

Abstract
The potent antiretroviral 4′-ethynyl-2-fluoro-2′-deoxyadenosine (EFdA) is a promising experimental agent for treating HIV infection. Pre-steady-state kinetics were used to characterize the interaction of EFdA-triphosphate (EFdA-TP) with human mitochondrial DNA polymerase γ (Pol γ) to assess the potential for toxicity. Pol γ incorporated EFdA-TP 4,300-fold less efficiently than dATP, with an excision rate similar to ddATP. This strongly indicates EFdA is a poor Pol γ substrate, suggesting minimal Pol γ-mediated toxicity, although this should be examined under clinical settings.
TEXT
Nucleoside reverse transcriptase inhibitors (NRTIs) are a critical component of highly active antiretroviral therapy for treating HIV infection. All FDA-approved NRTIs are nucleoside analogs lacking a 3′-hydroxyl group, forcing DNA chain termination upon incorporation by viral reverse transcriptase (RT). A central source of toxicity stems from the interaction of NRTIs with human mitochondrial DNA polymerase γ (Pol γ), the only human polymerase capable of using these drugs as substrates (2, 16, 18, 20). Incorporation of NRTIs can result in chain termination during replication, causing mitochondrial DNA depletion that can manifest in patients as myopathies, lipodystrophies, lactic acidosis, or liver failure (2–4, 12).
Current NRTIs can be plagued with toxicity and RT resistance, so there is a critical need for new antivirals. A promising new NRTI is 4′-ethynyl-2-fluoro-2′-deoxyadenosine (EFdA) (Fig. 1A) (17). Its 50% effective concentration (EC50) of 50 pM is one of the best reported for an NRTI, 440-fold better than zidovudine (AZT) and 66,000-fold better than tenofovir, and numerous NRTI-resistant strains of HIV also show sensitivity and even hypersensitivity to EFdA (13, 17, 24, 27). It is also effective in vivo, causing a significant decrease in viral load and low toxicity in a humanized HIV-infected mouse model (9).
Fig 1.
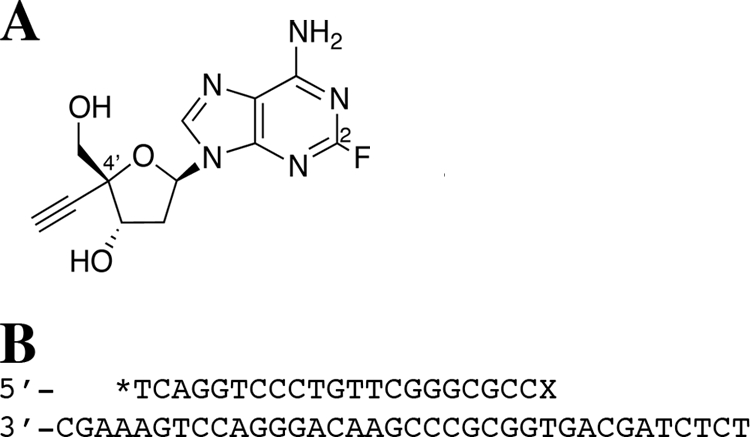
(A) Structure of EFdA. (B) DNA oligonucleotides used in the experiments. Shown are the D21 primer (radiolabeled, as indicated by an asterisk) and the D36 template. The primer “X” is the site of incorporation of the incoming dATP or EFdA-TP used in the single-nucleotide incorporation experiments or the location of the EFdA-MP to be removed in the excision studies.
Despite the 3′-hydroxyl group, EFdA acts as a chain terminator by preventing RT translocation (24). This is in contrast to KP-1212, another 3′-hydroxyl-containing NRTI that facilitates error-prone extension by RT (1, 25). The 4′-ethynyl group locks the sugar in a favorable position for incorporation, which, along with the 3′-hydroxyl, makes EFdA a better substrate for RT than native nucleotides (15, 24). Similarly, EFdA may be preferred by Pol γ, but kinetic studies are limited to a 50% inhibitory concentration (IC50) of 10 μM and a Ki of 25 μM for Pol γ (26, 27), indicating that EFdA serves as a substrate. In this study, we sought to expand our prior work with EFdA (26) to characterize the molecular mechanism of inhibition of Pol γ by EFdA. Such studies are critical to assess the safety of drugs in preclinical and clinical trials.
