

Abstract
CtsR is an important repressor that modulates the transcription of class III stress genes in Gram-positive bacteria. In Bacillus subtilis, a model Gram-positive organism, the DNA binding activity of CtsR is regulated by McsAB-mediated phosphorylation of the protein where phosphorylated CtsR is a substrate for degradation by the ClpCP complex. Surprisingly, the mcsAB genes are absent from many Gram-positive bacteria, including streptococci; therefore, how CtsR activity is modulated in those bacteria remains unknown. Here we show that the posttranslational modulation of CtsR activity is different in Streptococcus mutans, a dental pathogen. We observed that of all of the Clp-related proteins, only ClpL is involved in the degradation of CtsR. Neither ClpP nor ClpC had any effect on the degradation of CtsR. We also found that phosphorylation of CtsR on a conserved arginine residue within the winged helix-turn-helix domain is necessary for modulation of the repressor activity of CtsR, as demonstrated by both in vitro and in vivo assays. We speculate that CtsR is regulated posttranslationally by a different mechanism in S. mutans and possibly in other streptococci.
INTRODUCTION
Heat shock response is one of the most characterized physical stress adaptions of bacteria. Under heat shock stress, bacterial cells rapidly recruit a number of heat shock proteins that disaggregate, refold, and/or degrade misfolded proteins (35). This process is accomplished primarily by the Clp (caseinolytic protease) complex in the cell. In the Clp proteolytic system, ClpP protease is associated with an ATPase partner to form the functional complex (25). ClpP alone can degrade protein substrates but inefficiently (22). The function of Clp ATPases is to determine the specificity of the substrates and transfer them to the complex in which ClpP actually degrades the substrates (16, 28, 37, 48).
In Bacillus subtilis, the Gram-positive model organism, the heat shock response network is composed of multiple regulators such as the alternative sigma factor σB (19), the CssR two-component system (21), and two transcriptional repressors, CtsR and HrcA (9, 10, 40). While class I stress genes encode classical chaperones and class II stress genes are regulated mainly by the alternative sigma factor σB (38), CtsR is the major regulator that mainly regulates the expression of the class III stress genes that encode either Clp ATPases or ClpP protease by recognizing a tandemly repeated heptanucleotidic sequence known as the CtsR box (9, 10, 38).
In B. subtilis and other closely related bacteria, CtsR is encoded by the first gene of the clpC operon, which consists of four genes: ctsR, mcsA, mcsB, and clpC (30). The activity of CtsR is modulated mainly by McsB, which harbors arginine kinase activity (17). Under nonstress conditions, ClpC inhibits the kinase activity of McsB by sequestration of the protein (12, 29, 31). During heat shock, McsB is released from ClpC and activated by McsA. This activation leads to phosphorylation of CtsR by MscB on various conserved arginine residues, which prevents CtsR binding to its target DNA (13, 17, 29, 31). McsB also acts as an adaptor protein and delivers unbound phosphorylated CtsR to the ClpCP complex for degradation (27).
In addition to the above-described mechanism, CtsR can intrinsically sense the change in temperature via a glycine-rich loop (RGGGGY) present near the DNA binding winged helix-turn-helix (HTH) domain (10, 14, 24). Replacement of the glycine residue at position 64 with a more rigid residue greatly impairs the thermosensing ability of B. subtilis CtsR both in vitro and in vivo (14). Furthermore, the stability of the CtsR protein is also regulated by ClpE (a Clp family ATPase), which functions as a chaperone and is involved in overall protein quality control in the cell (36).
Surprisingly, the genomes of several low-GC Gram-positive organisms do not carry either the mcsA or the mcsB gene. Therefore, the nature of the posttranslational regulation of CtsR is not known in these bacteria. In this study, we have used Streptococcus mutans, a major etiological agent of dental caries, as a model organism to investigate the turnover of CtsR in McsAB-deficient bacteria. As in other streptococci, the S. mutans genome harbors five Clp ATPase-encoding genes: clpB, clpC, clpE, clpL, and clpX (1). Of these genes, only three, clpC, clpE, and clpX, are known to be regulated by the CtsR repressor in S. mutans (33, 47). We found that regulation of clpL gene expression is dependent on CtsR. We also found that ClpL is involved in the posttranslational degradation of CtsR. This is surprising, since in other Gram-positive organisms, ClpCP, and not ClpL, is the major protease complex that controls the stability of CtsR in the cell.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
The S. mutans strains and plasmids used in this study are listed in Table 1. Escherichia coli DH5α was grown in Luria-Bertani medium supplemented with 100 μg/ml ampicillin, 300 μg/ml erythromycin, or 50 μg/ml kanamycin when necessary. S. mutans strains were routinely grown in Todd-Hewitt medium (BBL, BD) supplemented with 0.2% yeast extract (THY medium). When necessary, 5 μg/ml erythromycin or 300 μg/ml kanamycin was added to THY medium.
Table 1.
S. mutans strains and plasmids used in this study
| Strain or plasmid | Descriptiona | Reference or source |
|---|---|---|
| Strains | ||
| A159 | Wild type, serotype c | 1 |
| IBS512 | UA159 derivative, ΔclpP | 2 |
| IBS514 | UA159 derivative, SMU1405::PclpP-gusA | 47 |
| IBS938 | IBS514 derivative, ΔctsR | 47 |
| IBSJ1 | UA159 derivative, ΔclpB | This study |
| IBSJ2 | UA159 derivative, ΔclpC | This study |
| IBSJ3 | UA159 derivative, ΔclpL | This study |
| IBSJ4 | UA159 derivative, ΔclpX | This study |
| IBSJ5 | UA159 derivative, ΔclpE | This study |
| IBSJ10 | IBSJ3 derivative, ΔclpL/pIBJ38 | This study |
| Plasmids | ||
| pGEM-T EZ | Commercial TA cloning vector, Apr | Promega |
| pCRII-TOPO | Commercial TA cloning vector, Apr Kmr | Invitrogen |
| pASK-IBA43+ | Commercial vector for protein expression, Apr | IBA |
| pCrePA | Thermosensitive plasmid that expresses Cre recombinase, Emr | 39 |
| pIBC36 | pGEMT-Easy carrying ctsR ORF | This study |
| pIBC37 | pASK-IBA43+ with wild type ctsR, Apr | This study |
| pIBC40 | pASK-IBA43+ with R62A ctsR, Apr | This study |
| pIBC41 | pASK-IBA43+ with R62K ctsR, Apr | This study |
| pIBC44 | pASK-IBA43+ with R62E ctsR, Apr | This study |
| pIB190 | Shuttle vector for protein expression in S. mutans, Emr | 7 |
| pIBJ1 | pIB190 with His-ctsR, Emr | This study |
| pIBJ2 | pIB190 with R62A His-ctsR, Emr | This study |
| pIBJ3 | pIB190 with R62K His-ctsR, Emr | This study |
| pIBJ4 | pIB190 with R62E His-ctsR, Emr | This study |
| pIBJ11 | pCRII-TOPO with loxP71-Kan-loxP66 cassette, Apr Kmr | This study |
| pIBJ12 | pGEM-T EZ derivative for deletion of clpB, Apr Kmr | This study |
| pIBJ13 | pGEM-T EZ derivative for deletion of clpC, Apr Kmr | This study |
| pIBJ18 | pGEM-T EZ derivative for deletion of clpX, Apr Kmr | This study |
| pIBJ19 | pGEM-T EZ derivative for deletion of clpE, Apr Kmr | This study |
| pIBJ20 | pGEM-T EZ derivative for deletion of clpL, Apr Kmr | This study |
| pIBJ38 | pIB184km derivative for clpL complementation, Kmr | This study |
Apr, ampicillin resistance; Kmr, kanamycin resistance; Emr, erythromycin resistance.
