

Abstract
Cysteine peptidases have been implicated in the development and pathogenesis of Eimeria. We have identified a single-copy cathepsin B-like cysteine peptidase gene in the genome database of Eimeria tenella (EtCatB). Molecular modeling of the predicted protein suggested that it differs significantly from host enzymes and could be a good drug target. EtCatB was expressed and secreted as a soluble, active, glycosylated mature enzyme from Pichia pastoris. Biochemical characterization of the recombinant enzyme confirmed that it is cathepsin B-like. Screening of a focused library against the enzyme identified three inhibitors (a nitrile, a thiosemicarbazone, and an oxazolone) that can be used as leads for novel drug discovery against Eimeria. The oxazolone scaffold is a novel cysteine peptidase inhibitor; it may thus find widespread use.
INTRODUCTION
The protozoan parasite Eimeria, an apicomplexan parasite (as is Plasmodium, the malaria parasite) belonging to the subclass of Coccidia (with other parasites such as Toxoplasma and Cryptosporidium), is responsible for coccidiosis, the most important death-causing agent in the poultry industry and thus of major economic importance. At least 11 species of Eimeria can infect chickens, but the most pathogenic species is Eimeria tenella. Coccidiosis is not limited to the poultry industry, and some Eimeria species can also affect cattle, sheep, and pigs; they thus constitute a potential threat for other intensive livestock industries. The anticoccidial drugs dominating the market in recent years for the treatment of avian Eimeria have been polyether ionophores (17). The advent of drug resistance and the limited efficacy of current drugs mean that identification of new drug targets and finding agents to exploit them are urgently required.
Eimeria is an obligate intracellular parasite. Invasion of host cells requires the regulated release from specialized secretory organelles (namely micronemes, rhoptries, and dense granules) that together form the apical complex characteristic of all apicomplexan parasites (16). Maturation, trafficking, and secretion of many of the secretory organelles' proteins rely upon proteolytic processing (9). After several cycles of asexual multiplication (schizogony or merogony), gametogony begins; macrogametocytes (female gamete) and microgametocytes (male) are formed and fuse to form oocysts that are released. The oocyst wall ensures the parasite survival in the external environment until the next host is found. The oocyst wall formation therefore constitutes an essential process for disease transmission. Peptidases have also been shown to be involved in the oocyst wall formation in Eimeria (5) by maturation of precursor proteins found in the wall-forming bodies. Thus, proteolytic cleavage is considered to be an essential component of at least two stages during this part of the life cycle.
Some cysteine peptidases from apicomplexan parasites have been identified as important factors for invasion of host cells (22). These enzymes include the cathepsin B-like toxopain-1 in Toxoplasma gondii (31) and falcipain-1 and falcipain-2 in Plasmodium falciparum (35). Furthermore, cysteine peptidases of parasites have attracted particular attention over recent years because of their importance in parasite survival, interaction with the host cells, and pathogenicity. Some of them seem to be promising targets for new selective inhibitors and antiparasite agents (2, 14, 40). Cysteine peptidases, however, comprise a very diverse group of enzymes (2). Enzymes designated cathepsin B, which belong to the clan CA, family C1 of peptidases together with cathepsin L, have a signal peptide, a prodomain, and a mature domain. The prodomain, which might possess in some cases information required for the correct folding of the protein, has been shown to be a strong inhibitor of the catalytic domain and therefore helps to ensure that the enzyme's activity is appropriately controlled (28, 42). The prodomain needs to be cleaved and then released for full activity of the enzyme.
We have identified a gene encoding a cathepsin B-like cysteine peptidase from E. tenella (designated EtCatB) which, however, differs significantly from mammalian counterparts; we expressed it in a highly active form and confirmed that it has the expected enzymatic activity. In order to discover inhibitors of the enzyme that might be leads in the search for a novel anticoccidial drug, we screened a focused library against the enzyme and identified three lead compounds, including a novel class of cysteine peptidase inhibitors.
MATERIALS AND METHODS
Reagents and parasites.
All chemicals were from Sigma-Aldrich, unless stated otherwise. The 7-amino-4-methylcoumarin (AMC) and para-nitroanilide (pNA) substrates were from Bachem. Oocysts of the E. tenella H strain were provided by Intervet Innovation GmbH, Schwabenheim, Germany.
Cloning of the cathepsin B gene EtCatB.
From a homology search using the BLASTN program at the Wellcome Trust Sanger Institute server and the cathepsin-B like sequence from T. gondii as a query (31), a contig containing a 1,467-bp open reading frame (ORF) coding for a protein with the cathepsin-B enzyme features was found in the E. tenella partial expressed sequence tag (EST) database and was named Etcb (Eimeria assembly, contig 5413). The complete putative ORF for the enzyme was amplified on genomic DNA of oocysts and cDNA synthesized from the mRNA of oocysts using the forward primer F1 (5′-TCC ACT TAG CTA CTA CGC TTT G-3′) and the reverse primer R1 (5′-GCC CTT GAA TCG CCT TTA GT-3′). The start methionine was confirmed by 5′ rapid amplification of cDNA ends (RACE; Gibco-BRL). Briefly, total mRNA from oocysts was isolated using the TRIzol reagent and was transcribed into single-stranded cDNA using the specific primer R2 (5′ AGC AGG TAC GGC AGC AAC TC 3′). The 5′ end of Etcb was then amplified from dC-tailed cDNA using the abridged anchor primer and the specific nested primer R3 (5′ TGC CGA CGG AAG TGA TCC CGC 3′), followed by a secondary amplification using the abridged universal anchor primer and another specific nested primer, R4 (5′ ACA ACC TCA TGG CCT CCT GG 3′).
Homology protein modeling.
The predicted sequence of the mature EtCatB was used to search the Brookhaven Protein Data Bank (PDB). To build the homology model, the crystal structure of bovine cathepsin B in complex with the irreversible epoxide inhibitor CA074 was used as a template (PDB entry identifier [ID] 1QDQ) (45). Homology models were calculated using the program Modeler implemented in the Insight II software package (Accelrys, Inc., San Diego, CA). All calculations were carried out under default conditions. For the alignment of the EtCatB sequence to the template, the BLOSUM 62 matrix implemented in Modeler's ALIGN123 module was taken. Four homology models were generated using the default conditions with the highest optimization level, and subsequently four additional structures were generated with a high loop refinement for each of the first four homology models. In summary, 16 homology models were built to ensure the generation of the highest possible structure quality. The corresponding model with the lowest value of probability density function (PDF) violations in combination with the lowest energy value was selected for validation. The quality of the models was validated using the ProStat and Profile-3D method implemented in the Insight II software package. Disulfide bridges were checked and assigned manually using the SYBYL, version 6.8, software package (Tripos, Inc., St. Louis, MO).
Covalent docking.
Version 5.0.1 of the GOLD docking suite (The Cambridge Crystallographic Data Centre) was used for covalent docking studies (27, 41). The resulting docking solutions were ranked based on the selected scoring function. In covalent mode, the program assumed that there is just one atom linking the ligand to the protein. Both protein and ligand files were set up with the link atom included. During docking runs, the link atom in the ligand is forced to fit onto the link atom in the protein. In order to ensure that the geometry of the bound ligand was correct, an angle-bending energy term for the link atom was included in the calculation of the fitness score (41). The above-described covalent docking mode of GOLD was applied for all docking runs using standard default settings. The scoring function GoldScore was used in its modified version for covalent docking. Protein and ligand structures were prepared according to the GOLD user manual. The sulfur atom of the C31 residue in the EtCatB homology model and the sulfur atom of the C29 residue in the Bos taurus CatB (BtCatB) X-ray structure were defined as the link atoms for the covalent bond. The three-dimensional (3D) illustration of the covalent docking result depicted in this paper was generated using MOLCAD (6).
Expression of recombinant promature EtCatB in E. coli.
For expression of the promature enzyme with an N-terminal His tag in Escherichia coli, the sequence coding for pro-Etcb was amplified from cDNA from oocysts using the forward primer F2 (5′-GTT GTC ATA TGC CCT CCG ATG ATT TGG G-3′) and the reverse primer R5 (5′-GCC TCT CGA GTT ATA GGT CCT GCG CTG ACG GCA G-3′) and cloned into the pET28a(+) vector (Invitrogen) using the NdeI and XhoI restriction sites. This construct was transformed in E. coli strain BL21(DE3) competent cells for expression. Cells were grown to an optical density at 600 nm (OD600) of 0.4 to 0.6 before being induced for 4 h with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at 37°C.