Assessing the potential for Pol γ-mediated toxicity requires discerning the individual rate constants of NRTI incorporation and excision using pre-steady-state kinetics. Since steady-state studies report only on the rate-limiting step, which for Pol γ is product release, pre-steady-state kinetics are required to determine NRTI affinity and rates of NRTI incorporation and excision by Pol γ, which provides a detailed kinetic mechanism of in vitro toxicity. Single-turnover conditions, in which the enzyme is in excess of the substrate, were used to generate kpol, the maximum rate of polymerization, and Kd, the binding affinity for the incoming nucleotide, as described previously (7, 11). Wild-type (WT) Pol γ (exonuclease deficient) catalytic subunit (Pol γA) and accessory subunit were purified and reconstituted as described elsewhere (22, 23, 29). A KinTek Instruments RQF-3 rapid chemical quench was used to mix Pol γ holoenzyme and substrate, a 5′-radiolabeled DNA primer annealed to a DNA template (D21/D36 substrate) (Fig. 1B), with magnesium chloride and various concentrations of dATP or EFdA-triphosphate (EFdA-TP). The reaction was quenched with EDTA, and reaction mixtures were separated on a 20% polyacrylamide denaturing gel and analyzed by phosphorimaging (Bio-Rad Molecular Imager FX). Plots of product formation versus time were fit to single-exponential (EFdA-TP) or double-exponential (dATP) equations to generate kobs (observed rate) values, which were plotted against nucleotide concentration and fit to a hyperbola to generate kpol and Kd (KaleidaGraph; Synergy) (Fig. 2).
Fig 2.
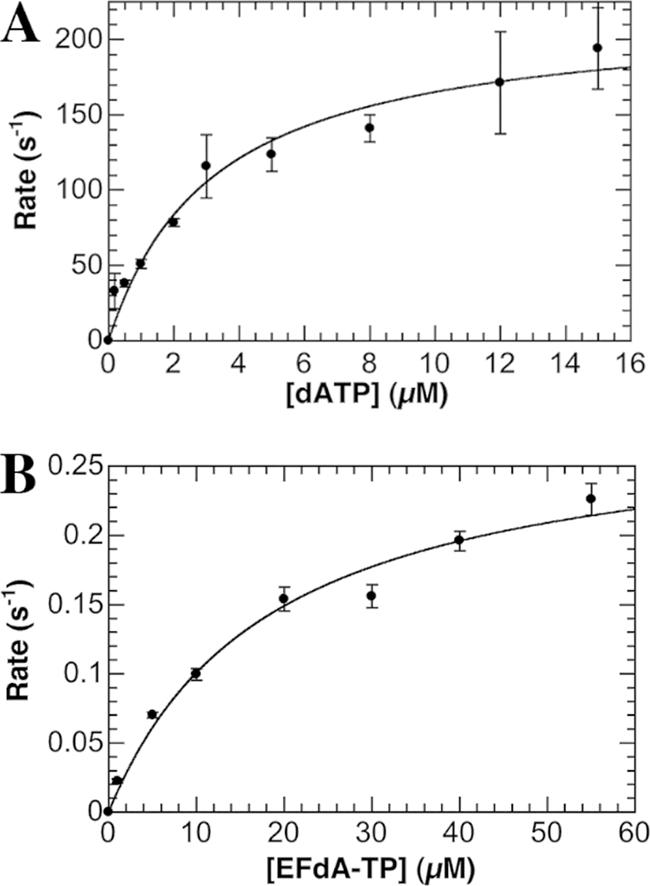
Concentration dependence of the observed rate of nucleotide incorporation by Pol γ. Each point in the plots represents the observed rate generated from fitting a time course with 10 different points using a double exponential equation (dATP) or a single exponential equation (EFdA-TP) (KaleidaGraph). The error bars in the plots represent the deviance from these exponential fits, and the standard errors associated with the determined rate constant values represent the deviance from the hyperbolic fits shown. (A) Observed rates of incorporation were plotted against dATP concentration and fit with a hyperbolic equation to generate a kpol value of 220 ± 16 s−1 and a Kd value of 3.2 ± 0.7 μM. (B) Observed rates of incorporation were plotted against EFdA-TP concentration and fit with a hyperbolic equation to generate a kpol value of 0.29 ± 0.02 s−1 and a Kd value of 18 ± 4 μM.