Whole-cell protein extraction from S. mutans and Western blot analysis.
For S. mutans whole-cell protein extraction, a culture grown overnight was used to inoculate THY medium and allowed to grow to exponential phase (optical density at 600 nm [OD600] = 0.4). Cells were harvested and resuspended in Tris-buffered saline and homogenized with a BeadBeater. Protein lysates were separated by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) or 12% Phos-tag (FMS Laboratory) SDS-PAGE (50 μM Mn2+-Phos-tag). The gels were blotted onto membranes, and Western blot assays were carried out using standard techniques with a antipolyhistidine monoclonal antibody (Sigma) as the primary antibody. The blots were developed with Pierce ECL plus reagent (Thermo Scientific), and the signals were detected by Typhoon FLA 9000 (GE Healthcare). All Western blot experiments were repeated at least twice to confirm the results.
RNA isolation and semiquantitative reverse transcription-PCR (sqRT-PCR).
A single colony was inoculated into 10 ml of THY medium and incubated overnight at 37°C. The grown culture was reinoculated into fresh THY medium with a dilution of 1:20 and incubated at 37°C to exponential phase (OD600 = 0.4). Ten milliliters of culture was centrifuged, the pellet was resuspended in 5 ml of RNAprotect reagent (Qiagen) and incubated at ambient temperature for 10 min, and the cells were lysed with a BeadBeater. Total RNA isolation was then performed using an RNeasy Mini Kit (Qiagen). DNA contaminants were removed by on-column DNase I treatment in accordance with the manufacturer's instructions. RNA samples were quantitated using a NanoDrop spectrophotometer. Reverse transcription with gene-specific primers was carried out in accordance with the technical manual of the Superscript II reverse transcriptase system (Invitrogen). Expression of the gyrA gene was chosen to serve as an internal control. The specific primers used for sqRT-PCR analyses are listed in Table 2.
Table 2.
Oligonucleotides used in this study
| Primer | Sequence (5′ to 3′) | Purpose |
|---|---|---|
| ClpB-UF | AATCATAGAAGCTGTCTCAG | Deletion of clpB gene |
| ClpB-UR | TCTCGAGGAAGAAGGATCCTGACTGGAAGAAATAGCAGG | Deletion of clpB gene |
| ClpB-DF | AGGATCCTTCTTCCTCGAGATTCTAGATGAGCTGAATCAC | Deletion of clpB gene |
| ClpB-DR | TTGTAGTACTTTTTGATAGGG | Deletion of clpB gene |
| ClpC-UF | CAAAAGAAGTGTTCTGGCAG | Deletion of clpC gene |
| ClpC-UR | TCTCGAGGAAGAAGGATCCTCTCTACCATTGCAAGAAGG | Deletion of clpC gene |
| ClpC-DF | AGGATCCTTCTTCCTCGAGACTTCAAGTACTTGATGATGG | Deletion of clpC gene |
| ClpC-DR | GAAGTAGGGTAAAGCGAGC | Deletion of clpC gene |
| ClpE-UF | TTCATATTTGTCAAGAACAGC | Deletion of clpE gene |
| ClpE-UR | TCTCGAGGAAGAAGGATCCTCTTCAAGCAACCCATTTGG | Deletion of clpE gene |
| ClpE-DF | AGGATCCTTCTTCCTCGAGAATATTGAAGATGCTATTACGG | Deletion of clpE gene |
| ClpE-DR | GTCTCACGATGACTGGAG | Deletion of clpE gene |
| ClpL-UF | AGACACGAAAGTACAATACG | Deletion of clpL gene |
| ClpL-UR | TCTCGAGGAAGAAGGATCCTGAGCTTGCTGTATTTCTGG | Deletion of clpL gene |
| ClpL-DF | AGGATCCTTCTTCCTCGAGATCTTCTCCTTCAAGTATTGG | Deletion of clpL gene |
| ClpL-DR | GCCTCTCAAATTGCATAG | Deletion of clpL gene |
| ClpX-UF | AGCTCTTTCTTGGTACAGG | Deletion of clpX gene |
| ClpX-UR | TCTCGAGGAAGAAGGATCCTAGAATGAACAATGCACTGTG | Deletion of clpX gene |
| ClpX-DF | AGGATCCTTCTTCCTCGAGAGAAATTCCGAGTCAAGAGG | Deletion of clpX gene |
| ClpX-DR | TATTCCATTTGCCACGAGG | Deletion of clpX gene |
| LoxPKanD7F | CTCCCGGCCGCCATGGCGGCCGC | Amplification of lox-Km-loc cassette |
| LoxPKanD7R | GGTCGACCTGCAGGCGGCCGCG | Amplification of lox-Km-loc cassette |
| RT-HisCtsR-F | AGAGGATCGCATCACCATC | qrt-PCR of ctsR |
| RT-HisCtsR-R | TCCAAGAACATCATCTCCTG | qrt-PCR of ctsR |
| gyrA-For | GACTATGCTATGAGTGTTATTGTTGCTCGGGC | qrt-PCR of gyrA |
| gyrA-Rev | GGCCATTCCAACAGCAATACCTGTCGCTCC | qrt-PCR of gyrA |
| Bam-CtsR-F | GGATCCATGACGTCAAAAAATACTTCAGAC | Amplification of ctsR ORF |
| Nco-CtsR-R | CCATGGTCATAGATGGTATCCTTTTCTAT | Amplification of ctsR ORF |
| Smu-CtsR-Ala-F | TTGAGAGCAAAGCCGGTGGTGGCGGTTATATTA | R62A substitution of CtsR |
| Smu-CtsR-Ala-R | GCCACCACCGGCTTTGCTCTCAACGGCATAACC | R62A substitution of CtsR |
| Smu-CtsR-Lys-F | TTGAGAGCAAAAAGGGTGGTGGCGGATATATTA | R62K substitution of CtsR |
| Smu-CtsR-Lys-R | TATCCGCCACCACCCTTTTTGCTCTCAACGGCATAACC | R62K substitution of CtsR |
| Smu-CtsR-R62E-F | TTGAGAGCAAAGAGGGTGGTGGCGGATATATTAGC | R62E substitution of CtsR |
| Smu-CtsR-R62E-R | ATATCCGCCACCACCCTCTTTGCTCTCAACGGCATAACC | R62E substitution of CtsR |
| Bam-ClpP-F6 | GAGCTGGATCCACGAGGTTAAGTCTGATAAATT | Amplification of PclpP |
| Xho-ClpP-Rout1 | AGTCGCTCGAGTGTTCAATAACTACAGGAATCAT | Amplification of PclpP |
| clpL-duet-L | CTTGGATCCGATGGCAAATTTTAATGGACGCG | clpL complementation |
| clpL-duet-R | CTTCTGCAGTTAAGCTTCTTCAATAATCAATTTGTC | clpL complementation |
Markerless clpB, clpC, clpE, clpL, or clpX gene deletion.
