. The rapCA hybrid gene was PCR amplified from pBS-RapCA1 using the oligonucleotide primers OMexp5′-OL3′Bam, digested with NdeI and BamHI, and cloned into similarly digested pET16cat.
Site-directed mutagenesis.
Site-directed mutagenesis was performed with the QuikChange site-directed mutagenesis kit according to the manufacturer's instructions (Stratagene). The pET16bcat vector carrying the rapA wild-type coding sequence (pMP9230cat) was used as a template. The alanine replacements were created using the mutagenic oligonucleotide primers indicated in Table S1 in the supplemental material. Every allele was sequenced in order to ensure the fidelity of the PCR. Transformation of these plasmids into B. subtilis results in chromosomal integration by single crossover, generating a full-length rapA gene expressed under its own promoter and a nonexpressed duplicated rapA coding sequence with the phrA gene. Transformants were selected for chloramphenicol resistance. RapA mutant proteins defective in the interaction with the PhrA inhibitor were identified by reactivating the phrA gene by means of plasmid pMP9258. This plasmid carries the rapA-phrA operon with an out-of-frame mutation at the BglII site within the rapA coding sequence. Upon integration in the chromosome by double crossover in the amyE gene, this construct promotes transcription of the phrA gene from the rapA-phrA promoter.
Kinetic analyses.
Kinetic parameters of the RapA-dependent Spo0F∼P dephosphorylation reaction were obtained by measuring the rates in the presence of 10 concentrations (0.33, 0.5, 1, 1.33, 2, 2.5, 3.33, 5, 7.5, and 10 μM) of Spo0F∼P. Seven time points (T0 to T6) were taken for each substrate concentration. The enzyme was used at a 1 μM final concentration. The rate of phosphatase activity at each substrate concentration was calculated from the slope of the plot of the percentages of remaining Spo0F∼P versus time, multiplied by the concentration of substrate according to the following equation: (100 − % remaining Spo0F∼P at T6)/time (s) × [spo0F∼P] = rate of phosphatase activity (μM/s).
The reactions were carried out in phosphatase buffer (50 mM HEPES [pH 7], 50 mM KCl, 10 mM dithiothreitol [DTT], 0.1 mM EDTA, 20% glycerol) and analyzed on 15% Tris-Tricine-SDS gels. Reactions were initiated by the addition of Spo0F∼P. An aliquot of each reaction was removed immediately after mixing, added to SDS loading dye, and frozen in a dry ice-ethanol bath. The radioactivity of Spo0F∼P in this sample from each reaction was used as the 100% value of remaining Spo0F∼P for normalization. Gels were dried and exposed to a PhosphorImager screen for quantitation with the ImageQuant software program.
The kinetics of PhrA inhibition of RapA was carried out by measuring the rates of dephosphorylation of 6 concentrations of Spo0F∼P (0.4, 1, 2, 3, 6, and 12 μM) in the presence of 4 concentrations of PhrA pentapeptide (0, 2, 10, and 50 μM). RapA was used at a 1 μM final concentration. For each reaction (24 reactions total), 6 time points were taken in addition to the time zero (T0) sample withdrawn immediately after the initiation of the reaction as described above. The SigmaPlot kinetic analysis software program (by Systat Software) was used to analyze the rates obtained and fit the data to various inhibition curves.
The assays for dephosphorylation activity and inhibition by the Phr peptides of the Rap hybrid proteins were also carried out in the phosphatase buffer as described above using proteins and peptide concentrations as reported in the figure legends.
Purification of Spo0F∼P.
The Spo0F His tag protein and its phosphorylated form were purified as previously described (18).
Protein purification.
E. coli BL21(DE3)pLysS was transformed with plasmid pET16cat derivatives carrying the rapA wild-type gene or the mutants and grown in LB medium containing ampicillin (100 μg/ml). Purification of the Rap proteins was carried out as previously described (18).
Native gel protein binding assay.
Analysis of complex formation between RapA wild-type (wt) or mutant proteins with a response regulator (Spo0F∼P or ComA) or the inhibitors PhrA/PhrC was carried out using native 10% Tris-Tricine EDTA gels as described previously (18). Proteins were incubated in 50 mM HEPES (pH 7.0), 20% glycerol, 10 mM DTT, and 10 mM EDTA for 5 min in ice. After the addition of loading dye, the samples were loaded on gels and run for approximately 7 to 8 h at 4°C.
Construction of a RapA homology model.
RapA and RapH sequences were aligned, and the RapA structural model was build on the RapH template structure (accession code 3q15) (27) utilizing the ProtMod protein structure modeling server, part of the FFAS Fold and Function Assignment System server (19, 20).
RESULTS
Kinetic analysis of RapA interaction with Spo0F∼P and PhrA.
The RapA protein of B. subtilis specifically interacts both with the Spo0F∼P substrate, promoting its dephosphorylation, and with its inhibitor, pentapeptide PhrA (ARNQT) (18, 29). As an initial step toward the molecular characterization of the mechanism of RapA interaction with the substrate and the inhibitor, we carried out a kinetic analysis of the RapA-dependent dephosphorylation reaction of Spo0F∼P and inhibition of RapA by PhrA. The main aim of this kinetic approach was to determine the mechanism of interaction of Spo0F∼P and PhrA with RapA.
The kinetic properties of RapA-dependent dephosphorylation of Spo0F∼P were first determined by measuring the Km of the reaction. A Spo0F∼P dephosphorylation assay was carried out with 10 concentrations of substrate in the presence of a constant amount of enzyme (RapA) as described in Materials and Methods. The rates of phosphatase activity of RapA were calculated from the slope obtained for each Spo0F∼P concentration, and they increased with increased substrate concentrations, reaching half-maximal velocity at a Km of ∼1.2 μM Spo0F∼P. Graphical representations of this analysis (Michaelis-Menten and Lineweaver-Burk plots) are shown in Fig. 1A and B.
Fig 1.

Kinetic analysis of RapA dephosphorylation of Spo0F∼P and inhibition by PhrA. (A and B) The rates of dephosphorylation were obtained at 10 concentrations of Spo0F∼P (0.33, 0.5, 1, 1.33, 2, 2.5, 3.33, 5, 7.5, and 10 μM). Seven time points were taken for each substrate concentration, and the remaining Spo0F∼P was measured by exposing the gels to a PhosphorImager screen and analyzing the data with the ImageQuant software program. The percentage of remaining Spo0F∼P was plotted versus time, and the slope of each reaction (calculated as shown in Materials and Methods) multiplied by the substrate concentration gave the rate. The rate at each substrate concentration was plotted as a Michaelis-Menten (A) or Lineweaver-Burk (B) graph. (C and D) Time points of Spo0F∼P desphosphorylation by RapA were collected in the presence of four concentrations of the PhrA inhibitor and six concentrations of the substrate. The rates for each reaction were calculated as described above and in Materials and Methods. The best-fit analysis was carried out with the SigmaPlot software program, and the Michaelis-Menten (C) and the Lineweaver-Burk (D) graphs of the inhibition equations that best fit the data are shown. The remaining graphs of the curve fit analysis are shown in Fig. S1 in the supplemental material.
