

Abstract
Hydrazones are natural and synthetic compounds containing a C=N-N moiety. Here we found that the opportunistic pathogen Pseudomonas aeruginosa PAO1 produced NAD+- or NADP+-dependent hydrazone dehydrogenase (HDH), which converts hydrazones to the corresponding hydrazides and acids rather than to the simple hydrolytic product aldehydes. Gene cloning indicated that the HDH is part of the group X aldehyde dehydrogenase (ALDH) family, which is distributed among bacteria, although the physiological roles of the ALDH family remain unknown. The PAO1 strain upregulated HDH in the presence of the hydrazone adipic acid bis(ethylidene hydrazide) (AEH). Gene disruption of the HDH-encoding hdhA (PA4022) decreased growth rates in culture medium containing AEH as the sole carbon source, and this effect was more obvious in the double gene disruption of hdhA and its orthologous exaC (PA1984), indicating that these genes are responsible for hydrazone utilization. Recombinant proteins of group X ALDHs from Escherichia coli, Paracoccus denitrificans, and Ochrobactrum anthropi also acted as HDHs in that they produced HDH activity in the cells and degraded hydrazones. These findings indicated the physiological roles of group X ALDHs in bacteria and showed that they comprise a distinct ALDH subfamily.
INTRODUCTION
Hydrazones are compounds with a functional group described as R1R2C=N-NR3R4, and they are distributed throughout nature. One example is the hemolytic toxin gyromitrin, which is produced by false morel mushrooms (9). An alkaloid containing a hydrazone has also been isolated from a marine sponge (18). Hydrazones also involve many chiral building blocks, and they are applied in the agrochemical, pharmaceutical, and chemical industries (15). Hydrazones with replacement of R2 and R3 with hydrogen atoms, as used herein, involve many synthetic and natural compounds (9, 15, 18), but most of their physiological roles and biosynthetic mechanisms are unknown. We recently found that the yeast Candida palmioleophila MK883 produces hydrazone dehydrogenase (HDH) and assimilates hydrazones, such as adipic acid bis(ethylidene hydrazide) (AEH) and valeric acid ethylidene hydrazide (VEH) (3). Hydrazone dehydrogenase, which belongs to the aldehyde dehydrogenase (ALDH) superfamily, attacked the C=N double bond of hydrazones and catalyzed NAD+-dependent oxidation to produce the relevant hydrazide and acid (Fig. 1A). Replacing the conserved Cys and Glu residues comprising the catalytic center of ALDH with Ser and Ala decreased HDH activity to <0.02% and 0.4%, respectively, of that of native HDH, indicating that they play a critical role in oxidizing the carbon atom of the substrate C=N moiety like conventional ALDH-oxidizing carbonyl (C=O) carbons (3). We also discovered that the reaction involves another step in addition to the oxidation reaction in which a hydrazone-enzyme thiolate intermediate is hydrolyzed to hydrazide and the relevant carboxylic acid (3). Thus, the reaction is considered to be “oxidative hydrolysis,” which is unique among known enzymes (Fig. 1B).
Fig 1.

Catalytic mechanism of HDH. (A) Oxidative degradation of AEH (scheme 1) and VEH (scheme 2) catalyzed by HDH. (B) Proposed catalytic mechanisms of oxidative hydrolysis of hydrazones by HDH. [I], intermediate complex; B, unidentified general base.
Phylogenetically, the C. palmioleophila HDH is a fungal-type protein of the ALDH superfamily that includes ALDH from yeasts and filamentous fungi. The ALDH superfamily comprises one of the most divergent families of proteins, and they are distributed across a vast variety of biological species (12, 19). To date, nearly 20,000 genes for predicted proteins in this superfamily that are listed in major protein databases have been phylogenetically classified into at least 13 families that are characterized by their physiological substrates. Bacteria produce proteins in the 14th ALDH family, called group X ALDH (12). Little is known about the physiological roles of proteins in this family except for the protein encoded by Rhodococcus sp. thcA, which might play a role in thiocarbamate catabolism (10). Known proteins of the ALDH superfamily were thought to exclusively catalyze the oxidation of substrate carbonyl carbons or its reverse reaction until a yeast HDH was identified that cleaves the C=N bond of hydrazones (3).
We isolated AEH-assimilating bacteria for which the 16S rRNAs were similar to those of Pseudomonas putida, Pseudomonas aeruginosa, Bacillus flexus, and Delftia acidovorans (3). These bacteria are Gram positive and negative, suggesting that the hydrazone degradation mechanism is widely distributed throughout the bacterial kingdom. A search of published databases did not reveal a fungal-type ALDH in predicted bacterial proteins, suggesting that bacteria do not produce such proteins, and thus the bacterial mechanism of hydrazone degradation has remained elusive. Here we show that P. aeruginosa PAO1 produces AEH-degrading HDH and that the encoding gene is critical for cleaving the C=N bond of AEH and for assimilating acetate liberated from it. The P. aeruginosa genes for HDH are involved in group X ALDH, and this study provides conclusive evidence of a physiological function of group X ALDH proteins. We found that some other bacterial group X ALDH proteins also function as HDHs and that they contribute to the bacterial degradation of hydrazones. We discovered novel functions of bacterial group X ALDHs, namely, the oxidation and assimilation of hydrazone compounds.
MATERIALS AND METHODS
Strains, cultures, and media.