Purification of the recombinant EtCatB produced in E. coli.
Pelleted cells from 100 ml of culture were resuspended in 2.5 ml of 50 mM Tris-HCl, pH 8.0, and sonicated on ice four times with six cycles (each) of 30 s on/30 s off. The supernatant of the cell lysate was applied to a 2.5-ml Ni-nitrilotriacetic acid (NTA) column (Qiagen) previously equilibrated in 50 mM Tris-HCl, pH 8.0. The column was washed with 5 column volumes of 50 mM Tris-HCl, pH 8.0, before being washed with 3 column volumes of 50 mM Tris-HCl (pH 8.0)–30 mM imidazole. The protein was eluted in three 1.5-ml volumes of 50 mM Tris-HCl (pH 8.0)–500 mM imidazole. Fractions containing the recombinant enzyme were dialyzed for 2 h at 4°C against 100 volumes of 50 mM Tris-HCl, pH 8.0. Expression in E. coli resulted in an abundant protein at 56 kDa corresponding to the expected size for the promature enzyme. No recombinant protein of the size of the putative mature enzyme (around 30 kDa) could be detected (results not shown). The vast majority of the recombinant protein was found to be in the soluble fraction of cell lysates, and about 20 mg of pure promature EtCatB could be obtained from 1 liter of E. coli culture. The pure soluble enzyme did not have any activity toward the substrates Z-F-R-AMC (benzyloxycarbonyl-Phe-Arg-AMC) and Z-R-R-AMC, despite extensive attempts to activate it using methods reported to be successful with other related enzymes. The protein was used for antibody production.
Expression of recombinant promature EtCatB in Pichia pastoris.
Signal peptide and a proregion prediction were performed using the program SignalP from the Center for Biological Sequence Analysis (http://www.cbs.dtu.dk/). A construct was designed for inducible expression of the promature enzyme, with subsequent release into the culture medium. The sequence was amplified on cDNA from oocysts using the forward primer F3 (5′-GCG GCT ACG TAA TGC CCT CCG ATG ATT TGG G-3′) containing the SnaBI restriction site and the reverse primer R6 (5′-GGT GCC TAG GTT ATA GGT CCT GCG CTG ACG G-3′) containing the AvrII restriction site and cloned in the pPIC9 vector (Invitrogen). The final construct pPIC9-Etcb and control empty vector were linearized with SacI prior to transfection in the host Pichia pastoris KM71 cells, according to the manufacturer's instructions (Invitrogen). Cells were spread on plates selective for histidine prototrophy (His+) and incubated for 3 to 4 days at 30°C until colonies developed, as described in the Pichia expression kit manual (21a). Insertions of the fragments were verified by PCR on genomic DNA extracted from positive clones as described in the Pichia expression kit manual, using the PCR primer pair α-factor and 3′ AOX1 or the pair 5′ AOX1 and 3′ AOX1. Positive colonies for each of the transfections were selected and tested for expression of cathepsin B. Cells were induced with 0.5% methanol (vol/vol; final concentration) every 24 h. Aliquots were taken, cells were separated from the supernatant of culture by centrifugation, and both were stored at −20°C before analysis. Samples of supernatant were precipitated with 10% (vol/vol) trichloroacetic acid (TCA) and analyzed on Coomassie-stained SDS-PAGE gels.
Purification of the recombinant EtCatB produced in P. pastoris.
Different aliquots of medium containing the recombinant EtCatB were incubated with various percentages of a saturating solution of ammonium sulfate for 2 h at 4°C with mild agitation to fractionate the proteins present in the culture supernatant according to their solubilities at various concentrations of ammonium sulfate. The samples were then centrifuged for 15 min at 13,000 × g, and the pellet was resuspended in the same volume of 50 mM sodium acetate, pH 5. The samples of resuspended pellets and supernatant were tested for activity and analyzed on Coomassie-stained SDS-PAGE gels.
EtCatB deglycosylation.
Supernatant of P. pastoris culture containing the recombinant EtCatB was treated with 40% followed by 70% saturation of ammonium sulfate, and aliquots (equivalent to about 20 μg of enzyme) were deglycosylated using 100 U of peptide N-glycosidase F (PNGase F; New England BioLabs) for 2.5 h at 37°C.
Peptidase assay and enzyme characterization.
The assays using the chromogenic substrates Z-F-R-pNA and Z-R-R-pNA consisted of 50 mM sodium acetate (pH 5), 2 mM EDTA, and 10 mM dithiothreitol (DTT), with 333 μM chromogenic substrate in a total volume of 600 μl. The buffer was incubated at 37°C with the enzyme sample for 10 min, and the reaction was started by addition of the substrate. The reaction was followed continuously for 10 min at 410 nm. Peptidase activity was also assessed using the fluorogenic substrates Z-F-R-AMC and Z-R-R-AMC at a concentration of 200 μM, unless otherwise stated, in 50 mM sodium acetate (pH 5), 5 mM EDTA, and 10 mM DTT. Reaction mixtures were in a total volume of 1 ml at 37°C and started by addition of the substrate after a preincubation of the buffer-enzyme mix for 10 min at 37°C. The appearance of 7-amino-4-methylcoumarin (AMC) was measured by the excitation wavelength at 350 nm and emission at 465 nm. Activities were calculated from the rate over the first 10 min. One unit of activity represents 1 μmol of substrate hydrolyzed per min and per mg of enzyme. All measurements were in triplicate. The pH dependence was determined using two different methods. The first comprised assaying the activity in a mix of four buffers (25 mM acetic acid, 25 mM morpholineethanesulfonic acid [MES], 75 mM Tris, and 25 mM glycine), adjusted to the appropriate pH, with 10 mM DTT. The reaction was started by addition of 30 μM Z-F-R-AMC. The second method involved using three different buffers separately, depending on their pH ranges. These buffers were 0.1 M acetic acid, 0.1 M sodium acetate, and 0.1 M potassium phosphate. All buffers were supplemented with 10 mM DTT. The reaction was started by addition of 30 μM Z-F-R-AMC. For testing of inhibitor effects, the inhibitor was added to the buffer-enzyme mix and incubated for 10 min at 37°C. The reaction proceeded as described for the peptidase assay.
Screening library.
The screening library used against EtCatB is a collection of 195 confirmed hits selected from in-house Leishmania mexicana cathepsin L (named CPB) and from human cathepsin K high-throughput screening (HTS) projects (resulting in 82 and 113 actives, respectively). The CPB HTS library was a collection of 74,339 small molecules acquired from commercial vendors (Asinex, Ltd., Moscow, Russia; Akos Consulting & Solutions GmbH, Basel, Switzerland; ChemBridge Corporation, San Diego, CA; Chemical Diversity Labs, Inc., San Diego, CA; Enamine, Ltd., Kiev, Ukraine; InterBioScreen, Moscow, Russia; LifeChemicals, Inc., Burlington, ON, Canada; Maybridge, Cambridge, United Kingdom; Otava, Kiev, Ukraine; Specs, Delft, Netherlands; TimTec Corp., Newark, NJ; and Vitas-M Laboratory, Ltd., Moscow, Russia). The 74,339 compounds were selected from an ∼2 × 106 vendor ISIS database (Symyx Technologies, Inc., Sunnyvale, CA) using the diverse solutions module implemented within the software tool SYBYL, version 6.8 (Tripos, Inc., St. Louis). Molecules that contained atoms other than C, O, H, N, S, P, F Cl, Br, or I and which had a molecular mass of >450 Da or which possessed more than eight rotatable bonds were removed from the data set. Filtering tasks and warhead-based selection of the confirmed hits resulting from the CPB and from the cathepsin K HTS project were done using the CACTVS software package (20). The purity of all screening compounds used was >90%.
Chemicals.