The type of inhibition exerted by the PhrA pentapeptide was determined by carrying out dephosphorylation assays using six concentrations of the substrate (Spo0F∼P) each in the presence of four concentrations of the inhibitor (PhrA) (see Materials and Methods). The rates of the reactions were calculated from the slope of dephosphorylation obtained for each substrate concentration and each inhibitor concentration for a total of 24 data points. The data were plotted as rate versus substrate concentration (Michaelis-Menten) or 1/rate versus 1/substrate concentrations (Lineweaver-Burk). These plots were analyzed with the SigmaPlot software program to identify which type of inhibitory mechanism best fits the data (see Fig. S1 in the supplemental material).
The results, summarized in Fig. 1C and D, demonstrate that PhrA inhibited RapA with noncompetitive partial kinetics, thus suggesting that the pentapeptide and the substrate Spo0F interact with the phosphatase at distinct sites.
Construction of RapAC and RapCA hybrid proteins.
The results of the kinetic analysis suggested distinct PhrA and Spo0F regulator binding sites on RapA and prompted us to test whether these binding sites could be physically separated. This was approached by means of hybrid proteins generated with swapped domains between the RapA and the RapC proteins. RapA (376 amino acids) and RapC (382 amino acids) conserve 44% of their residues (44% sequence identity) (Fig. 2) but specifically inhibit sporulation or ComA-dependent gene expression, respectively, when overexpressed from a multicopy plasmid (8, 33). These properties provided easy phenotypic screening for genetic analysis.
The multicopy plasmid pBS19 (34) was used to generate the first hybrid construct, pBS-RapAC1, expressing a protein with the amino-terminal 183 amino acids of RapA and the carboxy-terminal 196 amino acids of RapC. A reciprocal construct, pBS-RapCA1, which expressed a protein with the amino-terminal 186 amino acids of RapC and the carboxy-terminal 193 amino acids of RapA (Fig. 2), was generated. When transformed in the wild-type strain JH642, the pBS-RapAC1 plasmid gave rise to colonies with a strong sporulation-deficient phenotype, while the pBS-RapCA1 construct did not affect the sporulation efficiency of the colonies compared to the control strain carrying the vector pBS19 (Table 1).
Table 1.
Efficiency of sporulation of B. subtilis strains derivative of the parental strain JH642 carrying multicopy plasmids expressing the RapA-RapC hybrid proteins
| Straina | Protein expressed | Viable count (cells ml−1) | Spore count (ml−1) | % sporulationb |
|---|---|---|---|---|
| JH11349 | None | 2.0 × 108 | 7.0 × 107 | 35 |
| JH11169 | RapA | 1.2 × 108 | 4.2 × 106 | 3.5 |
| JH11084 | RapC | 2.3 × 108 | 8.9 × 107 | 38.6 |
| JH19275 | RapAC1 | 1.0 × 107 | 3.2 × 103 | 0.032 |
| JH19266 | RapCA1 | 1.1 × 108 | 4.5 × 107 | 40.9 |
| JH19276 | RapAC2 | 2.8 × 108 | 3.9 × 103 | 0.001 |
| JH19277 | RapAC3 | 2.3 × 108 | 2.8 × 103 | 0.001 |
| JH19267 | RapAC4 | 1.0 × 107 | 3.2 × 103 | 0.03 |
Strains were grown for 48 h at 37°C in Schaeffer's sporulation medium supplemented with chloramphenicol.
Data are representative of two independent experiments.
The pBS19 plasmid derivatives were also transformed into strain JH19278, which carries the rapC-lacZ fusion reporter construct, for the analysis of ComA-dependent gene expression, in a spo0A abrB rapC background. The spo0A abrB background was chosen to eliminate effects on competence gene expression exerted by these transcription regulators. The results shown in Fig. 3 indicated that the RapCA1 construct inhibited the expression of rapC while the RapAC1 construct did not have a significant effect.
Fig 3.
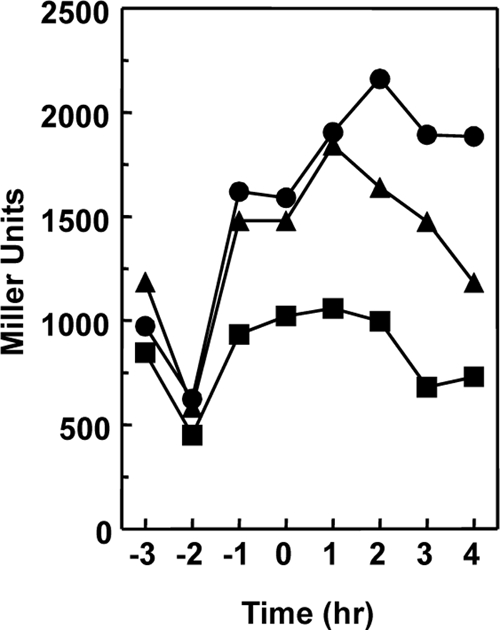
Effect of the multicopy plasmids expressing the RapAC1 or RapCA1 hybrid protein on rapC transcription. Strain JH19278 (spo0A abrB rapC amyE::rapC-lacZ) was transformed with the multicopy plasmids pBS19 (●), pBS19-RapAC1 (▲), and pBS19-RapCA1 (■). Cells were grown in Schaeffer's sporulation medium, and samples were taken at hourly intervals before and after the transition (T0) from the vegetative to the sporulation phase.
These results demonstrated that the domain for recognition of the substrate response regulator is within the amino-terminal half of the RapA and RapC proteins.
The substrate binding domain is in the N-terminal half of the Rap proteins.
We carried out in vitro experiments with purified RapAC1 and RapCA1 hybrid proteins to test their ability to bind to the Spo0F∼P or ComA response regulators. As shown in Fig. 4, the native gel binding assay indicated that the RapAC1 protein was able to form a complex with Spo0F∼P but not with ComA, while RapCA1 interacted with ComA but did not form a complex with Spo0F∼P.
Fig 4.

Interaction of the RapAC1 and RapCA1 hybrid proteins with the Spo0F and ComA response regulators. The 10% Tris-Tricine-EDTA native gel assay was carried out as described in Materials and Methods. Each protein was used at a 10 μM final concentration.
In vitro dephosphorylation assays of Spo0F∼P were also carried out to ensure that Rap enzymatic activity was maintained in the hybrid protein. These assays consistently demonstrated that the RapAC1 hybrid protein was capable of dephosphorylating Spo0F∼P (Fig. 5C). The RapCA1 protein was also tested in the Spo0F∼P dephosphorylation assay and found to be inactive (see Fig. S2 in the supplemental material).
Fig 5.