The MK841 strain was isolated from soil (3). P. aeruginosa PAO1 and Escherichia coli JM109 were obtained from laboratory stock belonging to one of the authors (N. Nomura). Paracoccus denitrificans NBRC13301 and Ochrobactrum anthropi ATCC 49188 were from the National Biological Resource Center (Japan) and the American Type Culture Collection, respectively. Typically, P. aeruginosa colonies were transferred to 5 ml of Luria-Bertani medium (1% tryptone, 0.5% yeast extract, 0.5% NaCl) and incubated at 30°C for 24 h, and then 1 ml was inoculated into 100 ml of minimal (MM) medium (10 mM NH4Cl, 10 mM potassium phosphate [pH 7.2], 0.05% MgSO4, 0.05% KCl, and 0.2% trace element solution) (3) containing 30 mM AEH (MMAEH medium) with shaking at 120 rpm at 30°C. Appropriate carbon sources replaced 30 mM AEH in some experiments.
Determination of hydrazone and its derivatives.
Levels of AEH, adipic acid ethylidene hydrazide (AMH), and adipic acid dihydrazide (ADH) were determined by using a high-performance liquid chromatographer (HP-1100; Hewlett-Packard, CA) equipped with a TSKgel ODS-80 column (4.6 by 150 mm; Tosoh, Tokyo, Japan) and by monitoring absorption at 210 nm. The mobile-phase solvent system comprised 50 mM potassium phosphate (pH 7.2)-acetonitrile (9:1, vol/vol) at a flow rate of 1.0 ml min−1. We separated VEH and valeric acid hydrazide (VH) using a solvent system comprising 50 mM potassium phosphate (pH 7.2)-acetonitrile (8:2, vol/vol). Acetate levels were determined by suppressor anion chromatography (Compact IC model 761; Metrohm, Switzerland) according to the manufacturer's instructions.
Enzyme assays.
Hydrazone dehydrogenase activity was measured in 50 mM potassium phosphate (pH 7.2) containing 5 mM AEH or VEH, 5 mM NAD+, and 1 mM dithiothreitol. The reaction was started by adding the enzyme, and increases in NADH were followed by monitoring absorbance at 340 nm at 25°C. The activity of ALDH was measured in a reaction mixture comprising 50 mM sodium pyrophosphate-potassium dihydrogen phosphate buffer (pH 9.0), 0.025 mM acetaldehyde, 1 mM NAD+, and 1 mM dithiothreitol. We used a molecular extinction coefficient of 6.22 mM−1 cm−1 for NADH. The protein concentration was determined using protein assay kits (Bio-Rad Laboratories, CA) with bovine serum albumin as the standard. Kinetic constants for hydrazone dehydrogenase activity were assessed from data obtained using variable concentrations of one compound with a fixed concentration (1 mM) of another. Apparent Km values for AEH and NAD(P)+ were determined by fitting each data set to the equation v/e = kcat [A]/(KA + [A]).
Preparation of HdhA.
P. aeruginosa PAO1 was incubated in 200 ml of Luria-Bertani medium at 30°C for 15 h. Cells were collected by centrifugation, transferred to 2 liters of MMAEH medium, and incubated at 30°C for 20 h. Cells (typically 25 g) obtained from 10 liters of culture were suspended in buffer A (50 mM potassium phosphate [pH 7.2], 10% glycerol, 0.1 mM dithiothreitol, 0.1 mM EDTA) and ultrasonically disrupted. The homogenates were centrifuged at 10,000 × g for 30 min, and the resulting supernatant (cell extract) was applied to a column containing DEAE cellulose (DE52; Whatman, United Kingdom) that was equilibrated with buffer A. Proteins were eluted from the column with 300 ml of buffer A at a flow rate of 0.8 ml min−1. Fractions with HDH activity were applied to a column containing butyl Sepharose (GE Healthcare, Chalfont St. Giles, Buckinghamshire, United Kingdom) that was equilibrated with buffer A containing 1 M ammonium sulfate and then eluted with a linear descending gradient of ammonium sulfate (1 to 0 M) in buffer A. All steps proceeded at 4°C. Fractions containing HDH activity were dialyzed against buffer A for 16 h and then applied to a Resource Q column (GE Healthcare) equilibrated with buffer A. Proteins were eluted with a linear gradient of KCl (0 to 0.5 M) in buffer A at a flow rate of 0.5 ml min−1. Fractions containing HDH were applied to a Superose 6 10/300 GL column (GE Healthcare) that was equilibrated with 50 mM potassium phosphate buffer (pH 7.2) and 150 mM NaCl and eluted at a flow rate of 0.5 ml min−1 to obtain purified HDH.
Quantitative PCR.
Total RNA was extracted from the bacteria using the RNAprotect bacterial reagent (Qiagen) according to the manufacturer's instructions. First-strand cDNA was synthesized using total RNA (0.25 μg) and the QuantiTect reverse transcription kit (Qiagen, Venlo, Netherlands). First-strand cDNA was amplified by quantitative PCR using iQ SYBR green supermix (Bio-Rad Laboratories, Hercules, CA) and MiniOpticon version 3.1 (Bio-Rad Laboratories) according to the manufacturer's instructions. The expression of HDH was normalized against that of the 16S rRNA genes. Data were calculated as relative expression levels. Table S1 in the supplemental material shows the primer sequences.
Preparation of recombinant HDH/ALDH.
To produce recombinant HdhA (rHdhA) and recombinant ExaC (rExaC), DNA fragments were amplified by PCR using sets of primers (see Table S1 in the supplemental material), digested with NdeI and HindIII, and cloned into pET28-b(+). The resultant plasmids were introduced into E. coli Origami 2 (DE3). Other ALDH genes were amplified using primers (see Table S1 in the supplemental material), cloned into pET28-a(+), and introduced into E. coli BL21(DE3). The recombinant proteins were prepared as described previously (3).