Markerless deletion of clpB, clpC, clpE, clpL, or clpX was achieved by adopting the Cre-lox protocol as described previously (2, 6). Briefly, a kanamycin resistance cassette with modified loxP sites (lox71-kan-lox66) was amplified and cloned into the pCRII-TOPO TA cloning vector to create plasmid pIBJ11. DNA fragments of about 500 bp each located up- and downstream of the target gene were amplified and fused together by overlapping PCR. The fusion fragment was inserted into pGEMT-EZ by TA cloning. The overlapping region of the fusion fragment also contained BamHI and XhoII restriction sites. The lox71-kan-lox66 cassette was isolated from pIBJ11 by BamHI and XhoII restriction and inserted into BamHI- and XhoII-restricted pGEMT-EZ derivatives. The final plasmids were linearized by NotI restriction and transformed into S. mutans UA159 for kanamycin resistance. Natural DNA transformation of S. mutans was performed as previously described (6). Clones with the target gene replaced with a lox71-kan-lox66 cassette were verified by PCR. To remove the lox71-kan-lox66 cassette, temperature-sensitive plasmid pCrePA (39) expressing Cre recombinase was transformed into the selected clones. The chromosomally integrated lox71-kan-lox66 cassette was excised by Cre at 30°C, and the pCrePA plasmid was cured from the resultant cells at 37°C. The names of the mutant strains and the primers used for gene deletions are listed in Tables 1 and 2, respectively.
Complementation of clpL.
The open reading frame (ORF) containing clpL was amplified from UA159 genomic DNA using primers clpL-duet-F and clpL-duet-R, and the 2-kb fragment was cloned into vector pIB184Km (7), which contains the P23 promoter, to create pIBJ38. Strain IBSJ3/pIBJ1 was transformed with pIBJ38 and screened on THY agar plates containing both erythromycin and kanamycin. The presence of the clpL and His-ctsR genes in the selected transformants was verified by PCR.
Construction of CtsR mutants.
The ORF containing ctsR (465 bp) was amplified from UA159 genomic DNA using primers Bam-CtsR-F and Nco-CtsR-R, and the fragment was cloned into vector pGEMT-EZ by TA cloning to create pIBC36. This plasmid was used as the template for site-directed mutagenesis to generate intermediate plasmids carrying three separate CtsR mutations (relevant primers are listed in Table-2). The ctsR fragments carrying various mutations were isolated from the intermediate plasmids by BamHI-NcoI digestion, gel purified, and cloned into vector pASK-IBA43, which was also restricted with BamHI-NcoI. The three CtsR mutations that were created are Arg62Ala (pIBC40), Arg62Lys (pIBC41), and Arg62Glu (pIBC44). The wild-type ctsR fragment was also isolated from pIBC36 by BamHI-NcoI digestion and cloned into BamHI-NcoI-digested vector pASK-IBA43 to generate pIBC37. For in vivo complementation studies, BamHI-NcoI-restricted fragments containing the ctsR ORF were isolated from pIBC36 and the intermediate plasmids (Table 1) and cloned into pIB190 (containing the P23 promoter [7]), which was also restricted with BamHI-NcoI. The resultant plasmids were named pIBJ1 (wild-type ctsR), pIBJ2 (Arg62Ala ctsR), pIBJ3 (Arg62Lys ctsR), and pIBJ4 (Arg62Glu ctsR). These plasmids were introduced into S. mutans by natural transformation (6).
Purification of CtsR protein and its derivatives.
E. coli DH5α with vector pASK-IBA43 containing either wild-type CtsR or the mutated CtsR derivatives was used for the overexpression of CtsR proteins. His-CtsR proteins were purified by Ni-nitrilotriacetic acid column chromatography (Qiagen) in accordance with the manufacturer's instructions. The protein was dialyzed overnight against a buffer containing 20 mM Tris-Cl (pH 8.0), 100 mM NaCl, 2 mM EDTA, and 10% (vol/vol) glycerol. The purity of the proteins was >95%, as determined by SDS-PAGE analysis. Protein concentrations were estimated using a Bradford protein assay kit (Bio-Rad) with bovine serum albumin as the standard.
Electrophoretic mobility shift assay (EMSA).
Interaction of CtsR (or its derivatives) with the clpP promoter was determined by EMSA as previously described (5). Briefly, primers Bam-ClpP-F6 and Xho-ClpP-Rout1 were used to amplify a 234-bp promoter region of clpP that contains the CtsR binding site. This clpP promoter fragment was labeled with [γ-32P]dATP using T4-PNK (NEB) in accordance with the manufacturer's protocol. For EMSA, the labeled fragment was incubated with different amounts of CtsR protein in a binding buffer [50 mM Tris-HCl (pH 8.0), 250 mM NaCl, 10 μg/ml poly(dI · dC), 5 mM CaCl2, 5 mM MgCl2, 5 mM dithiothreitol, 50% glycerol] for approximately 40 min at room temperature. Following incubation, 10 μl of the DNA-protein mixture was resolved in a 5% native polyacrylamide gel buffered with 0.05 M Na2HPO4-NaH2PO4 (pH 6.5) and run at 100 V for 1 h 30 min. The gel was then dried and exposed to a PhosphorImager plate and developed using a FLA9000 PhosphorImager (GE Healthcare). All EMSA experiments were repeated at least three times to confirm the results.
β-Glucuronidase (Gus) assay.
S. mutans cultures grown overnight were diluted 1:20 and grown to exponential phase. Before the culture was harvested, its OD600 was recorded. One milliliter of culture was harvested, washed in saline, and resuspended in 500 μl of Z buffer (60 mM Na2HPO4, 40 mM NaH2PO4, 10 mM KCl, 1 mM MgSO4, 20 mM dithiothreitol). S. mutans cells were homogenized by bead beating. A 200-μl volume of cell lysate was then transferred to a new test tube, 100 μl of p-nitrophenyl-β-d-glucoside (4 mg/ml in Z buffer) was added, and the mixture was incubated at 37°C until it turned yellow. The reaction was stopped by the addition of 200 μl of 1 M Na2CO3. The absorbance at 420 nm and the time period of the reaction were noted. Gus activity was defined as (1,000 × A420)/(time in minutes × OD600) in Miller units.
RESULTS
CtsR degradation is mediated by ClpL in S. mutans.