Time courses of Spo0F∼P dephosphorylation by RapA wt or RapAC1 and inhibition by Phr peptides. Purified Spo0F∼P (0.5 μM) was incubated alone or in the presence of RapA wt (A and B) or RapAC1 (C and D) (0.5 μM). The PhrA (A and C) or PhrC (B and D) peptides were added at a 1 μM final concentration. Samples were analyzed by 15% SDS-PAGE and quantitated by the ImageQuant software program after exposure to a PhosphorImager screen. Symbols: Spo0F∼P alone, ●; Spo0F∼P and Rap protein, ▲; Spo0F∼P, Rap protein, and Phr peptide, ■.
These results indicated that the amino-terminal half of the Rap proteins contains the necessary structural determinants for substrate binding and enzymatic activity.
Determining the minimal domain required for substrate recognition.
To test whether the substrate binding site of RapA could be narrowed down to a smaller domain within the amino-terminal half of the protein, a second RapA-RapC hybrid construct that expressed a protein with the amino-terminal 112 amino acids of RapA and the 266 carboxy-terminal amino acids of RapC was generated (Fig. 2). The multicopy plasmid carrying this construct, pBS-RapAC2, was transformed in the wild-type strain JH642, giving rise to colony transformants with a strong sporulation defect (Table 1).
Another construct, pBS-RapAC3, expressing a protein with 60 amino acids from the amino-terminal end of RapA and 321 amino acids from the C-terminal end of RapC, was also generated, and its transformation in JH642 also resulted in colonies with a sporulation-deficient phenotype (Fig. 2 and Table 1).
These results prompted us to test the activity of the RapAC2 and RapAC3 hybrid proteins in vitro by the native gel binding assay and dephosphorylation assay of Spo0F∼P (Fig. 6). Both proteins slightly interacted with Spo0F∼P but not as significantly as in the case of RapAC1 or RapA. Notably, neither RapAC2 nor RapAC3 interacted with the C-terminal domain of ComA, indicating that the first 60 amino acids of RapC are also required for the recognition of the response regulator DNA binding domain (data not shown).
Fig 6.
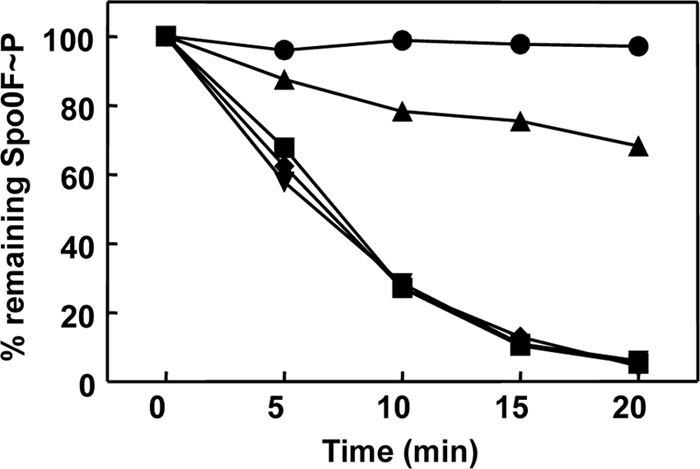
Activities of the RapAC2 and RapAC3 hybrid proteins. Time courses of dephosphorylation of Spo0F∼P by RapAC2 and RapAC3 were carried out in vitro as described in Materials and Methods. Proteins were used at a 1.5 μM concentration. The RapAC2 hybrid protein was also tested in the presence of PhrC (3 μM). The samples were run on a 15% SDS-PAGE gel, exposed to a PhosphorImager screen, and quantified by the ImageQuant software program. Symbols: ●, Spo0F∼P alone; ■, Spo0F∼P and RapA wt; ▼, Spo0F∼P and RapAC2; ◆, Spo0F∼P and RapAC3; ▲, Spo0F∼P, RapAC2, and PhrC.
In vitro dephosphorylation of purified Spo0F∼P by the RapAC2 and RapAC3 proteins followed essentially the same kinetics as that by the wild-type RapA protein at a 1:1 molar ratio of enzyme and substrate in the reaction, and both proteins were inhibited by PhrA (Fig. 6 and data not shown). These results suggested that the higher sensitivity of the native gel binding assay uncovered a functional deficiency that the in vivo multicopy expression system and the in vitro dephosphorylation assay failed to detect.
To further analyze the relative efficiency of RapA wt and RapAC1, RapAC2, and RapAC3 hybrid proteins in dephosphorylating Spo0F∼P, an additional in vivo assay was devised that analyzed the activity of the phosphatases when expressed in single copy rather than in multicopy from the native rapA promoter. For this assay, the pET16cat plasmid derivatives carrying the rapA wild-type gene and the rapAC1, rapAC2, and rapAC3 chimeric constructs were transformed in the wild-type strain JH642. Upon integration into the chromosome by single crossover, diagnostic PCR analysis was carried out to ensure that the colonies analyzed had the plasmid integrated at the rapA locus and not at the rapC locus, in the case of the hybrid constructs. The integration of such plasmids resulted in the inactivation of the phrA gene (see Fig. S3 in the supplemental material). In the presence of an active RapA or RapAC protein, inactivation of phrA results in a sporulation-deficient phenotype, thus providing an assay for quantitation of phosphatase activity. Quantitation of the sporulation efficiency of strains expressing the RapAC1, RapAC2, RapAC3, and wild type RapA proteins from these single-copy gene constructs (Table 2) indicated that RapAC1 was as active as the wild type in inhibiting sporulation while RapAC2 and RapAC3 were inactive.
Table 2.
Sporulation efficiency of strains derived from JH642 (parental) expressing the rapA wild type or the chimeric rapA genes in single copy from the chromosome
| Straina | Viable count (cells ml−1) | Spore count (ml−1) | % sporulation |
|---|---|---|---|
| JH12834ΔrapA | 3.2 × 108 | 2.25 × 108 | 70.3 |
| JH642 (parental) | 2.1 × 108 | 1.00 × 108 | 47.6 |
| JH642::rapAwt | 1.8 × 107 | 6.10 × 105 | 3.4 |
| JH642::rapAC1 | 7.5 × 105 | 1.15 × 104 | 1.5 |
| JH642::rapAC2 | 1.59 × 108 | 1.16 × 108 | 73.0 |
| JH642::rapAC3 | 7.8 × 107 | 5.50 × 107 | 70.5 |
Cells were grown for 48 h at 37°C in Schaeffer's sporulation medium. The data reported are representative of results from two independent experiments.
These results identified a minimal 60-amino-acid domain at the amino-terminal end of RapA as sufficient to recognize and dephosphorylate Spo0F∼P. However, an in vivo activity comparable to the activity of the wild-type protein when expressed from a single-copy gene required the amino-terminal 185 amino acids of RapA.
The carboxy-terminal half of Rap proteins contains the binding site for Phr peptides.