The cysteine peptidase inhibitor K11777 (N-methyl-piperazine-Phe-homoPhe-vinylsulfone-phenyl) was kindly provided by James H. McKerrow (San Francisco, CA). CP673854 was synthesized according to the method described by Rancovic et al. (32); the IUPAC name is 4-[(3-cyanophenyl)amino]-6-(1-piperidinyl)-1,3,5-triazine-2-carbonitrile (purity of >95%). CP725567 was ordered from ChemDiv, Inc. (San Diego, CA); the IUPAC name is 4-[(4-methoxyphenyl)methylene]-2-(4-propylcyclohexyl)-5(4H)-oxazolone (purity of >95%). CP989759 was ordered from Vitas-M Laboratory, Ltd. (Moscow, Russia); the IUPAC name is (2E)-2-[(3-chloro-5-methyl-1-phenyl-1H-pyrazol-4-yl)methylidene]hydrazinecarbothioamide (purity of >95%).
Determination of IC50 values.
The 50% inhibitory concentration (IC50) values of inhibitors of recombinant EtCatB and cathepsin B from bovine spleen (EC 3.4.22.1; Sigma-Aldrich Inc., St. Louis, MO) were measured in a homogenous fluorescence endpoint assay using Z-F-R-AMC as a substrate (38). The assay was carried out in black 384-well plates and read on a SPECTRAFluor Plus (Tecan Inc., Durham, NC) plate reader. Assays for EtCatB were performed at 25°C in a 40 mM sodium acetate buffer at pH 5.5 containing 10 mM DTT (Carl Roth GmbH, Karlsruhe, Germany), 2 mM EDTA (Merck, Darmstadt, Germany), 0.01% Pluronic (Merck, Darmstadt, Germany), 0.01% bovine serum albumin (BSA; PAA Laboratories GmbH, Pasching, Austria), 3.6% dimethyl sulfoxide (DMSO), and 400 μM Z-F-R-AMC. Prior to the addition of substrate, different concentrations of the inhibitor ranging from 30 μM to 0.3 nM were preincubated for 10 min with the enzyme to allow the establishment of the enzyme-inhibitor complex. The reaction was started by addition of the substrate and stopped after a 46-min reaction time at 25°C by addition of the cysteine peptidase inhibitor E64 [trans-epoxysuccinyl-l-leucylamido-(4-guanidino)butane] at 16.7 μM. The plates were centrifuged, and the enzyme activity was measured from the increase of fluorescence using excitation at 360 nm with emission at 465 nm. Assays for cathepsin B from bovine spleen were performed at 25°C in a 100 mM sodium acetate buffer, pH 5.5, containing 10 mM DTT (Carl Roth GmbH, Karlsruhe, Germany), 1 mM Titriplex II (Merck, Darmstadt, Germany), 0.01% Triton (Merck, Darmstadt, Germany), 0.01% BSA (PAA Laboratories GmbH, Pasching, Austria), 3.6% DMSO, and 100 μM Z-F-R-AMC. Prior to the addition of substrate, different concentrations of the inhibitor ranging from 30 μM to 0.3 nM were preincubated for 10 min with the enzyme to allow the establishment of the enzyme-inhibitor complex. The reaction was started by addition of substrate and stopped after a 35-min reaction time at 25°C by addition of stop buffer containing 41.7 mM sodium acetate and 41.7 mM sodium chloroacetate. The plates were centrifuged, and the enzyme activity was measured from the increase in fluorescence using excitation at 360 nm with emission at 465 nm. Enzyme activities were expressed as percentages of residual activity compared with an uninhibited control and were plotted versus increasing inhibitor concentrations. IC50 values were calculated using the four-parameter equation model 205 and the option “unlock” from the XLfit add-in (IDBS, Guildford, United Kingdom) in Excel (Microsoft Corporation, Redmond, WA). All values are mean values from at least three independent assays to ensure statistically significant results.
Kinetic analyses.
To verify whether the selected compounds are reversible or irreversible inhibitors, dilution experiments were performed. Enzyme and compound were incubated for 10 min at a 333-fold higher concentration than used for the standard IC50 determination. The mixtures were subsequently diluted 333-fold in substrate-containing assay buffer, and the residual EtCatB or bovine cathepsin B activity was immediately measured from the increase in fluorescence using excitation at 360 nm with emission at 465 nm. The irreversible cysteine peptidase inhibitor E64 was used at 1 μM as a control. The IC50 values were determined for at least three inhibitor concentrations as described above. Reversible inhibitors showed the same IC50s in this experiment as the standard IC50 determination, while the IC50s of irreversible inhibitors decreased.
Determination of Ki values.
Ki values for reversible inhibition of selected compounds toward EtCatB were measured in black 384-well plates and read on a SPECTRAFluor Plus (Tecan Inc., Durham, NC) plate reader. Positive controls contained enzyme, DMSO instead of compound, and substrate. The irreversible cysteine peptidase inhibitor E64 was used as a standard. In a first step, 5 μl of test compound or 5 μl of 10% DMSO in acetate buffer was added to each well. Subsequently, 25 μl of a premix was added to each well containing enzyme in 40 mM sodium acetate buffer (pH 5.5), 10 mM DTT, 2 mM EDTA, 0.01% Pluronic, 0.01% BSA, and 3.6% DMSO (final concentrations). The wells for the blanks contained the premix without enzyme. After preincubation for 10 min at 25°C, the reaction was started by addition of 20 μl of Z-F-R-AMC. The enzyme activity was measured from the increase in fluorescence using excitation at 360 nm with emission at 465 nm. Compound concentrations were 20, 10, 2, 1, 0.2, 0.1, 0.02, and 0.01 μM; Z-F-R-AMC concentrations were 1,200, 1,000, 800, 600, 400, 300, 200, and 100 μM (final concentrations). The assay procedure to determine Ki values for cathepsin B from bovine spleen was the same as described for the EtCatB determination except for the composition of the premix, which contained 100 mM sodium acetate buffer (pH 5.5), 10 mM DTT, 1 mM Titriplex II, 0.01% Triton, and 0.1 mg/ml BSA. In addition, the Z-F-R-AMC concentrations (500, 300, 200, and 100 μM) and compound concentrations (2, 1, 0.2, 0.1, 0.02, and 0.01 μM) used differed. Initial velocities were determined using linear regression in Excel (Microsoft Corporation, Redmond, WA). Ki values were then calculated using the enzyme kinetics module within SigmaPlot, version 8.0 (SYSTAT Software Inc., Chicago, IL). All values presented are graphically determined values from at least four independent assays to ensure statistically significant results.
Schizont maturation assay (SMA) to assess the anticoccidial activity of compounds.
Madin-Darby bovine kidney (MDBK) cells (2.5 × 104 cells/well) were seeded in 96-well microtiter plates (Costar Corning Corp., New York, NY) and incubated in Dulbecco's modified Eagle's medium (DMEM; PAN Biotech GmbH, Aidenbach, Germany) containing 10% (vol/vol) fetal calf serum for ∼24 h at 37°C with 5% (vol/vol) CO2. E. tenella sporocysts were excysted as described previously (43), and MDBK cells were infected with 7 × 104 sporozoites per well and incubated at 41°C with 10% (vol/vol) CO2 and 95% relative humidity. After incubation for ∼3 h, noninvaded sporozoites were removed by washing with Earle's medium 199 (Biochrom AG, Berlin, Germany), followed by the addition of 199 medium containing the respective cysteine peptidase inhibitor to reach final inhibitor concentrations per well of 50, 10, 2, 0.4, and 0.08 μM. Cytotoxicity was determined by exposure of the compounds to noninfected cells. Samples with no inhibitor served as controls. After 60 h of incubation at 41°C, completion of schizont development was visually confirmed by light microscopy. Cells were washed several times and fixed with 4% (vol/vol) formaldehyde (50 ml per well) for ∼45 min. Plates were washed three times with phosphate-buffered saline (PBS) containing 0.5% Tween 20, followed by a 45-min incubation with PBS containing 0.5% Tween 20 and 5% (wt/vol) milk powder (washing buffer). After several washing steps, an antimerozoite serum, which was produced in rabbits using standard procedures, was added and incubated for 1 h, followed by several washing steps and a 1 h of incubation with a fluorescein isothiocyanate (FITC)-conjugated AffiniPure Fab fragment goat anti-rabbit IgG(H+L) (Dianova GmbH, Hamburg, Germany). Cells were washed, and schizont maturation was determined by an enzyme-linked immunosorbent assay (ELISA) reader (Tecan Group Ltd., Männedorf, Switzerland) using an excitation wavelength of 485 nm and an emission wavelength of 535 nm. The 50% effective concentrations (EC50s) were calculated using the four-parameter equation 205 and the option unlock from the XLfit add-in (IDBS, Guildford, United Kingdom) in Excel (Microsoft, Redmond, WA). All values are mean values from at least two independent assays, each carried out in duplicate.