In several Gram-positive bacteria, such as B. subtilis, the clpC operon is composed of four genes, i.e., ctsR, mcsA, mcsB, and clpC (8, 30). However, mcsA and mcsB are absent from the genomes of some Gram-positive bacteria, including streptococci (Fig. 1A). Since McsB functions as an essential adaptor for ClpCP-dependent degradation of CtsR (27), we wondered how CtsR is degraded in mcsAB-deficient Gram-positive bacteria such as S. mutans. To identify the potential CtsR degradation pathway in S. mutans, we constructed a shuttle plasmid, pIBJ1, that constitutively expresses His-CtsR protein under the control of the P23 promoter (7). When pIBJ1 was introduced into S. mutans UA159, we observed that although the mRNA transcript was detected in large amounts (Fig. 1B), the His-CtsR protein was virtually undetected (Fig. 1C). We speculated that CtsR is rapidly degraded in the wild-type strain by posttranscriptional regulation. To investigate the mechanism of CtsR degradation, we first constructed single deletion mutants of all five of the Clp-related ATPases, i.e., clpB, clpC, clpL, clpX, and clpE. Plasmid pIBJ1 was then introduced into the above mutants to evaluate the fate of CtsR protein in vivo. Except for the clpL mutant, the CtsR protein level could not be detected in any of the Clp ATPase mutants (clpB, clpC, clpX, and clpE), while the mRNA was expressed steadily in all of the strains (Fig. 1B and C). We also included a clpP mutant strain (IBS512) as a control and observed no CtsR accumulation. To further verify the role of ClpL in the degradation of CtsR, we complemented the IBSJ3/pIBJ1 strain with a plasmid that expresses clpL under the control of the P23 promoter. The native ctsR-clpC promoter contains a CtsR binding consensus, and the transcription of ctsR is controlled by itself as previously reported. Therefore, we chose P23 as an independent promoter to study the posttranscriptional regulation of CtsR in order to prohibit the alteration of the protein level caused by transcriptional regulation. As shown in Fig. 1D, we observed no accumulation of CtsR protein, confirming that ClpL is crucial for the stability of CtsR. Since CtsR could not be detected in the clpP mutant (Fig. 1C), this suggests that the posttranscriptional regulation of CtsR is different in S. mutans since both ClpP and ClpC appear to have no effect on CtsR degradation. On the other hand, we observed that, CtsR was accumulated in large amounts inside the clpL mutant (Fig. 1C). In this mutant, we observed two different CtsR species; the faster-migrating species corresponds to monomeric CtsR, while the nature of the more slowly migrating species is unknown. Taken together, our results showed that ClpL plays a crucial role in the degradation of CtsR in S. mutans and the ClpCP complex does not have any effect on CtsR degradation.
Fig 1.

ClpL mediates the degradation of CtsR in S. mutans. (A) Genetic organization of the clpC operon in Gram-positive bacteria that contain (I) or do not contain (II) mcsAB. (B) The presence of His-ctsR mRNA expressed from plasmid pIBJ1 was verified by RT-PCR (bottom). Controls for DNA contamination and gyr mRNA are shown at the top and in the middle. Lanes 1 to 9: DNA molecular size markers, UA159/pIB190, UA159/pIBJ1, IBS512/pIBJ1, IBSJ1/pIBJ1, IBSJ2/pIBJ1, IBSJ3/pIBJ1, IBSJ4/pIBJ1, and IBSJ5/pIBJ1, respectively. (C) Amounts of His-CtsR protein in various clp mutants were evaluated by Western blotting with anti-His antibody. A 50-μg sample of total protein was loaded per lane. Samples in lanes 1 to 8: protein molecular size markers, UA159/pIBJ1, IBS512/pIBJ1, IBSJ1/pIBJ1, IBSJ2/pIBJ1, IBSJ3/pIBJ1, IBSJ4/pIBJ1, and IBSJ5/pIBJ1, respectively. (D) His-CtsR accumulation was not observed in the complemented strain (IBSJ10/pIBJ1, bottom). The presence of the clpL (top) and His-ctsR (middle) genes was verified by PCR.
CtsR regulates clpL transcription in S. mutans.
ClpL is a heat shock protein found mainly in Gram-positive bacteria (16). Although the detailed functional studies on ClpL have been limited, several previous reports have shown that the protein plays an important role as a chaperone during heat shock (32, 41, 42, 44). The protein was previously reported to be part of the CtsR regulon in several Gram-positive bacteria, such as Streptococcus thermophilus and Oenococcus oeni (18, 44). Our analysis of the promoter of S. mutans clpL also showed the presence of a weak CtsR binding consensus (Fig. 2A), and the sequence is similar to that of other known CtsR binding boxes in S. mutans (Fig. 2B). To verify whether CtsR can repress clpL transcription in S. mutans, we measured the clpL mRNA level by sqRT-PCR in IBS514 (an isogenic derivative of UA159 that contains PclpP-gusA), IBS938 (a ctsR deletion-containing strain derived from IBS514 [47]), and IBS938 containing plasmid pIBJ1. As expected, the clpL mRNA level was increased about 5-fold in the ctsR mutant compared to that in the wild type (determined by digital scanning of the image and quantitation by ImageQuant TL software), while the mRNA level in the complemented strain was similar to that in the wild-type strain (Fig. 2C and D). These data indicate that clpL is also part of the CtsR regulon and is regulated by a feedback mechanism.
Fig 2.
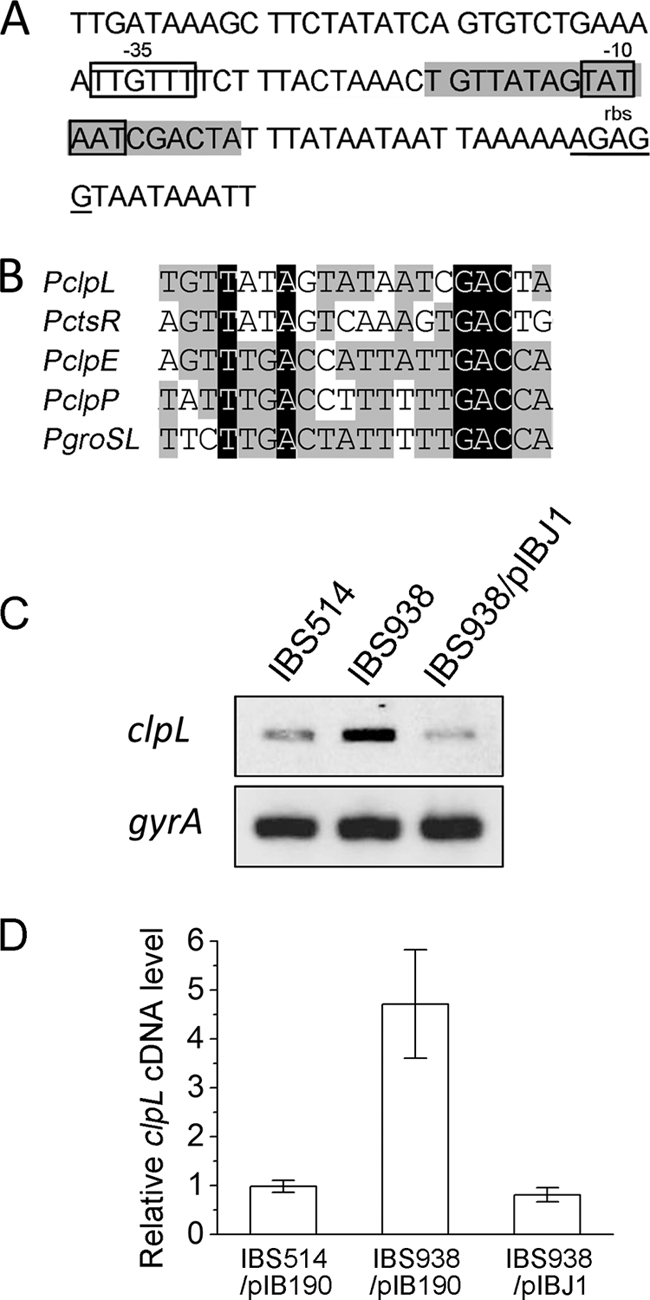
The clpL gene belongs to the CtsR regulon in S. mutans. (A) The sequence of the clpL promoter region in S. mutans is shown, with the predicted CtsR recognition consensus marked in the gray box. The Shine-Dalgarno sequence (rbs) and −35 and −10 regions are underlined. (B) Comparison of the CtsR binding consensus among the clpL promoter and other known CtsR-regulated promoters in S. mutans. (C) sqRT-PCR analysis of clpL. Total RNA was harvested from IBS514, IBS938, and IBS938/pIBJ1 and subjected to cDNA synthesis. Five nanograms of cDNA from each strain was used for sqRT-PCR. The gyrA gene was included to ensure that equal amounts of RNA were used for all reactions. (D) The average clpL mRNA levels in panel C are shown, and error bars show the standard deviations. n = 3 samples/group.