Gene disruption of P. aeruginosa PAO1.
We amplified 1,029- and 995-bp DNA fragments carrying the 5′ and 3′ regions of hdhA, respectively, fused with BamHI and EcoRI sites, by using the primer sets HdhA5s/HdhA5a and HdhA3s/HdhA3a (see Table S1 in the supplemental material). Both DNA fragments were fused with PCR using HdhA5s and HdhA3a, digested with BamHI and EcoRI, and cloned into pG19II (8), which was spliced beforehand with the same restriction enzymes. Plasmids for disrupting exaC (pG19exaC) were constructed in the same manner, except the 5′ and 3′ regions of exaC were amplified using the primers ExaC5s/ExaC5a and ExaC3s/ExaC3a, respectively (see Table S1 in the supplemental material), fused using ExaC5s and ExaC3a, and digested with EcoRI and XbaI before being introduced into pG19II. These plasmids were introduced into P. aeruginosa PAO1. Gentamicin-sensitive, sucrose-resistant clones were then selected from the conjugants (8, 21). The total DNA was Southern blotted using a digoxigenin (DIG) DNA labeling and detection kit (Boehringer Mannheim) according to the manufacturer's instructions. Fragments of DNA for hybridization probes were amplified using the primers (see Table S1 in the supplemental material).
Other methods.
We synthesized AEH and VEH as described previously (3). Purified HDH was excised from slab gels, digested with trypsin gold (Promega, Madison, WI), and analyzed by matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS) as described previously (17). Proteins were identified by peptide mass fingerprint analysis using the Mascot (Matrix Science Ltd.) search engine in the entire NCBI protein database. The maximum deviation permitted for matching the peptide mass values was set at 100 parts per million (ppm). Scores of >71 were considered significant (P < 0.005). Proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) as described previously (5), and other experiments proceeded as described in the supplemental material.
RESULTS AND DISCUSSION
Identification of hydrazone dehydrogenase from P. aeruginosa PAO1.
Our preliminary experiments indicated that P. aeruginosa PAO1 utilized a hydrazone (AEH), so this study investigated details of hydrazone utilization by strain PAO1, cultured in MM medium with AEH as the sole carbon source (MMAEH medium) (Fig. 2A). At the initial stage of the culture (∼20 h), AEH levels concomitantly decreased with increasing adipic acid ethylidene hydrazide (AMH) and adipic acid dihydrazide (ADH) levels. Further culture (∼30 h) decreased the accumulated AMH and increased ADH, indicating that the bacterium metabolized AEH to AMH and AMH to ADH (Fig. 2A). The reaction was stoichiometric regarding the amounts of the compounds, a result which supported the reaction described in scheme 1 (Fig. 1A). The results were essentially the same when the strain was cultured in MM medium containing the monoacyl hydrazone VEH, which generated valeric acid hydrazide (VH) (Fig. 2B). The optical density of the culture increased with AEH and VEH degradation, indicating that P. aeruginosa PAO1 assimilated the hydrazones.
Fig 2.
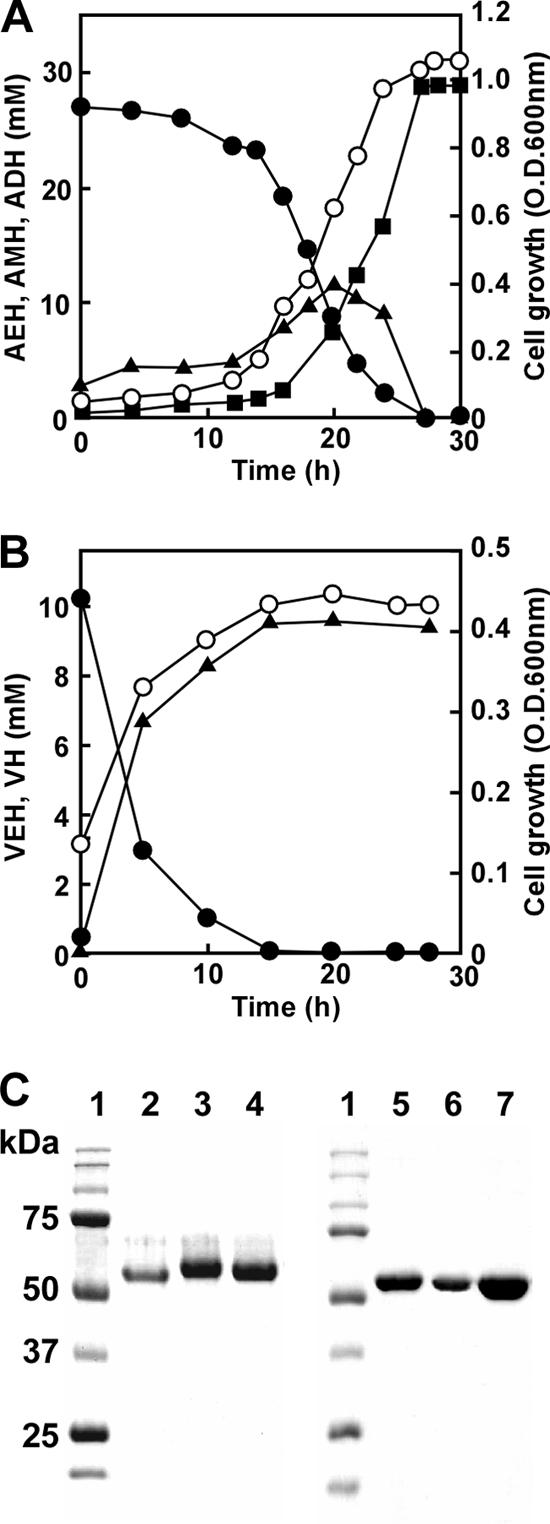
Degradation of hydrazones by P. aeruginosa PAO1. (A) P. aeruginosa PAO1 was cultured in MMAEH medium at 30°C, and concentrations of AEH (●), AMH (▲), ADH (■), and the optical density at 600 nm (OD600) (○) were measured. (B) P. aeruginosa PAO1 was cultured in MM medium containing 10 mM VEH at 30°C, and concentrations of VEH (●), VH (▲), and the optical density at 600 nm (OD600) (○) were measured. (C) Purified enzymes were resolved by SDS-PAGE on 10% polyacrylamide gels and stained with Coomassie brilliant blue. Lane 1, markers (Bio-Rad Precision Protein Standard kit); lane 2, HdhA purified from P. aeruginosa PAO1; lane 3, rHdhA; lane 4, rExaC; lane 5, rOaHdh; lane 6, rPaHdh; lane 7, rAldB.