Compound solubility measurements.
A 2-fold serial dilution of the compounds was performed (0.5 to 500 μM) in DMSO (Acros Organics, Fischer Scientific, Morris Plains, NJ) and added in duplicate to 96-well microtiter plates. Phosphate-buffered saline (PBS; pH 7.4) was added to give a total volume of 200 μl and a final DMSO concentration of 5% (vol/vol) in all wells. The plates were incubated at room temperature for 22 min, and the relative solubilities of the compounds were determined by measuring forward-scattered light using a NEPHELOstar laser-based microplate nephelometer (BMG Labtech GmbH, Offenburg, Germany). Wells containing only buffer and 5% (vol/vol) DMSO were used as controls. Data analysis was carried out using Excel (Microsoft Corporation, Redmond, WA).
NMR and LC-MS analyses.
Compound purity and molecular mass were confirmed by liquid chromatography-mass spectrometry (LC-MS) experiments which were performed on an Agilent LC7 MSD (mass-selective detector) 1100 LC-MS (Agilent Technologies, Santa Clara, CA). The liquid chromatography conditions were as follows: a Zorbax SB (stable bound) C18 column, 1.8-μm particle size, column dimensions of 4.6 by 30 mm, 0.1 μl/min flow rate, gradient of 10 to 100% eluent B over 3 min (eluent A was 95:5 H2O to CH3CN supplemented with 0.1% formic acid, and eluent B was CH3CN), and a column temperature of 313 K. MS detection was performed using an MSD 1100 electrospray ionization (ESI) and atmospheric pressure ionization (API) (Agilent Technologies, Santa Clara, CA) single quadrupole mass spectrometer. Nuclear magnetic resonance (NMR) spectroscopy was carried out on a Bruker Avance DRX 400 MHz NMR spectrometer (Bruker AXS, Madison, WI).
Western blotting and SDS-PAGE analyses.
Full-length, N-terminal His-tagged, nickel-agarose-purified, recombinant EtCatB produced in E. coli was used to raise antiserum in a rabbit (Biological Services, University of Glasgow), using standard protocols. Parasites were lysed, and approximately 5 × 107 cells were loaded per lane. Western blot analysis was normally performed with polyclonal immune rabbit serum diluted 1:8,000 in Tris-buffered saline containing 1% (wt/vol) low-fat dried milk and 0.1% Tween 20. Bound antibody was detected using horseradish peroxidase-coupled secondary antibodies diluted 1:5,000 (Santa Cruz Biotechnology) and ECL Western blotting detection reagents (Amersham Biosciences).
RESULTS
Cloning of the EtCatB gene.
Amplification by PCR of the complete putative sequence coding for the cathepsin-B like protein (MEROPS clan CA, family C1) on both genomic DNA and cDNA gave a single product of the same size, about 1.6 kb, which corresponds to the predicted size for the amplification product using the specific primers designed (data not shown). The amplified fragments were sequenced, and both fragments presented the same nucleotide sequences, confirming the absence of introns in the gene. The results of 5′ RACE showed the presence of an mRNA comprising a 40-bp 5′ untranslated region (5′ UTR), confirming the start methionine. Furthermore, the start methionine conforms to a Kozak eukaryotic consensus and the consensus assigned to protozoan parasites (23) favoring the presence of a purine at position −3 and +4 of the start ATG. The complete ORF of 1,535 bp (http://www.genedb.org/gene/ETH_00003570.1?actionName=%2FQuickSearchQuery) encodes a preproenzyme of 512 amino acids (Mr of 55,937). The amino acid sequence was analyzed with the SignalP algorithm (26), and a signal peptide (prepeptide) of 21 hydrophobic amino acids could be predicted. By comparison with cathepsin B enzymes from other organisms (Fig. 1), a prodomain of 214 amino acids could be predicted. As a comparison, the cathepsin B-like enzyme of T. gondii, known as toxipain-1 (31), has a prepeptide of 34 amino acids and a prodomain of 239 amino acids, and the mature enzyme is composed of 296 residues. The long prodomains of the two proteins are very similar; both enzymes lack the ERFNIN motif that is usually present in the prodomain of cathepsin L-like and cathepsin H-like enzymes but is absent from those of cathepsin Bs. The C-terminal end of the E. tenella enzyme contains a potential site for asparagine-linked glycosylation (N-X-S/T) at position 502. The complete preproenzyme of E. tenella has 42% identity with toxipain-1 of T. gondii, and the promature enzyme of 491 amino acids showed good similarity to the human cathepsin B (45%) and the cathepsin B enzymes of Leishmania major (39%), T. gondii (57%), and Schistosoma mansoni (44%) (Fig. 1). The positions of the catalytic cysteine, histidine, and asparagine in EtCatB are, respectively, 263, 445, and 465 (EtCatB numbering). The proenzyme contains 16 cysteine residues, which suggested the possibility of eight disulfide bridges. The positions for 12 of the cysteine residues in the homologous enzymes of the different organisms compared were conserved. The occluding loop characteristic of most cathepsin Bs and the two histidine residues implicated in exopeptidase activity are present (H352 and H353). By comparison with the human cathepsin B, a C-terminal extension of 8 amino acids is present. Such an extension is detected only in toxipain-1 (28 amino acids) and is absent from cathepsin Bs known from other protozoa including Trypanosoma (2).
Fig 1.


Protein sequence alignment of the prepro-EtCatB with cathepsin B enzymes from different organisms. (A) Preproregions. (B) Mature regions. EtCatB, E. tenella cathepsin B; TgCatB, T. gondii cathepsin B (31); SmCatB, S. mansoni cathepsin B (8); LmCatB, L. major cathepsin B (36); HsCatB, Homo sapiens liver cathepsin B (11); BtCatB, B. taurus cathepsin B (4). GenBank accession numbers are as follows: TgCatB, AY071839; SmCatB, AJ312106; LmCatB, U43705; HsCatB, M14221; BtCatB, L06075. Information on EtCatB is available on the GeneDB website (http://www.genedb.org/gene/ETH_00003570.1?actionName=%2FQuickSearchQuery). Highly conserved and conserved residues are highlighted by blue and orange backgrounds, respectively. (A) The predicted signal peptide (prepeptide) of EtCatB is marked by a rose bar, and the prodomain is marked by a yellow bar. (B) Start of the mature EtCatB is indicated by a vertical arrow. Residues of the S2 pocket are indicated by a green background. The K496E (K261E, mature enzyme numbering) exchange between EtCatB and BtCatB is marked with blue circles. The catalytic triad (C, H, and N) is indicated by magenta asterisks. The potential N-glycosylation site of EtCatB is marked with a gray circle. Cysteines of disulfide bridges are highlighted by yellow backgrounds. The six disulfide bridges of EtCatB are marked by black lines with different heads. The additional disulfide bridge of BtCatB is marked by a cyan line. Secondary structure elements are indicated schematically beneath the alignment. The seven β-sheets are indicated by green block arrows, and the four α-helices are indicated by red banners.
Homology modeling of the predicted mature EtCatB.