In vivo phosphorylation of CtsR in S. mutans.
Phosphorylation of the arginine residues in CtsR is considered one of the most important regulators of its activity in B. subtilis (23). In this organism, phosphorylation is catalyzed by McsB, which functions as an arginine kinase (17). Sequence alignment suggests that, irrespective of MscAB status, the CtsR protein is highly conserved at the N-terminal domain, which includes the HTH motif and the glycine-rich loop responsible for thermosensing. Several arginine residues considered to be sites for potential phosphorylation are also conserved, implying the possibility of phosphorylation of CtsR in mcsAB-deficient Gram-positive bacteria (Fig. 3A).
Fig 3.

(A) Alignment of the CtsR sequences with other Gram-positive bacteria. The asterisk indicates the conserved arginine residue used for the mutational study. Other conserved arginines as potential phosphorylation sites are indicated by arrowheads. Samples: SMU, S. mutans UA159; GAS, Streptococcus pyogenes M1; GBS, Streptococcus agalactiae NEM316; LLC, Lactobacillus lactis MG1363; LMO, Listeria monocytogenes EGD-e; BST, B. subtilis 168; SAU, Staphylococcus aureus N315. (B) Possible in vivo phosphorylation of His-CtsR in the ΔclpL mutant was detected by Phos-tag gel-based Western blot analysis. Purified His-CtsR was used as a nonphosphorylated control. Extract from IBSJ3/pIB190 was included as a negative control. IBSJ3/pIBJ1 cells were exposed to heat shock at 45°C for 0 (T0), 15 (T1), 30 (T2), 45 (T3), or 60 (T4) min before the preparation of crude extracts. (C) The shifted band in a Phos-tag gel-based Western blot assay was eliminated when the sample was treated with alkaline phosphatase. (D) Point mutations of Arg62 do not block phosphorylation. Extracts from IBSJ3/pIB190 (Control), IBSJ3/pIBJ1 (WT), IBSJ3/pIBJ2 (R62A), IBSJ3/pIBJ3 (R62K), and IBSJ3/pIBJ4 (R62E) were separated on a Phos-tag gel, transferred to a polyvinylidene difluoride membrane, and detected by Western blot analysis. Arrow indicates the phosphorylated form.
A novel type of polyacrylamide-bound Phos-tag electrophoresis (Mn2+-Phos-tag SDS-PAGE), which leads to a mobility shift detection of phosphoproteins, has been widely used to determine the phosphorylation status of many proteins (3, 26, 46). Analysis of CtsR by Phos-tag gel-based Western blotting revealed an additional much more slowly migrating band in the ΔclpL mutant strain (Fig. 3B). We speculate that the more slowly migrating band observed is a phosphorylated form of CtsR. To confirm this, we treated the crude extract from IBSJ3/pIBJ1 with alkaline phosphatase for up to 3 h. Western blotting showed that the shifted band was gradually eliminated during treatment (Fig. 3C), suggesting that the more slowly migrating band is a phosphorylated form of CtsR. However, unlike in B. subtilis, neither phosphorylation nor degradation of CtsR was induced by heat shock (Fig. 3B), implying that an alternative mode of CtsR regulation might operate in S. mutans. Interestingly, substitution mutations of a conserved arginine residue at position 62 (R62), which is one of the most important phosphorylation sites reported (17), did not abolish the overall phosphorylation status of the CtsR protein (Fig. 3D). This may not be too surprising, since multiple arginine residues of CtsR are known to be involved in phosphorylation, as reported previously (17).
Replacement of arginine 62 (R62) dramatically alters the DNA binding ability of S. mutans CtsR.
To explore whether the repressor activity of S. mutans CtsR would be affected by the phosphorylation of arginine residues, we conducted a mutational analysis of the protein by replacing arginine 62 with alanine (R62A), lysine (R62K), or glutamate (R62E), respectively. PclpP was chosen for the EMSA since it contains a typical CtsR binding consensus and was used for a CtsR binding experiment in a previous study (47). EMSA showed that CtsR efficiently bound to PclpP with high specificity, while a nonspecific competitor DNA fragment containing the promoter region of nlmA had no effect on the binding of CtsR to PclpP (Fig. 4A). EMSA analysis also showed that replacement of arginine with a neutral alanine residue or a positively charged lysine residue had no effect on the DNA binding activity of CtsR (Fig. 4B). However, replacement of the arginine residue with a phosphor-mimicking glutamate residue resulted in a drastic loss of the DNA binding ability of CtsR (Fig. 4B), indicating that phosphorylation may have an effect on DNA binding.
Fig 4.