The AEH-dependent NAD+ reduction (HDH activity) by cell extracts of P. aeruginosa PAO1 cultured in MMAEH medium was 0.25 μmol min−1 mg−1. We purified the HDH activity from cell extracts of P. aeruginosa PAO1 and confirmed the homogeneity of the purified preparation on SDS-PAGE (Fig. 2C; Table 1). The molecular masses of the enzyme, calculated from SDS-PAGE and gel filtration chromatography, were 55 and 110 kDa, respectively, indicating that the purified enzyme was dimeric. The specific activity of the purified HDH was 4.0 μmol min−1 mg−1 when NAD+ and AEH (5 mM each) were included. Enzymatic activity was maximal when cells were grown with AEH in the absence of glucose or succinate. The enzyme also used VEH as a substrate with a specific activity of 3.1 μmol min−1 mg−1. The purified HDH converted AEH and NAD+ to stoichiometric amounts of ADH, AMH, and acetate (see the supplemental Results section and Fig. S1 in the supplemental material). Similarly, NAD+ and VEH were converted to NADH, valeric acid hydrazide (VH), and acetate, indicating that the purified enzyme converted the hydrazone to the relevant hydrazides shown in Fig. 1A. This reaction is characterized by the involvement of another water molecule that hydrolyzes a possible reaction intermediate (I) to the hydrazide and a thioacyl-enzyme complex in addition to simply hydrating the thioacyl-enzyme complex (Fig. 1B). The HDH reaction is in contrast to the simple chemical reaction in which H+-dependent hydrolysis produces aldehydes instead of acids. This is the most unique enzymatic indicator of the HDH reaction being common to both fungal-type (3) and group X (see below) ALDH families (oxidative hydrolysis).
Table 1.
Summary of purification of HDH from P. aeruginosa PAO1
| Step | Total protein (mg) | Total activity (μmol min−1) | Sp act (μmol min−1 mg−1) | Yield (%) |
|---|---|---|---|---|
| Cell extract | 272 | 68.4 | 0.25 | 100 |
| DEAE cellulose | 68.8 | 47.5 | 0.69 | 69 |
| Butyl Sepharose | 11.7 | 16.3 | 1.4 | 24 |
| Resource Q | 1.7 | 4.7 | 2.7 | 6.7 |
| Superose 6 10/300 GL | 0.20 | 0.80 | 4.0 | 1.2 |
Hydrazone dehydrogenase belongs to the group X ALDH family.
Peptide mass fingerprint analysis of tryptic digests of purified HDH from the strain PAO1 identified it as a gene product of PA4022 (Fig. 3), which we designated hdhA. We identified the paralogous gene exaC (PA1984) with unknown function (16) in the P. aeruginosa PAO1 genome, and it encoded a protein that differed from HdhA by only eight amino acids. Identity between the purified HDH and HdhA was confirmed by detecting peptides specific to HdhA tryptic peptides (Fig. 3). We prepared recombinant HdhA (rHdhA) and ExaC (rExaC) as 6×His-tagged enzymes (Fig. 2C). The Km values of the rHdhA for VEH and NAD+ were similar to those of native HDH (Table 2). Recombinant HdhA also reduced NADP+ with a 37-fold higher Km value for NADP+ than for NAD+, indicating that rHdhA prefers NAD+. The levels of these parameters were similar for reactions by rExaC (Table 2). Double reciprocal plots of the HDH reaction followed a sequential mechanism (see Fig. S2 and Table S2 in the supplemental material), indicating that the enzyme forms a ternary complex with VEH and NAD(P)+ like that of C. palmioleophila HDH (3). Other enzymatic properties are shown in the supplemental material.
Fig 3.

Amino acid sequences of group X HDH and yeast HDH. Amino acid residues that differed between HdhA and ExaC from the PAO1 strain are underlined below the ExaC sequence. Tryptic fragments of P. aeruginosa PAO1 HdhA identified herein are indicated by lines above the HdhA sequence, and the molecular masses are given. Asterisks indicate fragments specific for HdhA. Asterisks under the alignment mark the conserved amino acids. cHdh, HDH from C. palmioleophila.
Table 2.