In order to gain insight into the protein's 3D structure, we generated a structural model of mature EtCatB using comparative modeling (10). Due to its excellent structure resolution and low B-factors (3), we used bovine cathepsin B (PDB ID 1QDQ) cocrystallized with the irreversible epoxide inhibitor CA074 as a template (44). The full-length sequence similarity of 47% and the sequence similarity of up to 80% in structurally conserved regions between EtCatB and bovine cathepsin B (BtCatB) are reasonable for the generation of a qualified homology model. Superimposition of the final EtCatB model and the crystal structure of BtCatB shows high similarity between the two proteins at the structural level (Fig. 2A). Similar to BtCatB, the homology model of EtCatB shows the structure of an α+β-type protein with four α-helices and seven β-strands. It also contains the occluding loop. This loop is a structural feature of cathepsin Bs that has been proposed to be important for their peptidyl-dipeptidase activity (21, 25). The model suggests that mature EtCatB has six disulfide bridges at residues C250-C280, C263-C310, C299-C371, C300-C305, C342-C375, and C350-C361 (EtCatB numbering of the whole preproenzyme) (Fig. 1). This is similar to the situation found for cathepsin Bs from chicken (13), mouse (11), human (34), and rat (39). The mature BtCatB is predicted to have an additional disulfide bridge at C227-C331 (BtCatB numbering of the whole preproenzyme).
Fig 2.

Modeling the structure of the mature EtCatB. (A) Homology model of EtCatB (gray) in ribbon-type rendering. For comparison, the BtCatB template structure (PDB ID 1QDQ; blue) was superimposed on the model structure. The superimposition exhibits almost identical folding patterns between the model and template structures. Disulfide bridges are depicted in capped-stick representation and colored by atom type (mature enzyme numbering). (B) The main residues of the active-site cleft of the EtCatB homology model. Numbers refer to the EtCatB mature sequence. Residues are shown as follows: S2 subsite in blue, S1 subsite in red, residues of the catalytic triad in yellow, S1′ subsite in magenta, and S2′ subsite in cyan. E245 (gray) from the BtCatB template structure (PDB ID 1QDQ) was superimposed on K261 (blue) of the model structure. The oppositely charged residues K261 and E245 might be addressed in lead optimization approaches to increase the inhibitor selectivity. (C) The top-ranked covalent-docking solution of CP989759 in the active site of EtCatB. The surface of the active site is colored by lipophilic potential. The color ramp ranges from red (highest lipophilic area of the surface) to blue (highest hydrophilic area of the surface). Relevant amino acids of the EtCatB active site and the inhibitor CP989759 are depicted in capped-stick representation and colored by atom type. The yellow arrow marks the covalent bond between C31 and CP989759, forming a tetrahedral transition state.
Comparison of the active-site pockets of EtCatB and BtCatB indicates high residue similarity for the S1′ and S1 pockets but also two significant residue differences between EtCatB and BtCatB (R81S and K261E; numbering is according to the mature enzymes) in the S2 pocket (Fig. 2B). The K261E variation could be of high importance for the selective inhibition of EtCatB as it involves a charge exchange, and the glutamate in the S2 pocket of BtCatB has previously been demonstrated to provide the enzyme with the ability to recognize substrates with positively charged P2 residues, e.g., R (19). This striking difference between the parasite and the host enzyme provides optimism for lead compound optimization approaches for the development of selective inhibitors that target the parasite protein.
Expression and purification of mature EtCatB in P. pastoris.
The sequence encoding the promature EtCatB was cloned in the pPIC9 vector and transfected in P. pastoris KM71 cells for inducible expression of the recombinant enzyme in the supernatant of culture. Time course analysis of expression showed that the recombinant protein was already present 24 h after the start of induction and that the amount increased slightly until 96 h after induction and then remained stable up to 192 h (data not shown). The cells were harvested 192 h after the start of induction. Analysis of expression showed a major protein at about 30 kDa in the supernatant of cells transfected with the pPIC9-Etcb plasmid but not in the supernatant of the cells transfected with the empty pPIC9 vector. This corresponds to the size expected for the mature EtCatB enzyme. The expression was estimated to be about 100 mg of protein per liter of culture. Ammonium precipitation resulted in the purification of two proteins of 30 and 32 kDa (Fig. 3, lane 1), with both proteins being specifically recognized by anti-pro-EtCatB antiserum (Fig. 3, lane 3). After treatment with PNGase F, a single protein at 30 kDa could be detected, corresponding to the mature deglycosylated EtCatB (Fig. 3, lanes 2 and 4).
Fig 3.
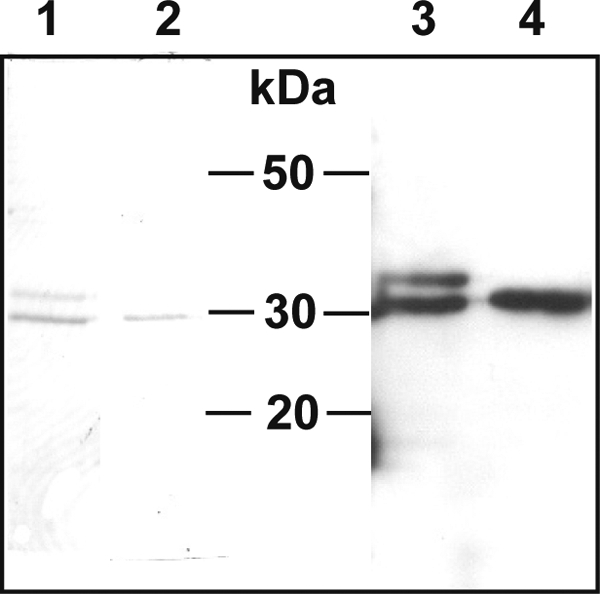
Deglycosylation of the EtCatB of E. tenella produced in P. pastoris. The supernatant from culture medium of expressing Pichia cells was treated with ammonium sulfate to 40% saturation. The supernatant resulting from this first treatment was then incubated in the presence of 70% ammonium sulfate. The precipitated fraction after this step was resuspended and analyzed using Coomassie-stained SDS–12% PAGE (lane 1) and by Western blotting (lane 3, 1:8,000 dilution of EtCatB antibody and 1:5,000 dilution of anti-rabbit antibody). The purified enzyme was treated with PNGase F and analyzed using SDS-PAGE (lane 2) and by Western blotting (lane 4) as described for the nondeglycosylated sample.
Activity of mature EtCatB produced using P. pastoris.
An activity of about 600 mU/ml (6 U/mg protein) toward the substrate Z-F-R-pNA (333 μM) could be detected in the sample of purified enzyme (concentration of 100 μg/ml). The enzyme activity was strictly dependent on the presence of DTT, with 10 mM being the minimum required for maximum activity. EDTA had a beneficial effect on the activity, with 5 mM being the minimum required for observation of maximum activity. The enzyme's activity was negligible at a pH below 4 or above 9, but there were two peaks of activity, at pH 5 and pH 8, in the mix of buffers (Fig. 4). The activity also presented two peaks using individual buffers, one between pH 4 and 5.5 and one at pH 7.5 (Fig. 4).
Fig 4.

Variation of the activity of EtCatB with pH. For the experiment shown in the left panel, pH dependence was determined using a mix of four buffers (25 mM acetic acid, 25 mM MES, 75 mM Tris, and 25 mM glycine) adjusted to the appropriate pH, with 10 mM DTT. The reaction was started by addition of 30 μM Z-F-R-AMC. At right, three different buffers were used separately, depending on their pH ranges. These buffers were 0.1 M acetic acid, 0.1 M sodium acetate, and 0.1 M potassium phosphate. All buffers were supplemented with 10 mM DTT. The reaction was started by addition of 30 μM Z-F-R-AMC. Reactions were measured continuously for 5 min at 37°C (excitation, 380 nm; emission, 465 nm).
The purified enzyme was active toward both the Z-F-R-AMC and Z-R-R-AMC substrates (Fig. 5A). The strong preference for a phenylalanine residue at the P2 site rather than an arginine is a feature of the cathepsin B class of enzymes (1) although characteristically they are active toward both. The preference was reflected by a large difference in Km values, with 168 ± 33 μM for Z-F-R-AMC and 1,400 ± 250 μM for Z-R-R-AMC (Fig. 5B). The very high Km for Z-R-R-AMC is consistent with the arginine residue at P2 being accommodated very poorly in the active site of EtCatB, as predicted by the model. The deglycosylated enzyme had very comparable characteristics to the enzyme preparation containing both the glycosylated and nonglycosylated proteins with respect to both activity and Km (Fig. 5).
Fig 5.