The results of the kinetic analysis and the localization of the substrate binding site to the amino-terminal half of the Rap proteins raised the question of whether the interaction site for the Phr peptides was contained within the carboxy-terminal half of the Rap proteins. In order to determine whether specificity to any peptide was maintained in the RapAC1 hybrid protein, in vitro dephosphorylation assays of Spo0F∼P were carried out in the presence of the PhrA or PhrC pentapeptide. The data (Fig. 5C and D) indicated that RapAC1 dephosphorylation of Spo0F∼P was inhibited by the PhrC pentapeptide while it was insensitive to the PhrA pentapeptide. Thus, the determinants for pentapeptide interaction must be contained in the carboxy-terminal half of the Rap proteins.
In order to further narrow down the region required for Phr peptide binding, two additional RapAC hybrid proteins were generated. The RapAC4 protein consisted of the N-terminal 288 amino acids of RapA and the C-terminal 92 amino acids of RapC, while the RapAC5 protein was made of the N-terminal 333 amino acids of RapA and the C-terminal 45 amino acids of RapC (Fig. 2). The RapAC4 and RapAC5 proteins were purified from overexpressing E. coli strains and tested in vitro for their ability to dephosphorylate Spo0F∼P and be inhibited by PhrA or PhrC. The results shown in Fig. 7 indicated that both proteins maintained the ability to dephosphorylate Spo0F∼P, as expected. However, only RapAC5 was able to respond to the PhrA pentapeptide, while RapAC4 was not inhibited by either PhrA or PhrC.
Fig 7.
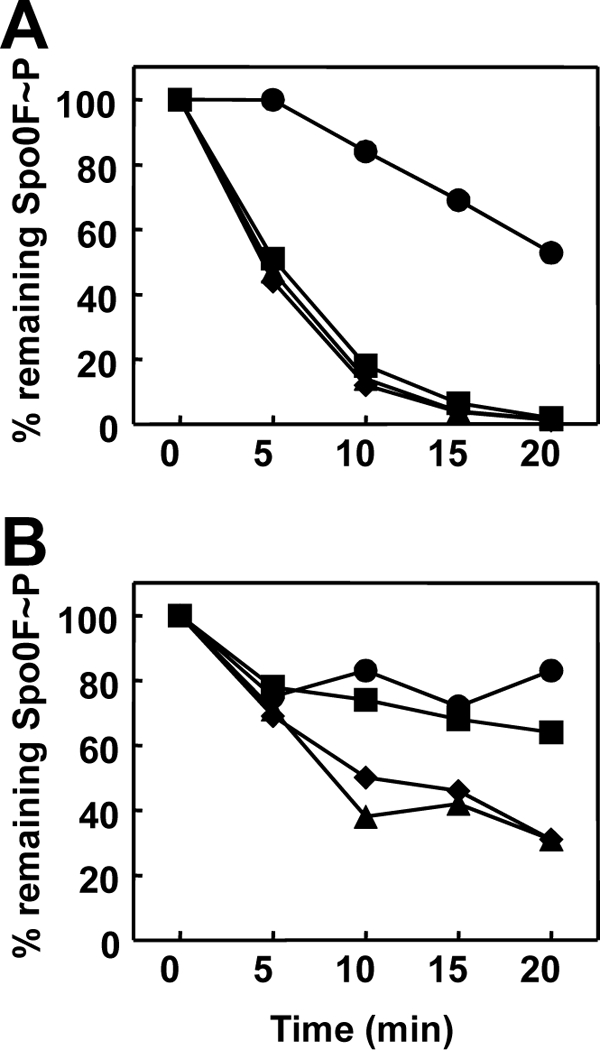
Time course of Spo0F∼P dephosphorylation by RapAC4 (A) or RapAC5 (B) and inhibition by PhrA or PhrC. Spo0F∼P (2.5 μM) was incubated in the absence (●) or presence (▲) of the RapAC proteins (2.5 μM) and the PhrA (■) or PhrC (◆) peptide at a 5 μM final concentration. Samples were analyzed by 15% SDS-PAGE and quantitated by the Image Quant software program after exposure to a PhosphorImager screen.
These results indicated that the determinants for Phr peptide binding specificity must be localized to the TPR3-5 region and may require the TPR5-6 connector region.
Identification of residues required for PhrA binding.
Two mutations in the rapA gene that resulted in proteins insensitive to PhrA but that maintained the phosphatase activity toward Spo0F∼P were originally identified (29, 33). Mapping of these mutant residues, D192N and P259L, on the RapA model structure showed that the two residues were located at opposite ends of the concave structure formed by the TPR domains, along the horizontal axis, and their side chains were protruding into the cavity (see Fig. 9). By drawing a line between the D192 and the P259 residues, we identified eight candidate residues whose side chains also protruded in the concave space generated by the TPR domains and thus could also be involved in the interaction with the pentapeptide: M193, Q194, Y224, N225, N228, K232, E253, and H260. Each residue was mutated to an alanine residue using plasmid pMP9230, and the activity of each mutant protein was tested in vivo by transforming the resulting plasmids in the wild-type strain JH642. All mutant proteins were essentially active in generating a sporulation-deficient phenotype equivalent to the one resulting from the transformation with plasmid pMP9230. However, when plasmid pMP9258 was transformed in these strains to reactivate PhrA production, the strains expressing the Y224A, N225A, N228A, and H260A mutations failed to recover sporulation proficiency, while the strains expressing the wild-type RapA protein or the M193A, Q194A, K232A, and E253A mutant proteins did (Table 3). This indicated that the Y224, Y225, N228, and H260 residues are critical for PhrA activity on RapA.
Fig 9.

Alanine scanning mutagenesis reveals the PhrA peptide binding pocket on RapA. (A) Shown is the structure of B. subtilis RapA (khaki), homology modeled on B. subtilis RapH in complex with Spo0F (gray). RapA alanine mutants unable to bind and respond to PhrA are in red, demonstrating that the PhrA peptide binding site is distal from the Spo0F binding site and on the concave face of the TPR domain. (B, C, and D Stereo view overlay (B) or side-by-side view (C and D) of TPR2-5 of RapA (khaki) with the equivalent of PlcR (blue); compare the position of alanine mutants unable to bind PhrA peptides (red) to the position of the PapR peptide ligand (magenta). Sites on PlcR that correspond to the RapA alanine mutants are in green. Hydrogen bonds between the peptide and the displayed PlcR residues are in yellow. Amino- and carboxy-terminal ends of the PapR peptide are as labeled.
Table 3.