Kinetic constants of HDH and group X ALDHa
| Enzyme | Strain |
Km (μM) for: |
kcat (min−1) for: |
kcat/Km (min−1 μM−1) for: |
|||||
|---|---|---|---|---|---|---|---|---|---|
| VEH | Acetaldehyde | NAD+ | NADP+ | VEH | Acetaldehyde | VEH | Acetaldehyde | ||
| HdhA | P. aeruginosa | 8.5 ± 2.2 | 19 ± 3 | 210 ± 40 | 1,700 ± 270 | 880 ± 80 | 980 ± 40 | 104 | 51.7 |
| rHdhA | P. aeruginosa | 52 ± 19 | 15 ± 3 | 18 ± 1 | 670 ± 140 | 1,500 ± 300 | 1,510 ± 100 | 29 | 104 |
| rHdhB | P. aeruginosa | 13 ± 2 | 15 ± 4 | 48 ± 8 | 640 ± 100 | 1,100 ± 100 | 1,100 ± 100 | 88 | 75 |
| rOaHdh | O. anthropi | 16 ± 5 | 2.7 ± 0.7 | 124 ± 41 | NA | 60 ± 4 | 54 ± 2 | 3.7 | 8.9 |
| rPdHdh | P. denitrificans | 65 ± 13 | 18 ± 6 | 900 ± 180 | NA | 91 ± 5 | 97 ± 7 | 1.4 | 5.3 |
| rAldB | E. coli | 69 ± 15 | 2.8 ± 0.6 | NA | 170 ± 20 | 170 ± 10 | 160 ± 10 | 2.5 | 57 |
| cHdhb | C. palmioleophila | 19.5 ± 3.6 | 14.3 ± 4 | 195 ± 23c | 697 ± 79c | 1,260 ± 66 | 1,180 ± 105 | 64.4 | 82.6 |
Enzyme activities were measured at fixed concentrations of either 1 mM NAD(P)+ or 1 mM VEH. Data are the means of four experiments (and the standard deviations for Km and kcat values). NA, not applicable due to low activity.
Data from Ito et al. (3).
Data obtained from double reciprocal plots.
Phylogenetic analysis located HdhA and ExaC in the class 1/2 trunk in the ALDH superfamily (Fig. 4), and they were related to group X ALDH according to the Hempel classification (12). A database search disclosed that HdhA is identical to Vibrio cholerae AldA-2 (72%) (11), Alcaligenes eutrophus ACDH-II (68%) (13), and Escherichia coli AldB (66%) (24), all of which oxidize acetaldehyde (Fig. 3). The amino acid identity between HdhA and C. palmioleophila HDH, involved in the fungal-type ALDH family (3), was at most 41%. The sequence 294FFNTGEVC is compatible with the motif for the catalytic Cys residue conserved among proteins in the ALDH superfamily (12, 19), and the sequence 236IAFTGSTPTG, which agrees with the consensus sequence for the NAD-binding Rossmann fold (7), was present (Fig. 3). Other residues for NAD+ binding (Glu262 and Phe404) and for the general base (Glu262) (19) were conserved in HDH. This study discovered that proteins in the group X ALDH family play crucial metabolic roles as HDH.
Fig 4.

Schematic illustration of phylogenetic relationships among ALDH families. The phylogenetic tree was constructed using the Clustal W and PHYLIP programs. An asterisk indicates an ALDH with HDH activity. ALDH, aldehyde dehydrogenase; BALDH, betaine aldehyde dehydrogenase; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GGSALDH, γ-glutamyl semialdehyde dehydrogenase; MMSALDH, methylmalonyl semialdehyde dehydrogenase; SSALDH, succinic semialdehyde dehydrogenase.
Group X and fungal-type ALDH families located in the same branch in class 1/2 trunks are phylogenetically related (Fig. 4). The amino acid sequence of P. aeruginosa PAO1 HdhA is 41% identical to those of the fungal-type C. palmioleophila HDH and S. cerevisiae Ald4p (YOR374W), both of which function as HDH (3). The highest identity to the predicted ALDH in the fungal ALDH family was 49%. The criterion that various ALDHs with sequences that have <40% identity belong to different families (22) suggests close relationships between these protein families and HDH activity. Proteins in the mammalian class 2 family are notably included in the same branch as group X ALDH and the fungal ALDH (Fig. 4). To date, ALDH functioning as HDH has not been determined in the mammalian class 2 family, although their sequence similarity and wide substrate range (4) suggest that they function as HDH. The probable oxidative degradation of hydrazones by liver ALDH would be a useful approach to understanding the xenobiotic responses of vertebrates.
The hdhA gene is critical for hydrazone utilization.
We conventionally constructed gene disruptants of hdhA (strain DA1), exaC (strain DB1), and both hdhA and exaC (strain DAB1) based on the double crossover between the DNA encoding the truncated genes and the chromosome of P. aeruginosa PAO1 (see Fig. S3 in the supplemental material). After exposure to AEH, the strain DA1 produced 52% of the HDH activity produced by the PAO1 strain (Fig. 5A). DB1 and DAB1 also produced less (74% and 28%) HDH activity, indicating defective HDH production in these strains. The remaining activity in DAB1 is due to other dehydrogenases in the cells. Strain DB1 grew at the same rate as PAO1 when AEH was the sole source of carbon, indicating that exaC is dispensable for assimilating AEH, whereas growth was partly defective in the DA1 strain (Fig. 5B). The AEH-dependent growth of DAB1 was the most impaired (Fig. 5B), a result which agreed with intracellular levels of HDH activity (Fig. 5A) and with the rates of AEH degradation to AMH and ADH (see Fig. S4 in the supplemental material). These results indicated that hdhA is responsible for assimilating AEH and that exaC partially compensates for the AEH assimilation deficiency that arises in the absence of intact hdhA.