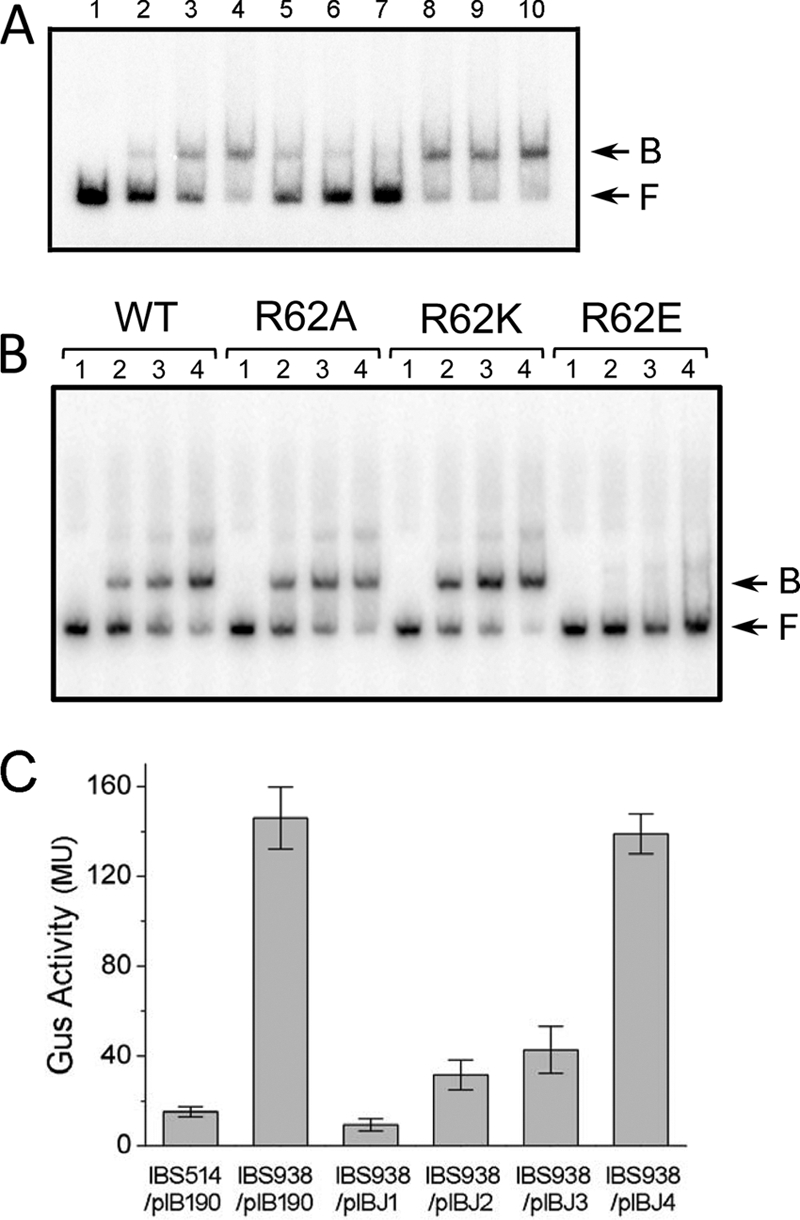
(A) CtsR specifically binds to a 32P-labeled 234-bp DNA fragment containing PclpP. Lanes 1 to 4: EMSA with 0 μg (lane 1), 0.05 μg (lane 2), 0.15 μg (lane 3), or 0.3 μg (lane 4) of His-CtsR protein added to 0.1 pmol of the 32P-labeled PclpP fragment. Lanes 5 to 7: EMSA with various amounts of the unlabeled PclpP fragment (lane 5, 0.3 pmol; lane 6, 0.8 pmol; lane 7, 1.5 pmol) and 0.3 μg of His-CtsR added to 0.1 pmol of the 32P-labeled PclpP fragment. Lanes 8 to 10: EMSA with various amounts of the PnlmA fragment as a nonspecific unlabeled competitor (lane 8, 0.3 pmol; lane 9, 0.8 pmol; lane 10, 1.5 pmol) and 0.3 μg of His-CtsR added to 0.1 pmol of the 32P-labeled PclpP fragment. (B) EMSA with various amount of different His-CtsR point mutation proteins added to 0.5 pmol of the 32P-labeled PclpP fragment. Samples: lane 1, 0 μg; lane 2, 0.4 μg; lane 3, 0.8 μg; lane 4, 1.6 μg. In panels A and B, the CtsR-PclpP DNA complexes (B) and free DNA (F) are indicated by arrows. WT, wild type. (C) Expression of the PclpP-gusA gene in the wild-type and ctsR mutant strains complemented with various ctsR derivatives. GusA activity was measured from the PclpP-gusA reporter construct that was present in the strains. The values shown are Miller units (MU) of Gus activity (n = 4 samples/group).
To verify the effect of phosphorylation of the R62 residue on in vivo S. mutans gene expression, we used a PclpP-gusA reporter fusion strain, IBS938, which was derived from IBS514 by deleting the ctsR gene locus (47). Plasmids pIBJ1, pIBJ2, pIBJ3, and pIBJ4, which express wild-type, R62A, R62K, and R62E CtsR, respectively, were introduced into IBS938. The Gus assay illustrated that expression of wild type, R62A, and R62E CtsR from the plasmids restored the repression of gusA transcription, while the expression of R62E CtsR failed to complement the ctsR deletion (Fig. 4C). Therefore, phosphorylation of S. mutans CtsR at R62 inhibits the repressor activity of the protein, whereas unphosphorylated CtsR binds to its target consensus sites with high affinity, a situation similar to those in Gram-positive bacteria that harbor MscAB.
DISCUSSION
In this study, we showed that ClpCP is not required for specific degradation of CtsR in S. mutans, an mcsAB-deficient, low-GC, Gram-positive bacterium. Furthermore, we found that ClpP protease activity is also not required for CtsR degradation. Surprisingly, we found that ClpL is critical for the stability of CtsR protein. ClpL has never been reported to be involved in a specific protein degradation process. ClpL belongs to the HSP100/Clp ATPase family and has been shown to be important for the stress tolerance response in some organisms (15, 41, 42, 45). Apart from its putative chaperone function and involvement in stress tolerance, very little is known about ClpL ATPase activity. Primary sequence analysis suggests that ClpL contains two nucleotide-binding domains with Walker motifs, one in the central region of the protein and the other in the C-terminal region. ClpL also contains a conserved domain termed the D2-small domain that was recently identified in the ClpB protein. However, ClpL does not contain the ClpP recognition tripeptide that is required for interaction with ClpP (25); therefore, ClpL appears to function independently of ClpP. Analysis of the ClpL sequence failed to identify any known protease-specific domain. We believe that ClpL possibly recruits some other proteases for the degradation of CtsR in vivo. It has been shown that overexpression of the Lactococcus lactis clpL gene in E. coli causes an overall increase in ATP-dependent proteolytic activity (20). Thus, the protease that interacts with ClpL may also be present in Gram-negative bacteria. The exact mechanism by which CtsR is degraded by ClpL is unknown and needs to be investigated further.
In the strain with clpL deleted, no obvious induction of CtsR degradation was observed during heat shock treatment, indicating the absence of any ClpL-independent degradation of CtsR. However, the ClpL-dependent degradation of CtsR may be increased after heat shock treatment since clpL expression is induced after heat shock. Since the proteolytic degradation of CtsR is so potent under normal growth condition, we speculate that ClpL-dependent degradation of CtsR does not play any specific role in heat shock induction.
Regulation of clpL expression appears to be complex and can vary, depending on the organism. In staphylococci, clpL is solely under the control of a secondary sigma factor, σB (15). In lactobacilli, the clpL promoter region contains a CIRCE sequence where HcrA, a transcriptional repressor, binds and regulates expression (41). In lactococci and some streptococci, clpL expression is regulated primarily by CtsR by binding to a consensus heptanucleotide repeat, A/GGTCAA/T, known as the CtsR box and located in the promoter region of the target genes. The exact placement of the CtsR box varies depending on the gene promoter. For S. thermophilus clpL, the CtsR box is located just upstream of the −35 region (44). We identified a weak CtsR box spanning the −10 region of the clpL promoter in S. mutans, and we have shown that clpL expression is indeed regulated by CtsR in this organism.