Pol γ incorporated EFdA-TP 760-fold more slowly and with 5.6-fold-lower affinity than the natural dATP substrate (Table 1). The efficiency of EFdA-TP incorporation (0.016 μM−1 s−1) is well within the range of NRTIs on the market, showing 340- and 1.5-fold improvement over stavudine and didanosine, respectively (11). Although lower efficiencies are seen with tenofovir (3.2-fold) and AZT (16-fold) (11), it is important to remember that EFdA in the steady state showed 440-fold- and 66,000-fold-higher potency than AZT and tenofovir, respectively (24). Importantly, in contrast to RT, which shows a 2-fold selectivity for EFdA-TP over dATP in steady-state studies (24), Pol γ shows a 4,300-fold preference for the natural dATP substrate over EFdA-TP (Table 1), and this coupled with the very low incorporation rate and low affinity for EFdA indicates a very low risk of Pol γ-mediated toxicity.
Table 1.
Pre-steady-state rate constants for dNTP incorporation by Pol γa
| Nucleotide | kpol (s−1) | Kd (μM) | Efficiency (μM−1 s−1)b | Discriminationc |
|---|---|---|---|---|
| dATP | 220 ± 16 | 3.2 ± 0.7 | 69 | |
| EFdA-TP | 0.29 ± 0.02 | 18 ± 4 | 0.016 | 4,300 |
At least 7 different time courses containing 10 time points each were used to generate kpol and Kd. The standard error estimates shown were derived from the deviance from the nonlinear regression fits determined using KaleidaGraph software.
Efficiency = kpol/Kd.
Discrimination = efficiency(dATP)/efficiency(EFdA-TP).
This rare EFdA incorporation event can be further mitigated via excision by Pol γ. To measure the rate of excision (kexo), WT HIV-1 RT (purified as described previously [6, 14]) incorporated a single EFdA-TP into a D21/D36 substrate (Fig. 1B) (11). Under single-turnover conditions, exonuclease-competent Pol γ holoenzyme (purification detailed elsewhere [23]) and the D21-EFdA/D36 substrate were manually mixed with magnesium chloride, followed by quenching and product separation (Fig. 3A) (28). A plot of the percent loss of substrate versus time was fit to a single exponential equation to generate kexo (Fig. 3B). EFdA was excised at 0.00057 ± 0.00003 s−1, yielding a half-life of ∼20 min, similar to ddAMP removal (0.0005 s−1) (11). Since DNA dissociates from Pol γ at 0.02 s−1 (10), it is likely that dissociation will occur before EFdA excision. The kexo for EFdA falls in range of currently available NRTIs, 1.4-fold higher than for d4T and AZT (10) and much higher than for zalcitabine, which is too low to be detected (<0.00002 s−1) (7), but 1.3-fold and 26-fold lower than for tenofovir (10) and lamivudine (7), respectively. Since dosages of EFdA are expected to be low due to the observed high potency, and because the Pol γ incorporation rate is very low with moderate EFdA excision rates, it is predicted that Pol γ-related toxicity will be limited. However, mitochondrial toxicity must be tested under clinical settings.
Fig 3.

Excision of EFdA by Pol γ. (A) Gel analysis of the intact D21-EFdA substrate and degradation products over a 3-h time course. (B) A single exponential fitting of a plot of the percent loss of substrate versus time yielded a kexo value of 0.00057 ± 0.00003 s−1. Each point represents a single experiment within the time course, and the standard error associated with the kexo value represents the deviance from this single exponential fitting as calculated by the KaleidaGraph software.
To probe the mechanism of the 4,300-fold increased efficiency of dATP incorporation over EFdA-TP, we used structural modeling to compare the dATP and EFdA-TP ternary complexes using the currently available Pol γ crystal structure (19). The active site residues of Pol γA are seen at topologically equivalent positions in other A family polymerases, including the large fragment of Pol I from Thermus aquaticus (KlenTaq), which has been reported with all four incoming ddNTPs (21). After superimposing Pol γA and KlenTaq using their active sites as a reference point, we docked 6 bp of DNA from the 3′ end of the primer, the ddATP, and two catalytic metal ions from the KlenTaq structure (PDB ID 1QSY [21]) into the Pol γ structure using Glide (Schrödinger Suite). Significant steric interactions involving residues M1057 to A1064, a region not found in other A family DNA polymerases (19), prevented energetically favorable docking of the template primer and ddATP at the Pol γA active site, and we deleted these residues because they are expected to undergo significant conformational changes in the ternary complex. Interestingly, similar changes are observed in N4 virion RNA polymerase, which undergoes a conformational change to displace a structurally equivalent region in order to accommodate duplex DNA (8).