Efficiency of sporulation of B. subtilis strains derivative of the parental strain JH642 expressing RapA wild-type or mutant proteins (from rapA genes integrated isotopically at the rapA locus) in the presence or absence of the PhrA protein (expressed from the phrA gene integrated ectopically at the amyE locus)
| Straina | Viable count (cells ml−1) | Spore count (ml−1) | % sporulationb |
|---|---|---|---|
| JH642 | 4.9 × 108 | 1.5 × 108 | 30.3 |
| JH642::rapAwt::phrA | 3.2 × 108 | 1.0 × 108 | 31.2 |
| JH642::rapAY224A::phrA | 5.1 × 108 | 1.1 × 107 | 2.15 |
| JH642::rapAN225A::phrA | 4.3 × 108 | 6.4 × 106 | 1.48 |
| JH642::rapAN228A::phrA | 4.5 × 108 | 1.3 × 107 | 2.8 |
| JH642::rapAH260A::phrA | 3.6 × 108 | 1.7 × 104 | 0.004 |
| JH642::rapAwt | 1.8 × 108 | 2.6 × 106 | 1.5 |
| JH642::rapAY224A | 2.0 × 108 | 2.1 × 106 | 1.0 |
| JH642::rapAN225A | 3.3 × 108 | 3.1 × 106 | 0.9 |
| JH642::rapAN228A | 1.9 × 108 | 5.6 × 106 | 2.9 |
| JH642::rapAH260A | 1.3 × 108 | 1.2 × 106 | 0.9 |
Strains were grown for 48 h at 37°C in Schaeffer's sporulation medium. The data reported are representative of two independent experiments.
The percentage of sporulation is the ratio between viable count and spore count.
In order to quantitate the enzymatic activity of the mutant proteins, in vitro dephosphorylation assays of Spo0F∼P were carried out using the four purified mutant proteins in the presence or absence of the PhrA pentapeptide. As shown in Fig. 8, the RapAY224A, RapAN225A, RapAN228A, and RapAH260A proteins dephosphorylated Spo0F∼P at the same rate as the wild-type protein, but none of them was inhibited by the pentapeptide. Consistently, the proteins were able to bind Spo0F∼P but did not bind the PhrA pentapeptide in the native gel binding assay (data not shown).
Fig 8.

Time courses of Spo0F∼P dephosphorylation by RapA wt or alanine mutant proteins and inhibition by Phr peptides. Purified Spo0F∼P (0.5 μM) was incubated alone or in the presence of RapA wt or RapA Y224A (A), RapA N225A (B), RapA N228A (C), or RapA H260A (D). The PhrA peptide was added at a 1 μM final concentration. Samples were analyzed by 15% SDS-PAGE and quantitated by the ImageQuant software program after exposure to a PhosphorImager screen. Symbols: no RapA, ●; RapAwt, ▲; RapA wt plus PhrA, ▵-; RapA mutant, ■-; RapA mutant plus PhrA, □-.
These results support our initial prediction that the PhrA pentapeptide likely binds to RapA in an extended conformation along the surface of the concave structure formed by the TPR domains spanning from residue D192 to residue P259 (Fig. 9) (31).
DISCUSSION
Structural features of Rap proteins.
Rap proteins of Gram-positive bacilli have been characterized as regulatory proteins that exert their function by dephosphorylating a response regulator or by binding to the DNA binding domain of a response regulator and inhibiting its activity. Rap proteins were shown to be inhibited by a peptide generated from the secretion, reimportation, and processing of a paired Phr protein (5, 8, 29, 33). Recognition of the target substrate and the inhibitor peptide by each Rap protein is highly specific regardless of the extensive amino acid sequence conservation shared by the members of this family.
Rap proteins are characterized by a structural organization in tetratricopeptide repeats (TPR) (31). The TPR is a highly versatile, all-helical structural motif identified in a wide variety of proteins from eukaryotic and prokaryotic organisms (11). The TPR domain is defined by the presence of 34 amino acids in the basic repeat that was shown to adopt a helix-turn-helix arrangement with a packing angle between the two helices of ∼24°. The TPR motif is usually present in tandem arrays of 3 to 16 motifs packed together in a parallel arrangement that generates a right-handed superhelical structure (12, 37).
The recently reported structure of the RapH-Spo0F complex confirmed the predictions made on the organization of Rap proteins (31). It showed that the N-terminal region of RapH contains a 3-helix-bundle (α1 to α3 in Fig. 2) connected to the C terminus of the protein by a flexible linker and a short α-helix (α4 in Fig. 2). Following helix α4 is the right-handed superhelical domain generated by six TPR domains, with TPR1-5 connected to TPR6 by two additional antiparallel α helices that resemble the TPR domain in their fold but do not contain the TPR motif signature residues. The structure also showed that the side chain of the conserved Q47 residue in RapH is required for activity, since it inserts into the active site of Spo0F, likely orienting a water molecule for hydrolytic attack in the dephosphorylation reaction (27).
The position and orientation of the catalytic Q47 residue of RapH is similar to the position and orientation of the phosphorylatable histidine (H30) of the Spo0B phosphotransferase of the phosphorelay previously shown to interact with Spo0F primarily through its four-helix bundle (27, 43). The Spo0F-Spo0B protein interface includes helices α1 and α2′ in Spo0B and helix α1 and all five β-α loops in Spo0F. Superimposition of the Spo0F-Spo0B and Spo0F-RapH structures (Fig. 10) clearly shows that the α1 and α2′ helices of Spo0B overlap the α2 and α3 helices of RapH. The α1′ helix of the Spo0B four-helix bundle and the α1 helix of RapH are also very similarly positioned. These observations confirm the prediction based on mutational analyses that the interface of the Rap proteins with Spo0F assumes a structural conformation with strong similarity to the helical conformation of the Spo0B binding interface with Spo0F (18, 42).
Fig 10.

The N-terminal helices of Rap phosphatases are structurally reminiscent of the four-helix bundle of the Spo0B protein. Previous alanine scanning mutagenesis of the entire surface exposed residues of Spo0F revealed an extensive overlap of mutants unable to interact with either Spo0B or the Rap phosphatases (42). Overlaying the Spo0F molecule of the Spo0F-Spo0B complex (blue) (43) and that of the Spo0F-RapH complex (yellow) (27) shows that the N-terminal helices α1, α2, and α3 of the Rap phosphatases (orange) structurally mimic the homodimeric four-helix bundle of Spo0B (turquoise), which forms extensive interactions with helix α1 of Spo0F. Helix α5, the first helix of the first TPR domain of the Rap phosphatase, completes the four-helix bundle-like structure; however, it is slanted by 40° in respect to helix α2 in Spo0B. Phosphorylation sites on Spo0B (H30) and on Spo0F (D54) and the catalytic residue on RapH (Q47) are displayed.
Modular organization of Rap proteins.