Environmental conditions also play an important role in clpL gene expression. Notably, growth under acidic conditions can induce clpL expression, as demonstrated in lactobacilli (45). The situation is probably similar in S. mutans, since the ClpL protein was produced in larger amounts when cells are grown at pH 5.0 than when they are grown at pH 7.0 (34). Osmotic stress, induced by the addition of 10% ethanol to the growth medium, can also activate clpL expression, as shown in O. oeni (4). Furthermore, clpL expression is induced by both heat shock and cold shock in S. thermophilus (44). Therefore, in addition to CtsR, other regulators may modulate clpL expression in bacteria.
Although the homologs of McsAB are absent from S. mutans, possible phosphorylation was detected in vivo by Phos-tag gel-based Western blot analysis when the CtsR protein was overexpressed in the cell. However, the overall phosphorylation level was low (less than 10%) and the level was unchanged after heat shock stimulation. The significance of this phosphorylation for the in vivo turnover of the CtsR protein needs to be investigated further. On the other hand, mutational analysis of a critical arginine residue at position 62 clearly showed that phosphorylation is very important both in vitro and in vivo. R62 is located within the winged HTH domain, and replacement of this residue with a negatively charged amino acid (R62E) dramatically abolished the DNA binding ability of CtsR. In contrast, replacement of this arginine residue with a neutral (R62A) or positively charged (R62K) residue did not alter the DNA binding ability of the protein. We speculate that the introduction of a negative charge at this specific site in the HTH domain adversely affects DNA binding. Our data are concordant with the results reported for Bacillus stearothermophilus CtsR (17).
In bacteria that contain McsAB, such as B. subtilis, CtsR responds to heat shock stimulation in both an McsB-independent and an McsB-dependent manner. CtsR intrinsically senses the temperature change through its glycine-rich loop (Fig. 3A) and is reversibly released from the target promoter at high temperature (11, 14), thereby activating gene expression. Under heat shock stress, McsB is also activated and consequently phosphorylates CtsR, which prevents the protein from binding to the target promoters (17, 29), and the unbound phosphorylated CtsR protein is subsequently degraded by ClpCP (27, 31). The situation seems to be different in mcsAB-deficient bacteria. A recent report suggests that ClpEP contributes to CtsR stability during heat shock in L. lactis; however, the details of the mechanism are unknown (14). Heat shock-induced phosphorylation of CtsR has not been reported for MscAB-deficient bacteria, and the induction of CtsR-regulated genes is relatively weak after heat shock in these bacteria. For instance, expression from the clpP promoter was increased about 15-fold in B. subtilis (31) after heat shock while the induction was approximately 2-fold in S. mutans (33, 47) and less than 2-fold in S. pneumoniae (32). Some recent studies suggest that CtsR can respond to other stresses such as oxidative stress and ethanol (13, 43) and strongly support the notion that alternate posttranscriptional regulation may exist for CtsR than thermal-stress-mediated phosphorylation.
In summary, we have studied the posttranslational regulation of CtsR in S. mutans that does not contain mcsAB. We found that phosphorylation of S. mutans CtsR significantly alters the repressor activity of the protein. However, phosphorylation and degradation of CtsR apparently are not induced by heat shock. Surprisingly, we found that ClpL mediates normal CtsR turnover in this organism. This is in contrast to MscAB-positive bacteria, which employ ClpCP for CtsR degradation. These results suggest that posttranslational modulation of CtsR is totally different in MscAB-positive and -negative bacteria.
ACKNOWLEDGMENT
This work was supported by NIDCR grant DE021664 to I.B.
Footnotes
Published ahead of print 13 January 2012
REFERENCES
- 1. Ajdíc D, et al. 2002. Genome sequence of Streptococcus mutans UA159, a cariogenic dental pathogen. Proc. Natl. Acad. Sci. U. S. A. 99:14434–14439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Banerjee A, Biswas I. 2008. Markerless multiple-gene-deletion system for Streptococcus mutans. Appl. Environ. Microbiol. 74:2037–2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barbieri CM, Stock AM. 2008. Universally applicable methods for monitoring response regulator aspartate phosphorylation both in vitro and in vivo using Phos-tag-based reagents. Anal. Biochem. 376:73–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Beltramo C, Grandvalet C, Pierre F, Guzzo J. 2004. Evidence for multiple levels of regulation of Oenococcus oeni clpP-clpL locus expression in response to stress. J. Bacteriol. 186:2200–2205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Biswas I, Drake L, Biswas S. 2007. Regulation of gbpC expression in Streptococcus mutans. J. Bacteriol. 189:6521–6531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Biswas I, Drake L, Johnson S, Thielen D. 2007. Unmarked gene modification in Streptococcus mutans by a cotransformation strategy with a thermosensitive plasmid. Biotechniques 42:487–490 [DOI] [PubMed] [Google Scholar]
- 7. Biswas I, Jha JK, Fromm N. 2008. Shuttle expression plasmids for genetic studies in Streptococcus mutans. Microbiology 154:2275–2282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Derré I, Rapoport G, Devine K, Rose M, Msadek T. 1999. ClpE, a novel type of HSP100 ATPase, is part of the CtsR heat shock regulon of Bacillus subtilis. Mol. Microbiol. 32:581–593 [DOI] [PubMed] [Google Scholar]
- 9. Derré I, Rapoport G, Msadek T. 1999. CtsR, a novel regulator of stress and heat shock response, controls clp and molecular chaperone gene expression in gram-positive bacteria. Mol. Microbiol. 31:117–131 [DOI] [PubMed] [Google Scholar]
- 10. Derré I, Rapoport G, Msadek T. 2000. The CtsR regulator of stress response is active as a dimer and specifically degraded in vivo at 37 degrees C. Mol. Microbiol. 38:335–347 [DOI] [PubMed] [Google Scholar]
- 11. Elsholz AK, Gerth U, Hecker M. 2010. Regulation of CtsR activity in low GC, Gram+ bacteria. Adv. Microb. Physiol. 57:119–144 [DOI] [PubMed] [Google Scholar]
- 12. Elsholz AK, et al. 2011. Activity control of the ClpC adaptor McsB in Bacillus subtilis. J. Bacteriol. 193:3887–3893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Elsholz AK, et al. 2011. CtsR inactivation during thiol-specific stress in low GC, Gram+ bacteria. Mol. Microbiol. 79:772–785 [DOI] [PubMed] [Google Scholar]
- 14. Elsholz AK, Michalik S, Zuhlke D, Hecker M, Gerth U. 2010. CtsR, the Gram-positive master regulator of protein quality control, feels the heat. EMBO J. 29:3621–3629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Frees D, et al. 2004. Clp ATPases are required for stress tolerance, intracellular replication and biofilm formation in Staphylococcus aureus. Mol. Microbiol. 54:1445–1462 [DOI] [PubMed] [Google Scholar]