Fig 5.

Role of HDH isozymes on hydrazone and ethanol utilization. (A) P. aeruginosa PAO1 (W), DA1 (ΔhdhA), DB1 (ΔexaC), and DAB1 (ΔhdhA ΔexaC) were precultured in LB medium and transferred to MMAEH medium, and HDH activity in cell extracts was measured after 16 h in culture. *, P < 0.005; **, P < 0.03. (B and C) Growth of the P. aeruginosa strains in MM containing 30 mM AEH (B) and 100 mM ethanol (C). Strains are PAO1 (●), DA1 (▲), DB1 (■), and DAB1 (○).
Although the growth rates of the PAO1, DA1, DB1, and DAB1 strains did not significantly differ when cultured in MM media containing glucose or acetate as the sole carbon source (data not shown), growth of the DAB1 strain was delayed in MM medium containing ethanol as a carbon source (Fig. 5C). This defect was not evident in the DA1 and DB1 strains, indicating that either hdhA or exaC is required for assimilating ethanol. P. aeruginosa oxidizes ethanol to acetate through the successive reactions of alcohol dehydrogenase and ALDH and then assimilates it (16). We found here that purified rHdhA and rExaC both oxidize acetaldehyde. The kcat and Km values for the ALDH reaction and HDH reactions were comparable (Table 2), indicating that rHdhA and rExaC function as ALDH as efficiently as they function as HDH. Thus, hdhA and exaC are involved in the assimilation mechanism of ethanol as well as hydrazone, although hdhA contributes to hydrazone assimilation more than exaC does.
Several isozymes of ALDH have been found in eubacteria, fungi, and higher eukaryotes (12), but their physiological significance is not fully understood. P. aeruginosa PAO1 possesses 26 genes that encode ALDH-like proteins (19), and betB, mmsA, and exaC have been characterized as betaine aldehyde dehydrogenases, methylmalonate semialdehyde dehydrogenase, and acetaldehyde dehydrogenase for ethanol utilization, respectively (2, 20, 23). This study uncovered genetic evidence that hdhA and exaC oxidize hydrazones and acetaldehyde to different degrees. Considering their similar kinetic properties (Table 2) and amino acid sequences (see Fig. S2 in the supplemental material), this difference was not caused by their enzymatic properties but rather by a difference in their gene expression. That is, ethanol and the hydrazones upregulate exaC and hdhA transcription, respectively, causing the bacterium to efficiently assimilate carbon sources (see Fig. S5 in the supplemental material). We plan to determine in future studies precisely how the expression of these genes is regulated.
Distribution of HDH/group X ALDH among bacteria.
Proteins similar to HdhA were searched among predicted sequences in bacterial genomes using the genomic BLAST program (http://www.ncbi.nlm.nih.gov/sutils/genom_table.cgi). Phylogenetic analysis indicated that most alpha-, beta-, and gammaproteobacteria and actinobacteria contain proteins in group X of ALDHs, indicating a wide distribution of group X ALDHs in these groups of bacteria. Figure 6 shows representative proteins. Other proteobacteria and cyanobacteria contained fewer or no group X ALDHs. Most analyzed firmicute genomes contained few genes for group X ALDHs, but those of Brevibacillus and Geobacillus contained some exceptions. The amino acid sequences of the group X ALDHs of Paracoccus denitrificans (Pden_2366 in Fig. 3), Ochrobactrum anthropi (Oant_0271), and E. coli (AldB) were 69%, 69%, and 66% identical to P. aeruginosa HdhA (Fig. 3), and each ALDH was a unique group X type in the genome of each strain. Recombinant proteins for these ALDHs that were prepared using E. coli (rPaHdh, rOaHdh, and rAldB, respectively) (Fig. 3) oxidized acetaldehyde with kcat and Km values comparable to those of the known ALDH (Table 2), indicating that these enzymes function as ALDHs. They were also active as HDH and reacted with VEH to generate VH. The Km for VEH was comparable to that for acetaldehyde as well as those of P. aeruginosa HdhA and ExaC for VEH and acetaldehyde (Table 2). The kcat for the reaction was as high as that for acetaldehyde oxidation. Recombinant PaHdh and rOaHdh used only NAD+, whereas E. coli AldB used NADP+ (2), indicating a more distinct preference for nucleotide cofactors than P. aeruginosa HDH. These results indicated that these group X ALDHs oxidized VEH as efficiently as acetaldehyde.
Fig 6.

Distribution of group X ALDH/HDH among bacteria. Phylogenetic tree constructed using the Clustal W and PHYLIP programs. Accession numbers and protein names are provided together with species names. Alpha, alphaproteobacteria; Beta, betaproteobacteria; Gamma, gammaproteobacteria.
Reverse transcription-PCR (RT-PCR) indicated that the bacteria exposed to hydrazones transcribed the group X ALDH genes (Fig. 7A). They produced HDH activity in cell extracts (Fig. 7B). O. anthropi as well as P. aeruginosa cells degraded VEH with relatively higher titers and thrived when VEH was the sole carbon source (Fig. 7B and C). Their HDH gene expression was upregulated by the hydrazone (Fig. 7A), indicating that bacteria both responded to and assimilated VEH. In contrast, neither Paracoccus denitrificans nor E. coli could grow in the presence of VEH (Fig. 7C) as the sole source of carbon. Incubation with VEH generated cellular HDH activity (Fig. 7B). These strains were unable to grow in the presence of the hydrazone (Fig. 7C), a result which agreed with a lower rate of VEH degradation (Fig. 7B) and impaired cell growth in the presence of the toxic hydrazone (Fig. 7C). We did not examine any bacterial cultures with AEH except for that of the PAO1 strain since AEH inhibited their growth more severely than VEH.