In this study, by means of genetic and biochemical approaches, we have defined the modular organization of Rap proteins and established that distinct modules are involved in binding to the substrate response regulator or to the Phr peptide inhibitor. Chimeras generated between RapA, which dephosphorylates Spo0F∼P, and RapC, which binds to the DNA binding domain of ComA, allowed us to determine that the first 60 amino acids in the N terminus of the Rap proteins are sufficient to confer specificity toward the substrate, although an additional 120 amino acids are necessary for full activity in vivo. The RapAC2 and RapAC3 proteins with, respectively, the N-terminal 112 and 60 amino acids belonging to RapA can dephosphorylate Spo0F∼P as efficiently as wild-type RapA in vitro and generate a sporulation-deficient phenotype when overexpressed in vivo. However, they both showed reduced affinity for the substrate in the protein binding assay and were inactive in vivo when expressed in single copy from the rapA promoter. Additionally, neither RapAC2 nor RapAC3 interacted with the C-terminal domain of ComA (Fig. 6), indicating that the N-terminal 60 amino acids of Rap proteins are also required for interaction with the DNA binding domain, despite drastic structural differences from the response regulator domain. The data did not resolve whether binding interfaces on Rap proteins for these two proteins overlap.
These in vivo and in vitro results confirmed the conclusions drawn from the crystal structure of the RapH-Spo0F complex indicating that the α3 helix, containing Q47, and the α2-α3 loop in the N-terminal domain are mainly involved in the interaction and are sufficient for phosphatase activity; however, residues in TPR1, TPR2, and the TPR2-TPR3 loop (corresponding to residues L96, D134, E137, and Y1750) also affected enzymatic activity (27). Therefore, the lack of in vivo activity of single-copy-expressed RapAC2 and RapAC3 is likely the result of reduced affinity of these Rap chimeras for Spo0F. This in turn favors the phosphorylation reaction by sporulation histidine kinases and phosphotransfer to Spo0B, resulting in the sporulation proficiency of the strains expressing these proteins from single-copy genes. On the contrary, when overexpressed, RapAC2 and RapAC3 are present at concentrations sufficiently high to efficiently compete with kinases and Spo0B for the Spo0F/Spo0F∼P molecules, thus generating a sporulation defect. This is consistent with the observation that a catalytically inert RapH protein inhibited phosphotransfer from KinA to Spo0F and from Spo0F∼P to Spo0B (27).
Furthermore, while the manuscript was in review, Baker and Neiditch reported the crystal structure of the RapF-ComA complex and showed that the ComA DNA-binding domain binds to the RapF protein at a site distinct from the site of Spo0F binding to RapH but within the same first 78 amino acids of the N-terminal domain (3). This explains how the same domain of a Rap protein can interact with two structurally distinct protein substrates.
The central 3-TPR module mediates specificity toward the Phr peptide.
Because TPR motifs are known to mediate protein-protein and protein-peptide interactions, we originally proposed that the TPR core domain of Rap proteins had to be involved in the interaction with the Phr peptide. This assumption was supported by two PhrA peptide-insensitive RapA mutant (D192N and P259L) proteins, both of which maintained full phosphatase activity toward the substrate Spo0F∼P. The hybrid RapA-RapC proteins generated in this study allowed us to demonstrate that a region of the central core comprising TPR3, TPR4, and TPR5 contains the determinants for Phr peptide binding and inhibition, thus demonstrating that RapA binding to Spo0F and to PhrA occurs at two independent sites. Our results are also consistent with the kinetic analyses that suggested a noncompetitive partial mechanism of inhibition of RapA by PhrA (Fig. 1).
The involvement of three TPR of RapA in its interaction with PhrA is in agreement with previous observations that established that the 3-TPR module is the most common in nature and the minimal functional binding unit (11).
Residues in the concave side of the 3-TPR module are involved in Rap-Phr interaction.
Further analysis of the determinants for PhrA binding to RapA was achieved by means of a partial alanine mutagenesis strategy focused on the concave surface formed by the TPR3-5 domains. Identification of residues aligned between the D192 and P259 residues in the RapA structural model originally described by Perego and Brannigan (31) detected four additional residues likely involved in peptide binding. One of these residues, N225, in α helix 1 of TPR4, is extremely conserved among Rap proteins, with one exception being the BA3790 protein in Bacillus anthracis, which instead contains a threonine residue. Also highly conserved is the D192 residue, with substitutions by serine and glutamate in Bacillus halodurans BH4014 or B. subtilis RapG, respectively. These two residues are unlikely to determine the specificity that characterizes the Rap-Phr interaction, but their side chains could be involved in the interaction with the peptide backbone. Two other residues among the ones newly identified, Y224 and N228, are more variable among Rap proteins, often replaced by hydrophobic residues, as observed also with the P259 residue, and therefore these could be more likely to be involved in specific interactions with residues in the Phr peptide. Finally, the H260 residue is highly variable among Rap proteins (replaced by R, A, G, K, Q, or N) and therefore is a strong candidate for being involved in conferring specificity to peptide binding.
Pivotal role of a TPR conserved asparagine in Rap-Phr interaction.
In an attempt to better define the role of the six RapA amino acid residues that affect PhrA binding when mutated, we compared the crystal structure of the PlcR regulator of Bacillus cereus in complex with its activating peptide PapR (LPFEF) to the structure of RapA modeled using the structure of the RapH-Spo0F complex (Fig. 9) (6, 13, 27). PlcR is the major virulence regulator of B. cereus and Bacillus thuringiensis, and its interaction with the PapR peptide stimulates its transcriptional activity (39).
Like Rap proteins, PlcR is structurally organized in TPR domains: the five TPR domains in this protein are preceded by an N-terminal helix-turn-helix domain, involved in DNA binding, and are followed by a capping C-terminal helix. In the crystal, the PapR pentapeptide binds to the concave side of the TPR domain, interacting mainly with the first helix of TPR3, TPR4, and TPR5. Additional studies indicated that the physiological peptide is a heptamer (ADLPFEF), and modeling in the PlcR-PapR structure showed a perfect fit into the groove formed by the TPR domains (6). Upon PapR binding, the curvature of the PlcR TPR domain increases, and this results in a separation of the N-terminal helix-turn-helix (HTH) domains in the dimer, positioning them appropriately for DNA binding. A similar mechanism of peptide-binding activation was also proposed for the pheromone-binding PrgX protein of Enterococcus faecalis, which is not recognized as a TPR-containing protein but folds in a very similar manner (27, 38).
The PlcR-PapR structure shows that the PapR peptide main chain is fixed to PlcR by hydrogen bonds from residues N159 (in helix α1 of TPR3), N201, and K204 (in helix α1 of TPR4). Notably, N201 of PlcR corresponds to N225 of RapA when the two protein sequences are aligned (see Fig. S4 in the supplemental material), and these residues are in the same orientation when the two structures are superimposed (Fig. 9). This additional observation supports the prediction that RapAN225 is likely a key residue for establishing an interaction with the PhrA backbone. Furthermore, K204 of PlcR makes a hydrogen bond with the backbone of residue L1 in the PapR; its corresponding residue in RapA is N228, which, in the structure, is positioned such that it is likely to also interact with the alanine in the N-terminal position of PhrA. A key residue that can possibly confer specificity to the RapA-PhrA interaction is H260, because in addition to being highly variable, as mentioned above, its side chain could be in close contact with the side chain of an amino acid of the peptide, likely to be the Q in position 4. In fact, in the PlcR-PapR structure, the corresponding residue, Q237, is less than 4 Å away from the E residue in position 4 of PapR.