- 16. Frees D, Savijoki K, Varmanen P, Ingmer H. 2007. Clp ATPases and ClpP proteolytic complexes regulate vital biological processes in low GC, Gram-positive bacteria. Mol. Microbiol. 63:1285–1295 [DOI] [PubMed] [Google Scholar]
- 17. Fuhrmann J, et al. 2009. McsB is a protein arginine kinase that phosphorylates and inhibits the heat-shock regulator CtsR. Science 324:1323–1327 [DOI] [PubMed] [Google Scholar]
- 18. Grandvalet C, Coucheney F, Beltramo C, Guzzo J. 2005. CtsR is the master regulator of stress response gene expression in Oenococcus oeni. J. Bacteriol. 187:5614–5623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Haldenwang WG, Losick R. 1979. A modified RNA polymerase transcribes a cloned gene under sporulation control in Bacillus subtilis. Nature 282:256–260 [DOI] [PubMed] [Google Scholar]
- 20. Huang DC, Huang XF, Novel G, Novel M. 1993. Two genes present on a transposon-like structure in Lactococcus lactis are involved in a Clp-family proteolytic activity. Mol. Microbiol. 7:957–965 [DOI] [PubMed] [Google Scholar]
- 21. Hyyryläinen HL, et al. 2001. A novel two-component regulatory system in Bacillus subtilis for the survival of severe secretion stress. Mol. Microbiol. 41:1159–1172 [DOI] [PubMed] [Google Scholar]
- 22. Jennings LD, Lun DS, Medard M, Licht S. 2008. ClpP hydrolyzes a protein substrate processively in the absence of the ClpA ATPase: mechanistic studies of ATP-independent proteolysis. Biochemistry 47:11536–11546 [DOI] [PubMed] [Google Scholar]
- 23. Jung K, Jung H. 2009. A new mechanism of phosphoregulation in signal transduction pathways. Sci. Signal. 2:pe71. [DOI] [PubMed] [Google Scholar]
- 24. Karatzas KA, et al. 2003. The CtsR regulator of Listeria monocytogenes contains a variant glycine repeat region that affects piezotolerance, stress resistance, motility and virulence. Mol. Microbiol. 49:1227–1238 [DOI] [PubMed] [Google Scholar]
- 25. Kim YI, et al. 2001. Molecular determinants of complex formation between Clp/Hsp100 ATPases and the ClpP peptidase. Nat. Struct. Biol. 8:230–233 [DOI] [PubMed] [Google Scholar]
- 26. Kinoshita E, Kinoshita-Kikuta E, Takiyama K, Koike T. 2006. Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol. Cell. Proteomics 5:749–757 [DOI] [PubMed] [Google Scholar]
- 27. Kirstein J, Dougan DA, Gerth U, Hecker M, Turgay K. 2007. The tyrosine kinase McsB is a regulated adaptor protein for ClpCP. EMBO J. 26:2061–2070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kirstein J, Molière N, Dougan DA, Turgay K. 2009. Adapting the machine: adaptor proteins for Hsp100/Clp and AAA+ proteases. Nat. Rev. Microbiol. 7:589–599 [DOI] [PubMed] [Google Scholar]
- 29. Kirstein J, Zuhlke D, Gerth U, Turgay K, Hecker M. 2005. A tyrosine kinase and its activator control the activity of the CtsR heat shock repressor in B. subtilis. EMBO J. 24:3435–3445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Krüger E, Hecker M. 1998. The first gene of the Bacillus subtilis clpC operon, ctsR, encodes a negative regulator of its own operon and other class III heat shock genes. J. Bacteriol. 180:6681–6688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Krüger E, Zuhlke D, Witt E, Ludwig H, Hecker M. 2001. Clp-mediated proteolysis in Gram-positive bacteria is autoregulated by the stability of a repressor. EMBO J. 20:852–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kwon HY, et al. 2003. Effect of heat shock and mutations in ClpL and ClpP on virulence gene expression in Streptococcus pneumoniae. Infect. Immun. 71:3757–3765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lemos JA, Burne RA. 2002. Regulation and physiological significance of ClpC and ClpP in Streptococcus mutans. J. Bacteriol. 184:6357–6366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Len AC, Harty DW, Jacques NA. 2004. Proteome analysis of Streptococcus mutans metabolic phenotype during acid tolerance. Microbiology 150:1353–1366 [DOI] [PubMed] [Google Scholar]
- 35. Liberek K, Lewandowska A, Zietkiewicz S. 2008. Chaperones in control of protein disaggregation. EMBO J. 27:328–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Miethke M, Hecker M, Gerth U. 2006. Involvement of Bacillus subtilis ClpE in CtsR degradation and protein quality control. J. Bacteriol. 188:4610–4619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Molière N, Turgay K. 2009. Chaperone-protease systems in regulation and protein quality control in Bacillus subtilis. Res. Microbiol. 160:637–644 [DOI] [PubMed] [Google Scholar]
- 38. Nair S, Derré I, Msadek T, Gaillot O, Berche P. 2000. CtsR controls class III heat shock gene expression in the human pathogen Listeria monocytogenes. Mol. Microbiol. 35:800–811 [DOI] [PubMed] [Google Scholar]
- 39. Pomerantsev AP, Sitaraman R, Galloway CR, Kivovich V, Leppla SH. 2006. Genome engineering in Bacillus anthracis using Cre recombinase. Infect. Immun. 74:682–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schulz A, Schumann W. 1996. hrcA, the first gene of the Bacillus subtilis dnaK operon encodes a negative regulator of class I heat shock genes. J. Bacteriol. 178:1088–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Suokko A, Poutanen M, Savijoki K, Kalkkinen N, Varmanen P. 2008. ClpL is essential for induction of thermotolerance and is potentially part of the HrcA regulon in Lactobacillus gasseri. Proteomics 8:1029–1041 [DOI] [PubMed] [Google Scholar]
- 42. Tran TD, et al. 2011. Decrease in penicillin susceptibility due to heat shock protein ClpL in Streptococcus pneumoniae. Antimicrob. Agents Chemother. 55:2714–2728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. van Bokhorst-van de Veen H, et al. 2011. Short- and long-term adaptation to ethanol stress and its cross-protective consequences in Lactobacillus plantarum. Appl. Environ. Microbiol. 77:5247–5256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Varcamonti M, et al. 2006. Expression of the heat shock gene clpL of Streptococcus thermophilus is induced by both heat and cold shock. Microb. Cell Fact. 5:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wall T, et al. 2007. The early response to acid shock in Lactobacillus reuteri involves the ClpL chaperone and a putative cell wall-altering esterase. Appl. Environ. Microbiol. 73:3924–3935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yamada S, et al. 2007. Separation of a phosphorylated histidine protein using phosphate affinity polyacrylamide gel electrophoresis. Anal. Biochem. 360:160–162 [DOI] [PubMed] [Google Scholar]
- 47. Zhang J, Banerjee A, Biswas I. 2009. Transcription of clpP is enhanced by a unique tandem repeat sequence in Streptococcus mutans. J. Bacteriol. 191:1056–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zolkiewski M. 2006. A camel passes through the eye of a needle: protein unfolding activity of Clp ATPases. Mol. Microbiol. 61:1094–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]