Analysis of the activity of the nondeglycosylated and deglycosylated EtCatB produced in P. pastoris toward the Z-F-R-AMC and Z-R-R-AMC substrates. (A) Representation of the activity (U/mg protein) of the different enzyme fractions against the two substrates Z-F-R-AMC and Z-R-R-AMC. Enzyme samples were tested for activity in a final volume of 1 ml. The enzyme sample was incubated for 10 min at 37°C in the assay buffer before the reaction was started by addition of the substrate (200 μM; for the Km determinations, concentrations of 10 μM to 1.5 mM were used). Excitation and emission wavelengths were, respectively, 380 and 465 nm. The assays were continuous for 10 min at 37°C. (B) Representation of the Km values (μM) for the different enzyme fractions against the two substrates Z-F-R-AMC and Z-R-R-AMC.
Various peptidase inhibitors were tested on both the nondeglycosylated and deglycosylated enzymes (Table 1). As expected, those compounds known to inhibit clan CA, family C1 cysteine peptidases (E64, K11777, Z-F-R-diazomethylketone [DMK], leupeptin, and TLCK [Nα-p-tosyl-l-lysine chloromethyl ketone]) strongly inhibited the enzymes. The sulfhydryl-blocking agent, iodoacetamide, showed strong inhibition too, while N-ethylmaleimide (NEM) did not. The metallopeptidase inhibitor 1,10-phenanthroline, the serine peptidases inhibitors phenylmethylsulfonyl fluoride (PMSF) and benzamidine, and the aspartyl peptidase inhibitor pepstatin A did not show any effect unless used at very high concentrations. The inhibition at high concentration might be due to interfering interactions with other amino acids involved in the catalytic reaction. The aminopeptidase inhibitor bestatin showed strong inhibition. This is in contrast with its lack of effect on papain, the classical clan CA, family C1 cysteine peptidase, and deserves further investigation.
Table 1.
Inhibition of the purified recombinant CatB of E. tenellaa
| Enzyme and inhibitor | Inhibitor concn (μM) | Activity (%) |
|---|---|---|
| Purified enzyme | ||
| E64 | 100 | 0 |
| 10 | 2 ± 0.2 | |
| 1 | 7 ± 2.4 | |
| K11777 | 10 | 0 |
| 1 | 1.3 ± 0.3 | |
| ZFRDA | 100 | 0 |
| 10 | 1.8 ± 0.3 | |
| Leupeptin | 100 | 0.4 ± 0.1 |
| 10 | 3.3 ± 0.2 | |
| TLCK | 100 | 1.3 ± 0.2 |
| 10 | 4.3 ± 0.6 | |
| Iodoacetamide | 100 | 0.7 ± 0.1 |
| 10 | 14 ± 2.5 | |
| Bestatin | 100 | 3 ± 0.2 |
| 10 | 59 ± 5 | |
| 1,10-Phenanthroline | 1,000 | 50 ± 8 |
| 100 | 97 ± 5 | |
| PMSF | 1,000 | 66 ± 4 |
| 100 | 99 ± 7 | |
| Benzamidine | 1,000 | 72 ± 6 |
| 100 | 98 ± 6 | |
| Pepstatin A | 10 | 99 ± 2 |
| 1 | 100 ± 5 | |
| NEM | 100 | 86 ± 3 |
| 10 | 91 ± 2 | |
| 2-Pyrrolidinone | 100 | 95 ± 3 |
| 10 | 100 ± 1 | |
| Purified deglycosylated enzyme | ||
| E64 | 100 | 0 |
| 10 | 0 | |
| 1 | 4 ± 1.5 | |
| K11777 | 10 | 0.2 ± 0.1 |
| 1 | 1.6 ± 0.3 | |
| ZFRDA | 100 | 0 |
| 10 | 2.1 ± 0.1 | |
| Leupeptin | 100 | 0 |
| 10 | 2.1 ± 0.8 | |
| TLCK | 100 | 0.6 ± 0.4 |
| 10 | 5.6 ± 0.9 |
For testing the efficacy of inhibitors, the compound was added to the buffer-enzyme mix and incubated for 10 min at 37°C. The reaction was started by addition of the substrate Z-F-R-AMC at 200 μM and monitored continuously for 5 min (excitation at 380 nm; emission at 465 nm). ZFRDA, Z-F-R-DMK.
Screening recombinant EtCatB for inhibitors.
To identify inhibitors of EtCatB that could be lead candidates for further optimization in an anticoccidial drug discovery program, we tested a focused compound library in an in vitro assay system. The library was set up as depicted in Fig. 6. Starting from a database of ∼2 × 106 commercially available compounds, we used BCUT metrics developed from the work of Burden and Pearlman (7, 29, 30) to select a structurally diverse subset. This subset was further filtered by application of a property filter (see Materials and Methods for details) which decreased the number of compounds in the screening library to 74,339 entities. This screening library was used to screen a recombinant cathepsin L-like cysteine peptidase of Leishmania mexicana known as CPB (37) with cathepsin B from bovine spleen (24) as a counter screen (data not shown). In addition, we considered active compounds found in an in-house human cathepsin K screening project. By application of a warhead filter (see Materials and Methods for details), we selected 82 actives from the CPB screen and 113 actives from the human cathepsin K screen. The 195 active compounds selected were of the nitrile, thiosemicarbazone, Michael acceptor, hydrazone, and mercaptoacyl structural classes (structural classes are named according to the warhead that interacts with the catalytic cysteine). This hit enrichment from two different cysteine peptidase screening projects was applied to archive a manageable, diverse set of compounds reflecting the most frequently observed cysteine peptidase warheads. This focused library was used for the EtCatB screen, with cathepsin B from bovine spleen (BtCatB) as a counter screen to provide information on the selectivity of the inhibitors. The assay resulted in 16 compounds with IC50 values of <4.5 μM. These hits were then subjected to an in vitro lead compound confirmation procedure. Thus, IC50 determination was replicated by using freshly dissolved compound from either in-house resynthesized solid stock or solid stock taken from the supplier in order to eliminate potential false positives caused by, for example, degradation products. Subsequently, the molecular structure of the relevant hit compounds was confirmed by 1H-NMR spectroscopy and LC-MS. Only compounds without contradictory findings passed this confirmation step. In the next step, the compound's aqueous solubility was determined using nephelometry. The lower solubility limit for acceptance of a compound as a confirmed in vitro lead was chosen to be 500 μM. Ki values were determined for the three most active compounds (one for each warhead class) passing all confirmation steps. The compounds were also proved for their binding mode on EtCatB and BtCatB by results of dilution experiments (12) (see Materials and Methods for details). All three binders were characterized as reversible binding inhibitors, with Ki values on EtCatB in the micromolar to nanomolar range. The activity data of these three inhibitors toward EtCatB are listed in Table 2 together with the data toward BtCatB. The compounds belong to three different compound classes, namely, nitriles (CP673854), thiosemicarbazones (CP989759), and oxazolones (CP725567). Only CP673854 exhibited activity on cathepsin B from bovine spleen in the low-micromolar range; CP989759 and CP725567 had IC50s of >30 μM toward bovine cathepsin B.
Fig 6.

Discovering the lead inhibitors. Setup of the EtCatB focused library and filtering steps during hit enrichment. Three confirmed leads were identified in the drug discovery workflow, two resulting from the vendor library and one from the cathepsin K screening project. LmCatL, L. mexicana cathepsin L.
Table 2.
Confirmed lead compound structures inhibiting EtCatBa

Three confirmed lead compounds against EtCatB were identified, belonging to the structural classes nitrile, thiosemicarbazone, and oxazolone. Cathepsin B from bovine spleen (BtCatB) was used as the counter-assay to test the selectivity profile of the confirmed leads. Results are expressed as mean IC50 ± standard deviation from three independent experiments and as graphically determined Ki values ± standard deviation from four independent experiments.
ND, not determined.