The pivotal role of the N225 residue of RapA or N201 of PlcR is reiterated by structural/functional studies of TPR domains of eukaryotic proteins that bind the C termini of the chaperone heat shock proteins Hsp90 and Hsp70 (37). These studies show that an asparagine residue (see N43 in Fig. S4 in the supplemental material) in the first helix of the second TPR domain within the 3-TPR module essential for peptide binding is involved, with its carbonyl side chain directly contacting the backbone amide of the C-terminal residue of the binding peptide. This asparagine corresponds to N225 of RapA and N201 of PlcR when the sequences are aligned (see Fig. S4). Also, the alanine in position 46 of the TPR1 module is interacting with the isoleucine of the ligand peptide (GASSGPTIEEVD) that corresponds to the alanine in the N-terminal position of PhrA. Furthermore, the only peptide side chain in these complexes that is recognized via an electrostatic interaction is the side chain carboxylate of the peptide C-terminal residue with a lysine residue in TPR1 that corresponds to H260 in RapA. This observation supports our hypothesis that specificity may be conferred at least in part by the interactions established by this residue (9, 10, 37).
Residues D192 and Y224 could also be within a 5-Å distance from residues in the peptide based on the position of the corresponding residues in PlcR, E167 and Y200, respectively. Nevertheless, D192 may have a more structural role in accommodating the peptide given its position toward the end of α-helix 1 of TPR3 and its interaction with the conserved T155 at the beginning of α-helix 2 of TPR2 (27). Residue P259 of RapA may also have a structural function rather than a direct role in interacting with the PhrA peptide given its conformational characteristics, its vicinity to H260, and the fact that its corresponding amino acid in PlcR (G236) is not within a 5-Å distance from the PapR peptide.
Structural-functional role for α-helix 15 and TPR6.
The PlcR-PapR structure shows that interactions are established by the peptide with residues in the terminal helix of PlcR that determine the specificity of the interaction, in particular with residue A278. This helix structurally corresponds to helix α15 in the TPR5-6 connector region of Rap proteins (Fig. 2). Our analysis of the RapAC chimeras indicated that this region connecting TPR5 to TPR6 is required for Phr activity, since RapAC5, in which RapA extends to contain the TPR5-6 connector, responded to PhrA, while RapAC4, which contains the TPR5-6 connector from RapC, did not (Fig. 7).
Furthermore, a random mutagenic analysis obtained by error-prone PCR (our unpublished data) identified mutations in α-helix 15 that affected RapA activity. In particular, a change to arginine in L300, which corresponds to A278 in PlcR, as well as L296S and Y308D substitutions resulted in proteins inactive in vivo and unable to bind in vitro to Spo0F∼P and PhrA. These proteins were also unstable both in B. subtilis and in E. coli, suggesting that the C-terminal extension of Rap proteins may have an important structural-functional role, as suggested for the C-terminal extension of PlcR (6).
The role of TPR6 in the structure/function of RapA is at this time unclear. The inhibition of RapAC5 by PhrA indicates that TPR6 is not involved in pentapeptide binding and/or specificity. However, TPR6 may be required for structural stability, because its deletion also results in a protein subject to degradation in both E. coli and B. subtilis (our unpublished data).
Overall, these results show how the versatility of the TPR module has been adapted by microorganisms of the genus Bacillus such that, on a common folded framework, a variety of modifications have occurred allowing for the grafting of specific ligand- or substrate-binding capabilities and regulatory effects on highly diversified physiological pathways.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by grants GM55594 and GM19416 from the Institute of General Medical Sciences, National Institutes of Health. Oligonucleotide synthesis and DNA sequencing were supported in part by the Stein Beneficial Trust. Michela Morelli was supported in part by a fellowship from the Istituto Giuseppe Toniolo, Piacenza, Italy.
Darshini Metha is acknowledged for the construction of plasmids pBS-RapAC1 and pBS-RapCA1; Florence Bertin is acknowledged for the construction of pET16-RapAC5.
Footnotes
Published ahead of print 20 January 2012
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1. Anagnostopoulos C, Spizizen J. 1961. Requirements for transformation in Bacillus subtilis. J. Bacteriol. 81:741–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Auchtung JM, Lee CA, Monson RE, Lehman AP, Grossman AD. 2005. Regulation of a Bacillus subtilis mobile genetic element by intercellular signaling and the global DNA damage response. Proc. Natl. Acad. Sci. U. S. A. 102:12554–12559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baker MD, Neiditch MB. 2011. Structural basis of response regulator inhibition by a bacterial anti-activator protein. PLoS Biol. 9:e1001226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bongiorni C, Ishikawa S, Stephenson S, Ogasawara N, Perego M. 2005. Synergistic regulation of competence development in Bacillus subtilis by two Rap-Phr systems. J. Bacteriol. 187:4353–4361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bongiorni C, Stoessel R, Shoemaker D, Perego M. 2006. Rap phosphatase of virulence plasmid pXO1 inhibits Bacillus anthracis sporulation. J. Bacteriol. 188:487–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bouillaut L, et al. 2008. Molecular basis for group-specific activation of the virulence regulator PlcR by PapR heptapeptides. Nucleic Acids Res. 36:3791–3801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Burbulys D, Trach KA, Hoch JA. 1991. The initiation of sporulation in Bacillus subtilis is controlled by a multicomponent phosphorelay. Cell 64:545–552 [DOI] [PubMed] [Google Scholar]
- 8. Core LJ, Perego M. 2003. TPR-mediated interaction of RapC with ComA inhibits response regulator-DNA binding for competence development in Bacillus subtilis. Mol. Microbiol. 49:1509–1522 [DOI] [PubMed] [Google Scholar]
- 9. Cortajarena AL, Kajander T, Pan W, Cocco MJ, Regan L. 2004. Protein design to understand peptide ligand recognition by tetratricopeptide repeat proteins. Protein Eng. Des. Sel. 17:399–409 [DOI] [PubMed] [Google Scholar]
- 10. Cortajarena AL, Wang J, Regan L. 2010. Crystal structure of a designed tetratricopeptide repeat module in complex with its peptide ligand. FEBS J. 277:1058–1066 [DOI] [PubMed] [Google Scholar]
- 11. D'Andrea LD, Regan L. 2003. TPR proteins: the versatile helix. Trends Biochem. Sci. 28:655–662 [DOI] [PubMed] [Google Scholar]