The modeled KlenTaq–DNA–EFdA-TP ternary complex is shown in Fig. 4A and B for comparison with the Pol γA–DNA–EFdA-TP complex shown in Fig. 4C and D. The 4′-ethynyl group of EFdA-TP is reasonably accommodated in a pocket defined by KlenTaq residues I614, E615, R753, and H784 (corresponding to Q894, E895, R853, and H1134 in Pol γ), which are 4.2, 3.5, 3.3, and 4.6 Å, respectively, away from this group, consistent with a relatively small change in affinity for EFdA-TP versus dATP (Table 1). A measureable Ki value (25 μM) has been determined for EFdA-TP (26), supporting the hypothesis that the binding pocket is similar for EFdA-TP and dATP. To probe this idea further, we employed a pre-steady-state competition assay in which the rate of dATP incorporation by the Pol γ holoenzyme was measured in the presence of various concentrations of EFdA-TP. EFdA-TP was able to compete with dATP for the binding pocket, as indicated by a linear decrease in the kobs, although concentrations higher than the Kd for EFdA binding (18 μM) were required to lower the rate of incorporation of 3 μM dATP (the Kd of dATP) (data not shown). To achieve a 4.8-fold drop in the rate of dATP incorporation, a concentration of EFdA-TP at nearly 6 times its Kd was required (data not shown). Overall, this competition experiment supports our model of a similar binding pocket for EFdA-TP and dATP, leading to only minor changes in Kd, as well as our finding that EFdA-TP is a poor competitor of dATP due to its inefficient incorporation.
Fig 4.

(A) KlenTaq–DNA–EFdA-TP. (B) KlenTaq–DNA–EFdA-TP in stereo view, highlighting interactions of the 4′-ethynyl group of EFdA-TP with the neighboring amino acid residues. (C) Pol γA–DNA–EFdA-TP. (D) Pol γA–DNA–EFdA-TP in stereo view, highlighting the interactions of 4′-ethynyl group of EfdA-TP with proximal amino acid residues. For (A) and (C), the ternary complex model of the polymerase subunit with template (cyan), primer (yellow), and EFdA-TP (colored by atom type as follows: carbon, gray; nitrogen, blue; oxygen, purple; phosphorous, orange). The enzyme is rendered as a Connolly surface representation with the thumb, palm, finger, and exonuclease domains colored in green, red, blue, and orange, respectively. For clear visualization of EFdA-TP, some residues at the tip of the fingers and thumb have been removed. However, these side chains were part of the computations. The metal ions are depicted as white spheres. For panels B and D, the amino acid residues (colored by atom type as follows: carbon, green; nitrogen, blue; oxygen, red) important for interaction with EFdA-TP (using the same color scheme, except carbon is colored gray and phosphorous purple) are highlighted. The template is shown in cyan. The thin lines depict the possible interactions with the amino acid residues in the vicinity of the 4′-ethynyl group of EFdA-TP. These interactions are not necessarily the hydrogen bonds. Interaction lengths range from 2.9 to 4 Å. Panels A and C were generated by PyMOL (http://www.pymol.org/), and panels B and D were generated by Maestro 9.1 (Schrödinger Inc., NY).
The 4′-ethynyl group of EFdA appears to be in the proximity of E895 in Pol γ (E615 in KlenTaq) and may disturb hydrogen bonding with Y955 (Y671 in KlenTaq), which is required for efficient catalysis in the Pol I family (Fig. 4). This may explain the severe defects in kpol observed for EFdA incorporation. Interestingly, incorporation of 4′-alkyl-modified nucleoside analogs by the Klenow fragment of DNA Pol I from Escherichia coli showed a lower kpol (∼60-fold), with a small loss of affinity (∼1.5-fold), similar to that for EFdA (5).
In summary, we have investigated the effects of a novel NRTI, EFdA, on Pol γ. EFdA has shown great promise in preclinical trials for the treatment of HIV infection because of its high potency and low toxicity. We show that this low toxicity is due in part to extremely inefficient EFdA incorporation and, to a lesser extent, subsequent EFdA excision by human Pol γ. Structural modeling suggests that the relatively small change in affinity for EFdA-TP relative to dATP is due to similar accommodation within a pocket, while the severe decrease in kpol may be due to hydrogen bond disruption by EFdA. Characterizing the ability of Pol γ to incorporate NRTIs into a DNA template is critical to assess the safety of new drugs in this important class of antivirals.