To understand the selectivity profile of EtCatB inhibitors compared to BtCatB inhibitors, the thiosemicarbazone CP989759 was covalently docked into the active site of the EtCatB homology model and into the active site of the BtCatB X-ray structure (PDB ID 1QDQ). The obtained interaction models provide insight into the activity differences of CP989759 toward the two enzymes. The top-ranked docking solution of CP989759 with EtCatB is shown in Fig. 2C. The phenyl portion of CP989759 is oriented toward the deep S2 pocket, and the 3-chloro-5-methyl pyrazole moiety fits into the shallow S1 pocket. The thiosemicarbazone scaffold was assumed to interact via a 1,2-polar addition of the catalytic C31 to the C=S group of CP989759 (42). Thus, the resulting tetrahedral thio-acyl transition state was used as the initial conformation of the thiosemicarbazone scaffold for covalent docking. Because the C=S group represents a prochiral center and because it is a priori not known which isomer resembles the most active transition state, both possible stereo-isomers were generated for the covalent docking procedure. In Fig. 2C the top-ranked S-isomer of CP989759 is shown, and the covalent bond between the sulfur of C31 and the thiocarbonyl group of the thiosemicarbazone moiety is marked by a yellow arrow. The NH2 group of the thiosemicarbazone scaffold fits close to the carbonyl of G29. The distance between the nitrogen in the thiosemicarbazone and the oxygen of the G29 carbonyl was calculated to be 2.36Å, which accounts for a favorable hydrogen bond. The reaction of C31 to the C=S group would be further assisted by the transfer of the H210 proton to the thiosemicarbazone sulfur (Fig. 2C). When the top-ranked S-isomer of CP989759 is covalently docked into the active site of BtCatB, none of the top-ranked orientations resulting from 10 different docking runs are similar to those of EtCatB. Most solutions orient the phenyl portion of CP989759 toward the S2′ pocket, which suggests that the phenyl group does not fit into the S2 pocket of BtCatB due to the K261/E245 exchange. This is consistent with the substantial difference in the activity of CP989759 toward EtCatB and BtCatB.
The three inhibitors were tested for anticoccidial activity using an E. tenella schizont maturation cell culture assay. CP673854 (EC50 of 9 μM) and CP725567 (EC50 of 14 μM) inhibited parasite schizont development at micromolar concentrations, whereas CP989759 had no activity in this assay (EC50 of >50 μM). Toxicity toward the host cell was observed for all three compounds only at concentrations above 50 μM.
DISCUSSION
Peptidases have been shown to take part in essential events of the life cycle in Eimeria, such as the formation of the oocyst wall (5) and invasion of the host cell (18). By database mining using the cathepsin B sequence of T. gondii (31), a single complete sequence coding for a cathepsin B-like enzyme was identified in the E. tenella EST database. The sequence was amplified on genomic DNA and cDNA and did not have any introns. The predicted enzyme contained the cysteine, asparagine, and histidine catalytic triad and showed good similarity with the cathepsin B of T. gondii (42%) and other cathepsin Bs from different organisms (Fig. 1).
We successfully expressed active mature recombinant EtCatB in a P. pastoris inducible system (Fig. 3). The enzyme was released in the culture supernatant at a concentration of about 100 μg/ml and was readily purified by sequential ammonium sulfate saturation precipitation followed by deglycosylation (Fig. 3). The characteristics of the enzyme conform to what is expected for a cathepsin B; it is strictly dependent on reducing agents, it cleaves F-R and R-R substrates with a preference for F-R (Fig. 5), and it is inhibited strongly by general cysteine inhibitors like E64 and iodoacetamide, as well as by the specific inhibitor K11777 (Table 1). Interestingly, it is active over a wide spectrum of pHs, whereas most cathepsin Bs are mostly active at acidic pH. This might reflect a broad distribution of the enzyme in various locations in the cell. The large amount of active recombinant enzyme produced by this system means that future structural studies to analyze inhibitor binding in detail as part of a drug discovery program are now feasible. This was not, however, included as part of this study.
Cysteine peptidases from E. tenella appear to have a role in host cell invasion as invasion by sporozoites can be inhibited by cysteine peptidase inhibitors (S. Brown et al., unpublished data); thus, this could be one part played by EtCatB of the parasite and would potentially make it a suitable drug target. It should be noted that specificity between Eimeria and host enzymes might not be critical in the path toward drug development as short-term inhibition of host enzymes might not cause serious toxicity. Moreover, as infection involves parasites in the intestinal tract, therapy toward these with a compound nonabsorbed by the host might be effective and could avoid harmful inhibition of many host enzymes. Nevertheless, bioinformatics analysis, comparative homology modeling, and covalent docking studies revealed striking differences between the EtCatB of Eimeria and the bovine counterpart (notably the K261E exchange), which further suggested that the EtCatB of E. tenella might present a good new drug target to combat coccidiosis.
Because it was not feasible to carry out gene deletion in Eimeria and therefore genetic validation of the EtCatB as a drug target was not possible, we adopted a more direct approach using HTS to detect good inhibitors and then to chemically validate the target enzyme by the application of these specific inhibitors in cell culture assays in vitro. Thus, we screened a focused chemical library against active EtCatB and subsequently tested the most promising hits in a schizont maturation assay. Based on the results from these screens, three compounds, CP673854, CP989759, and CP725567, were selected as confirmed in vitro leads. The nitrile (CP673854) and the thiosemicarbazone (CP989759) are members of classes of compounds already known to contain cysteine peptidase inhibitors (15, 33), although not previously tested against EtCatB. To the best of our knowledge, the oxazolone scaffold of CP725567 is a novel cysteine peptidase inhibitor scaffold that has not been reported before. Together with the thiosemicarbazone, the oxazolone showed high selectivity for EtCatB compared with the corresponding bovine enzyme (Table 2). Moreover, CP725567 inhibited parasite schizont development at micromolar concentrations. Thus, it is hoped that these three lead inhibitors can be used effectively in the drive toward novel drugs against Eimeria and that the oxazolone scaffold may find more widespread application as a cysteine peptidase inhibitor.
ACKNOWLEDGMENTS
We thank A. Strunk for her technical support in performing the HTS and the kinetic analyses. We are grateful to S. Koch and T. Newton for the synthesis of selected compounds. We gratefully thank Z. Rancovíc and X. Fradera for providing selected compounds and corresponding data from their cathepsin K project.
The Wellcome Trust Centre for Molecular Parasitology is supported by core funding from the Wellcome Trust (085349).