- 12. Das AK, Cohen PTW, Barford D. 1998. The structure of the tetratricopeptide repeats of protein phosphatase 5: implications for TPR-mediated protein-protein interactions. EMBO J. 17:1192–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Declerck N, et al. 2007. Structure of PlcR: insights into virulence regulation and evolution of quorum sensing in Gram-positive bacteria. Proc. Natl. Acad. Sci. U. S. A. 104:18490–18495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Diaz AR, et al. 2008. Functional role for a conserved aspartate in the Spo0E signature motif involved in the dephosphorylation of the Bacillus subtilis sporulation regulator Spo0A. J. Biol. Chem. 283:2962–2972 [DOI] [PubMed] [Google Scholar]
- 15. Fujita M, Losick R. 2005. Evidence that entry into sporulation in Bacillus subtilis is governed by a gradual increase in the level and activity of the master regulator Spo0A. Genes Dev. 19:2236–2244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gatto GJ, Jr, Geisbrecht BV, Gould SJ, Berg JM. 2000. Peroxisomal targeting signal-1 recognition by the TPR domains of human PEX5. Nat. Struct. Biol. 7:1091–1095 [DOI] [PubMed] [Google Scholar]
- 17. Horton RM, Cai ZL, Ho SN, Pease LR. 1990. Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. Biotechniques 8:528–535 [PubMed] [Google Scholar]
- 18. Ishikawa S, Core LJ, Perego M. 2002. Biochemical characterization of aspartyl phosphate phosphatase interaction with a phosphorylated response regulator and its inhibition by a pentapeptide. J. Biol. Chem. 277:20483–20489 [DOI] [PubMed] [Google Scholar]
- 19. Jaroszewski L, Li Z, Cai XH, Weber C, Godzik A. 2011. FFAS server: novel features and applications. Nucleic Acids Res. 39:W38–W44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jaroszewski L, Rychlewski L, Li Z, Li W, Godzik A. 2005. FFAS03: a server for profile-profile sequence alignments. Nucleic Acids Res. 33:W284–W288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jiang M, Grau R, Perego M. 2000. Differential processing of propeptide inhibitors of Rap phosphatases in Bacillus subtilis. J. Bacteriol. 182:303–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jiang M, Shao W, Perego M, Hoch JA. 2000. Multiple histidine kinases regulate entry into stationary phase and sporulation in Bacillus subtilis. Mol. Microbiol. 38:535–542 [DOI] [PubMed] [Google Scholar]
- 23. LeDeaux JR, Yu N, Grossman AD. 1995. Different roles for KinA, KinB and KinC in the initiation of sporulation in Bacillus subtilis. J. Bacteriol. 177:861–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mirouze N, Parashar V, Baker MD, Dubnau DA, Neiditch MB. 2011. An atypical Phr peptide regulates the developmental switch protein RapH. J. Bacteriol. 193:6197–6206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ogura M, Shimane K, Asai K, Ogasawara N, Tanaka T. 2003. Binding of response regulator DegU to the aprE promoter is inhibited by RapG, which is counteracted by extracellular PhrG in Bacillus subtilis. Mol. Microbiol. 49:1685–1697 [DOI] [PubMed] [Google Scholar]
- 26. Ohlsen KL, Grimsley JK, Hoch JA. 1994. Deactivation of the sporulation transcription factor Spo0A by the Spo0E protein phosphatase. Proc. Natl. Acad. Sci. U. S. A. 91:1756–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Parashar V, Mirouze N, Dubnau DA, Neiditch MB. 2011. Structural basis of response regulator dephosphorylation by Rap phosphatases. PLoS Biol. 9:e1000589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Perego M. 1993. Integrational vectors for genetic manipulation in Bacillus subtilis, p 615–624. In Sonenshein AL, Hoch JA, Losick R. (ed), Bacillus subtilis and other gram-positive bacteria: biochemistry, physiology, and molecular genetics. American Society for Microbiology, Washington, DC [Google Scholar]
- 29. Perego M. 1997. A peptide export-import control circuit modulating bacterial development regulates protein phosphatases of the phosphorelay. Proc. Natl. Acad. Sci. U. S. A. 94:8612–8617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Perego M. 2001. A new family of aspartyl-phosphate phosphatases targeting the sporulation transcription factor Spo0A of Bacillus subtilis. Mol. Microbiol. 42:133–144 [DOI] [PubMed] [Google Scholar]
- 31. Perego M, Brannigan JA. 2001. Pentapeptide regulation of aspartyl-phosphate phosphatases. Peptides 22:1541–1547 [DOI] [PubMed] [Google Scholar]
- 32. Perego M, Glaser P, Hoch JA. 1996. Aspartyl-phosphate phosphatases deactivate the response regulator components of the sporulation signal transduction system in Bacillus subtilis. Mol. Microbiol. 19:1151–1157 [DOI] [PubMed] [Google Scholar]
- 33. Perego M, et al. 1994. Multiple protein aspartate phosphatases provide a mechanism for the integration of diverse signals in the control of development in Bacillus subtilis. Cell 79:1047–1055 [DOI] [PubMed] [Google Scholar]
- 34. Perego M, Hoch JA. 1987. Isolation and sequence of the spo0E gene: its role in initiation of sporulation in Bacillus subtilis. Mol. Microbiol. 1:125–132 [DOI] [PubMed] [Google Scholar]
- 35. Perego M, Hoch JA. 1996. Cell-cell communication regulates the effects of protein aspartate phosphatases on the phosphorelay controlling development in Bacillus subtilis. Proc. Natl. Acad. Sci. U. S. A. 93:1549–1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schaeffer P, Millet J, Aubert JP. 1965. Catabolic repression of bacterial sporulation. Proc. Natl. Acad. Sci. U. S. A. 54:704–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Scheufler C, et al. 2000. Structure of TPR domain-peptide complexes: critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell 101:199–210 [DOI] [PubMed] [Google Scholar]
- 38. Shi K, et al. 2005. Structure of peptide sex pheromone receptor PrgX and PrgX/pheromone complexes and regulation of conjugation in Enterococcus faecalis. Proc. Natl. Acad. Sci. U. S. A. 102:18596–18601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Slamti L, Lereclus D. 2002. A cell-cell signaling peptide activates the PlcR virulence regulon in bacteria of the Bacillus cereus group. EMBO J. 21:4550–4559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Smits WK, et al. 2007. Temporal separation of distinct differentiation pathways by a dual specificity Rap-Phr system in Bacillus subtilis. Mol. Microbiol. 65:103–120 [DOI] [PubMed] [Google Scholar]
- 41. Tortosa P, Dubnau D. 1999. Competence for transformation: a matter of taste. Curr. Opin. Microbiol. 2:588–592 [DOI] [PubMed] [Google Scholar]
- 42. Tzeng Y-L, Feher VA, Cavanagh J, Perego M, Hoch JA. 1998. Characterization of interactions between a two-component response regulator, Spo0F, and its phosphatase, RapB. Biochemistry 37:16538–16545 [DOI] [PubMed] [Google Scholar]
- 43. Zapf JW, Sen U, Madhusudan Hoch JA, Varughese KI. 2000. A transient interaction between two phosphorelay proteins trapped in a crystal lattice reveals the mechanism of molecular recognition and phosphotransfer in signal transduction. Structure 8:851–862 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.