Fig 7.
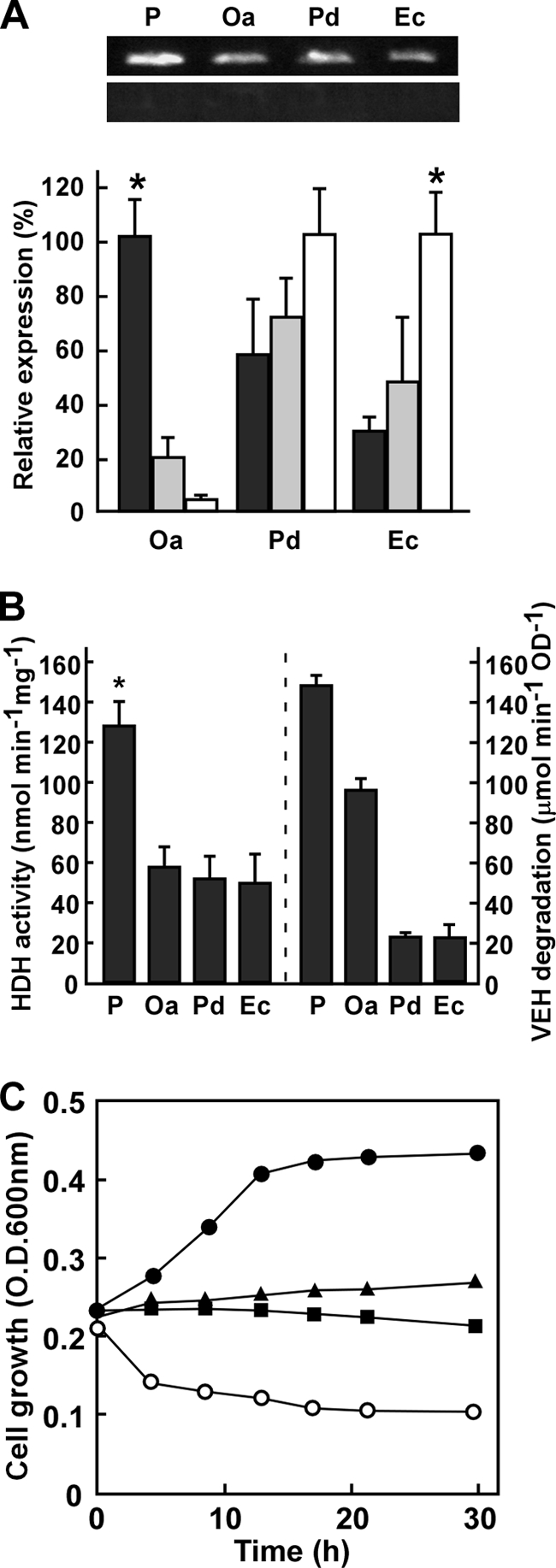
Hydrazone degradation by bacteria. (A) Transcripts of group X ALDH genes from P. aeruginosa (hdhA), O. anthropi (Oa), Paracoccus denitrificans (Pd), and E. coli (Ec) were analyzed by RT-PCR (top) and quantitative PCR (bottom). Bacteria were cultured in LB medium for 16 h and then incubated in MM medium containing 30 mM AEH (black), 100 mM glucose (gray), and 100 mM ethanol (white) at 30°C for 12 h. *, P < 0.001. (B) Cell-free HDH activity (left) and HDH in vivo (right) were assayed in bacteria cultured in MMAEH medium as described above (Materials and Methods). *, P < 0.01. (C) Time-dependent change in OD600 in MM medium containing 10 mM VEH. ●, P. aeruginosa PAO1; ▲, O. anthropi; ■, E. coli; ○, Paracoccus denitrificans.
Overall, the present findings demonstrated four bacterial group X ALDH proteins that function as HDH. The predicted group X ALDH proteins were distributed among proteobacteria and actinobacteria, which include known environmental and pathogenic types. For example, Paracoccus denitrificans and O. anthropi strains are distributed in the environment (1, 25) and P. aeruginosa, O. anthropi, and E. coli involve opportunistic pathogens of animals (6, 14). These imply that many natural and xenobiotic hydrazone compounds are metabolized by these bacteria in nature.
Supplementary Material
ACKNOWLEDGMENTS
We thank Norma Foster for critical reading of the manuscript.
This study was partly supported by the Bio-oriented Technology Research Advancement Institution and a Grant-in-Aid for Scientific Research from the Ministry of Education, Science, Culture and Sports, Japan.