Footnotes
Published ahead of print 5 December 2011
REFERENCES
- 1. Alves LC, et al. 2001. Analysis of the S(2) subsite specificities of the recombinant cysteine proteinases CPB of Leishmania mexicana, and cruzain of Trypanosoma cruzi, using fluorescent substrates containing non-natural basic amino acids. Mol. Biochem. Parasitol. 117:137–143 [DOI] [PubMed] [Google Scholar]
- 2. Atkinson HJ, Babbitt PC, Sajid M. 2009. The global cysteine peptidase landscape in parasites. Trends Parasitol. 25:573–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baudys M, Meloun B, Gan-Erdene T, Pohl J, Kostka V. 1990. Disulfide bridges of bovine spleen cathepsin B. Biol. Chem. Hoppe-Seyler 371:485–491 [DOI] [PubMed] [Google Scholar]
- 4. Bechet DM, et al. 1991. Expression of lysosomal cathepsin B during calf myoblast-myotube differentiation. Characterization of a cDNA encoding bovine cathepsin B. J. Biol. Chem. 266:14104–14112 [PubMed] [Google Scholar]
- 5. Belli SI, Smith NC, Ferguson DJ. 2006. The coccidian oocyst: a tough nut to crack! Trends Parasitol. 22:416–423 [DOI] [PubMed] [Google Scholar]
- 6. Brickmann J, Exner TE, Keil M, Marhöfer RJ. 2000. Molecular graphics: trends and perspectives. J. Mol. Mod. 6:328–340 [Google Scholar]
- 7. Burden FR. 1989. Molecular identification number for substructure searches. J. Chem. Infect. Comput. Sci. 29:225–227 [Google Scholar]
- 8. Caffrey CR, et al. 2002. SmCB2, a novel tegumental cathepsin B from adult Schistosoma mansoni. Mol. Biochem. Parasitol. 121:49–61 [DOI] [PubMed] [Google Scholar]
- 9. Carruthers VB, Sherman GD, Sibley LD. 2000. The Toxoplasma adhesive protein MIC2 is proteolytically processed at multiple sites by two parasite-derived proteases. J. Biol. Chem. 275:14346–14353 [DOI] [PubMed] [Google Scholar]
- 10. Cavasotto CN, Phatak SS. 2009. Homology modeling in drug discovery: current trends and applications. Drug Discov. Today 14:676–683 [DOI] [PubMed] [Google Scholar]
- 11. Chan SJ, San Segundo BS, McCormick MB, Steiner DF. 1986. Nucleotide and predicted amino acid sequences of cloned human and mouse preprocathepsin B cDNAs. Proc. Natl. Acad. Sci. U. S. A. 83:7721–7725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chatterjee S, et al. 1997. Synthesis and biological activity of a series of potent fluoromethyl ketone inhibitors of recombinant human calpain I. J. Med. Chem. 40:3820–3828 [DOI] [PubMed] [Google Scholar]
- 13. Dong AS, et al. 1995. Avian cathepsin B cDNA: sequence and demonstration that mRNAs of two sizes are produced in cell types producing large quantities of the enzyme. Biochim. Biophys. Acta 1251:69–73 [DOI] [PubMed] [Google Scholar]
- 14. Dou Z, Carruthers VB. 2011. Cathepsin proteases in Toxoplasma gondii. Adv. Exp. Med. Biol. 712:49–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Du X, et al. 2002. Synthesis and structure-activity relationship study of potent trypanocidal thiosemicarbazone inhibitors of the trypanosomal cysteine protease cruzain. J. Med. Chem. 45:2695–2707 [DOI] [PubMed] [Google Scholar]
- 16. Dubremetz JF. 1998. Host cell invasion by Toxoplasma gondii. Trends Microbiol. 6:27–30 [DOI] [PubMed] [Google Scholar]
- 17. Dutton CJ, Banks BJ, Cooper CB. 1995. Polyether ionophores. Nat. Prod. Rep. 12:165–181 [DOI] [PubMed] [Google Scholar]
- 18. Fuller AL, McDougald LR. 1990. Reduction in cell entry of Eimeria tenella (Coccidia) sporozoites by protease inhibitors, and partial characterization of proteolytic activity associated with intact sporozoites and merozoites. J. Parasitol. 76:464–467 [PubMed] [Google Scholar]
- 19. Hasnain S, Hirama T, Huber CP, Mason P, Mort JS. 1993. Characterization of cathepsin B specificity by site-directed mutagenesis. Importance of Glu245 in the S2-P2 specificity for arginine and its role in transition state stabilization. J. Biol. Chem. 268:235–240 [PubMed] [Google Scholar]
- 20. Ihlenfeldt WD, Voigt JH, Bienfait B, Oellien F, Nicklaus MC. 2002. Enhanced CACTVS browser of the Open NCI Database. J. Chem. Infect. Comput. Sci. 42:46–57 [DOI] [PubMed] [Google Scholar]
- 21. Illy C, et al. 1997. Role of the occluding loop in cathepsin B activity. J. Biol. Chem. 272:1197–1202 [DOI] [PubMed] [Google Scholar]
- 21a. Invitrogen 2010. Pichia expression kit user manual. Invitrogen, Carlsbad, CA: http://tools.invitrogen.com/content/sfs/manuals/pich_man.pdf [Google Scholar]
- 22. Kim K. 2004. Role of proteases in host cell invasion by Toxoplasma gondii and other apicomplexa. Acta Trop. 91:69–81 [DOI] [PubMed] [Google Scholar]
- 23. Kozak M. 1999. Initiation of translation in prokaryotes and eukaryotes. Gene 234:187–208 [DOI] [PubMed] [Google Scholar]
- 24. Meloun B, Baudys M, Pohl J, Pavlík M, Kostka V. 1988. Amino acid sequence of bovine spleen cathepsin B. J. Biol. Chem. 263:9087–9093 [PubMed] [Google Scholar]
- 25. Musil D, et al. 1991. The refined 2.15 Å X-ray crystal structure of human liver cathepsin B: the structural basis for its specificity. EMBO J. 10:2321–2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nielsen H, Engelbrecht J, Brunak SS, von Heijne G. 1997. A neural network method for identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Int. J. Neural. Syst. 8:581–599 [DOI] [PubMed] [Google Scholar]
- 27. Nissink JWM, et al. 2002. A new test set for validating predictions of protein-ligand interaction. Proteins Struct. Funct. Genet. 49:457–471 [DOI] [PubMed] [Google Scholar]
- 28. Pandey KC, Sijwali PS, Singh A, Na BK, Rosenthal PJ. 2004. Independent intramolecular mediators of folding, activity, and inhibition for the Plasmodium falciparum cysteine protease falcipain-2. J. Biol. Chem. 279:3484–3491 [DOI] [PubMed] [Google Scholar]
- 29. Pearlman RS, Smith KM. 1998. Novel software tools for chemical diversity. Perspect. Drug Discov. Design 9:339–353 [Google Scholar]
- 30. Pearlman RS, Smith KM. 1999. Metric validation and the receptor relevant subspace concept. J. Chem. Infect. Comput. Sci. 39:28–35 [Google Scholar]
- 31. Que X, et al. 2002. The cathepsin B of Toxoplasma gondii, toxopain-1, is critical for parasite invasion and rhoptry protein processing. J. Biol. Chem. 277:25791–25797 [DOI] [PubMed] [Google Scholar]
- 32. Rankovic Z, Cai J, Cumming I. February 2005. Preparation of 2-cyano-1,3,5-triazine-4,6-diamine derivatives for the treatment of osteoporosis and atherosclerosis. International patent WO 2005011703 A1
- 33. Rankovic Z, et al. 2010. Design and optimization of a series of novel 2-cyano-pyrimidines as cathepsin K inhibitors. Bioorg. Med. Chem. Lett. 20:1524–1527 [DOI] [PubMed] [Google Scholar]
- 34. Ritonja A, Popovic T, Turk V, Wiedenmann K, Machleidt W. 1985. Amino acid sequence of human liver cathepsin B. FEBS Lett. 181:169–172 [DOI] [PubMed] [Google Scholar]
- 35. Rosenthal PJ. 2011. Falcipains and other cysteine proteases of malaria parasites. Adv. Exp. Med. Biol. 712:30–48 [DOI] [PubMed] [Google Scholar]
- 36. Sakanari JA, et al. 1997. Leishmania major: comparison of the cathepsin L- and B-like cysteine protease genes with those of other trypanosomatids. Exp. Parasitol. 85:63–76 [DOI] [PubMed] [Google Scholar]
- 37. Sanderson SJ, et al. 2000. Expression and characterization of a recombinant cysteine proteinase of Leishmania mexicana. Biochem. J. 347:383–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Selzer PM, et al. 1999. Cysteine protease inhibitors as chemotherapy: lessons from a parasite target. Proc. Natl. Acad. Sci. U. S. A. 96:11015–11022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Takio K, Towatari T, Katunuma N, Teller DC, Titani K. 1983. Homology of amino acid sequence of rat liver cathepsin B and H with that of papain. Proc. Natl. Acad. Sci. U. S. A. 80:3666–3670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Teixeira C, Gomes JR, Gomes P. 2011. Falcipains, Plasmodium falciparum cysteine proteases as key drug targets against malaria. Curr. Med. Chem. 18:1555–1572 [DOI] [PubMed] [Google Scholar]
- 41. Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. 2003. Improved protein-ligand docking using GOLD. Proteins 52:609–623 [DOI] [PubMed] [Google Scholar]
- 42. Wang SX, et al. 2006. Structural basis for unique mechanisms of folding and hemoglobin binding by a malarial protease. Proc. Natl. Acad. Sci. U. S. A. 103:11503–11508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Whitmire WM, Kyle JE, Speer CA, Burgess DE. 1988. Inhibition of penetration of cultured cells by Eimeria bovis sporozoites by monoclonal immunoglobulin G antibodies against the parasite surface protein P20. Infect. Immun. 56:2538–2543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yamamoto A, et al. 2000. Molecular dynamics simulations of bovine cathepsin B and its complex with CA074. Chem. Pharm. Bull. 48:480–485 [DOI] [PubMed] [Google Scholar]
- 45. Yamamoto A, et al. 2000. Substrate specificity of bovine cathepsin B and its inhibition by CA074, based on crystal structure refinement of the complex. J. Biochem. 127:635–643 [DOI] [PubMed] [Google Scholar]