Footnotes
Published ahead of print 20 January 2012
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1. Doi Y, Takaya N, Takizawa N. 2009. Novel denitrifying bacterium Ochrobactrum anthropi YD50.2 tolerates high levels of reactive nitrogen oxides. Appl. Environ. Microbiol. 75:5186–5194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ho KK, Weiner H. 2005. Isolation and characterization of an aldehyde dehydrogenase encoded by the aldB gene of Escherichia coli. J. Bacteriol. 187:1067–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Itoh H, Suzuta T, Hoshino T, Takaya N. 2008. Novel dehydrogenase catalyzes oxidative hydrolysis of carbon-nitrogen double bonds for hydrazone degradation. J. Biol. Chem. 283:5790–5800 [DOI] [PubMed] [Google Scholar]
- 4. Klyosov AA. 1996. Kinetics and specificity of human liver aldehyde dehydrogenases toward aliphatic, aromatic, and fused polycyclic aldehydes. Biochemistry 35:4457–4467 [DOI] [PubMed] [Google Scholar]
- 5. Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685 [DOI] [PubMed] [Google Scholar]
- 6. Lister PD, Wolter DJ, Hanson ND. 2009. Antibacterial-resistant Pseudomonas aeruginosa: clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin. Microbiol. Rev. 22:582–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liu ZJ, et al. 1997. The first structure of an aldehyde dehydrogenase reveals novel interactions between NAD and the Rossmann fold. Nat. Struct. Biol. 4:317–326 [DOI] [PubMed] [Google Scholar]
- 8. Maseda H, et al. 2004. Enhancement of the mexAB-oprM efflux pump expression by a quorum-sensing autoinducer and its cancellation by a regulator, MexT, of the mexEF-oprN efflux pump operon in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 48:1320–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Michelot D, Toth B. 1991. Poisoning by Gyromitra esculenta—a review. J. Appl. Toxicol. 11:235–243 [DOI] [PubMed] [Google Scholar]
- 10. Nagy I, et al. 1995. Degradation of the thiocarbamate herbicide EPTC (S-ethyl dipropylcarbamothioate) and biosafening by Rhodococcus sp. strain NI86/21 involve an inducible cytochrome P-450 system and aldehyde dehydrogenase. J. Bacteriol. 177:676–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Parsot C, Mekalanos JJ. 1991. Expression of the Vibrio cholerae gene encoding aldehyde dehydrogenase is under control of ToxR, the cholera toxin transcriptional activator. J. Bacteriol. 173:2842–2851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Perozich J, Nicholas H, Wang BC, Lindahl R, Hempel J. 1999. Relationships within the aldehyde dehydrogenase extended family. Protein Sci. 8:137–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Priefert H, Krüger N, Jendrossek D, Shimidt B, Steinbüchel A. 1992. Identification and molecular characterization of the gene coding for acetaldehyde dehydrogenase II (acoD) of Alcaligenes eutrophus. J. Bacteriol. 174:899–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ramos JM, Román A, Fernández-Roblas R, Cabello A, Soriano F. 1996. Infection caused by Ochrobactrum anthropi. Clin. Microbiol. Infect. 1:214–216 [DOI] [PubMed] [Google Scholar]
- 15. Rothgery EF. 2004. Hydrazine and its derivatives, p 562–607 Kirk-Othmer encyclopedia of chemical technology, vol 13 Wiley-Interscience, New York, NY [Google Scholar]
- 16. Schobert M, Görish H. 1999. Cytochrome c550 is an essential component of the quinoprotein ethanol oxidation system in Pseudomonas aeruginosa: cloning and sequencing of the genes encoding cytochrome c550 and an adjacent acetaldehyde dehydrogenase. Microbiology 145:471–481 [DOI] [PubMed] [Google Scholar]
- 17. Shimizu M, Fujii T, Masuo S, Fujita K, Takaya N. 2009. Proteomic analysis of Aspergillus nidulans cultured under hypoxic conditions. Proteomics 9:7–19 [DOI] [PubMed] [Google Scholar]
- 18. Shimogawa H, Kuribayashi S, Teruya T, Suenaga K, Kigoshi H. 2006. Cinachyramine, the novel alkaloid possessing a hydrazone and two animals from Cinachyrella sp. Tetrahedron Lett. 47:1409–1411 [Google Scholar]
- 19. Sophos NA, Vasiliou V. 2003. Aldehyde dehydrogenase gene superfamily: the 2002 update. Chem. Biol. Interact. 143–144:5–22 [DOI] [PubMed] [Google Scholar]
- 20. Steele MI, Lorenz D, Hatter K, Park A, Sokatch JR. 1992. Characterization of the mmsAB operon of Pseudomonas aeruginosa PAO encoding methylmalonate-semialdehyde dehydrogenase and 3-hydroxyisobutyrate dehydrogenase. J. Biol. Chem. 267:13585–13592 [PubMed] [Google Scholar]
- 21. Toyofuku M, et al. 2008. Influence of the Pseudomonas quinolone signal on denitrification in Pseudomonas aeruginosa. J. Bacteriol. 190:7947–7956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vasiliou V, Bairoch A, Tipton KF, Nebert DW. 1999. Eukaryotic aldehyde dehydrogenase (ALDH) genes: human polymorphisms, and recommended nomenclature based on divergent evolution and chromosomal mapping. Pharmacogenetics 9:421–434 [PubMed] [Google Scholar]
- 23. Velasco-García R, González-Segura L, Muñoz-Clares RA. 2000. Steady-state kinetic mechanism of the NADP+- and NAD+-dependent reactions catalysed by betaine aldehyde dehydrogenase from Pseudomonas aeruginosa. Biochem. J. 352:675–683 [PMC free article] [PubMed] [Google Scholar]
- 24. Xu J, Johnson RC. 1995. aldB, an RpoS-dependent gene in Escherichia coli encoding an aldehyde dehydrogenase that is repressed by Fis and activated by Crp. J. Bacteriol. 177:3166–3175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zumft WG. 1997. Cell biology and molecular basis of denitrification. Microbiol. Mol. Biol. Rev. 61:533–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.