ACKNOWLEDGMENTS
This work was supported by NIH grants R01 GM049551 (to K.S.A.), F32 GM099289 (to C.D.S.), AI094715, AI076119, AI079801, and AI074389 (to S.G.S.) and by the Intramural Research Program of the NIH, NIEHS, ES 065080 (to W.C.C.).
Additionally, C.D.S. and K.S.A. thank Ligong Wang and Krasimir Spasov for the purification of the Pol γ accessory subunit and RT, respectively.
Footnotes
Published ahead of print 12 December 2011
REFERENCES
- 1. Anderson JP, Daifuku R, Loeb LA. 2004. Viral error catastrophe by mutagenic nucleosides. Annu. Rev. Microbiol. 58:183–205 [DOI] [PubMed] [Google Scholar]
- 2. Brinkman K, Kakuda TN. 2000. Mitochondrial toxicity of nucleoside analogue reverse transcriptase inhibitors: a looming obstacle for long-term antiretroviral therapy? Curr. Opin. Infect. Dis. 13:5–11 [DOI] [PubMed] [Google Scholar]
- 3. Brinkman K, Smeitink JA, Romijn JA, Reiss P. 1999. Mitochondrial toxicity induced by nucleoside-analogue reverse-transcriptase inhibitors is a key factor in the pathogenesis of antiretroviral-therapy-related lipodystrophy. Lancet 354:1112–1115 [DOI] [PubMed] [Google Scholar]
- 4. Dalakas MC, et al. 1990. Mitochondrial myopathy caused by long-term zidovudine therapy. N. Engl. J. Med. 322:1098–1105 [DOI] [PubMed] [Google Scholar]
- 5. Di Pasquale F, et al. 2008. Opposed steric constraints in human DNA polymerase beta and E. coli DNA polymerase I. J. Am. Chem. Soc. 130:10748–10757 [DOI] [PubMed] [Google Scholar]
- 6. Feng JY, Anderson KS. 1999. Mechanistic studies comparing the incorporation of (+) and (-) isomers of 3TCTP by HIV-1 reverse transcriptase. Biochemistry 38:55–63 [DOI] [PubMed] [Google Scholar]
- 7. Feng JY, Johnson AA, Johnson KA, Anderson KS. 2001. Insights into the molecular mechanism of mitochondrial toxicity by AIDS drugs. J. Biol. Chem. 276:23832–23837 [DOI] [PubMed] [Google Scholar]
- 8. Gleghorn ML, Davydova EK, Rothman-Denes LB, Murakami KS. 2008. Structural basis for DNA-hairpin promoter recognition by the bacteriophage N4 virion RNA polymerase. Mol. Cell 32:707–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hattori S, et al. 2009. Potent activity of a nucleoside reverse transcriptase inhibitor, 4′-ethynyl-2-fluoro-2′-deoxyadenosine, against human immunodeficiency virus type 1 infection in a model using human peripheral blood mononuclear cell-transplanted NOD/SCID Janus kinase 3 knockout mice. Antimicrob. Agents Chemother. 53:3887–3893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Johnson AA, Johnson KA. 2001. Exonuclease proofreading by human mitochondrial DNA polymerase. J. Biol. Chem. 276:38097–38107 [DOI] [PubMed] [Google Scholar]
- 11. Johnson AA, et al. 2001. Toxicity of antiviral nucleoside analogs and the human mitochondrial DNA polymerase. J. Biol. Chem. 276:40847–40857 [DOI] [PubMed] [Google Scholar]
- 12. Kakuda TN, Brundage RC, Anderson PL, Fletcher CV. 1999. Nucleoside reverse transcriptase inhibitor-induced mitochondrial toxicity as an etiology for lipodystrophy. AIDS 13:2311–2312 [DOI] [PubMed] [Google Scholar]
- 13. Kawamoto A, et al. 2008. 2′-Deoxy-4′-C-ethynyl-2-halo-adenosines active against drug-resistant human immunodeficiency virus type 1 variants. Int. J. Biochem. Cell Biol. 40:2410–2420 [DOI] [PubMed] [Google Scholar]
- 14. Kerr SG, Anderson KS. 1997. RNA dependent DNA replication fidelity of HIV-1 reverse transcriptase: evidence of discrimination between DNA and RNA substrates. Biochemistry 36:14056–14063 [DOI] [PubMed] [Google Scholar]
- 15. Kirby KA, et al. 2011. The sugar ring conformation of 4′-ethynyl-2-fluoro-2′-deoxyadenosine and its recognition by the polymerase active site of HIV reverse transcriptase. Cell. Mol. Biol. (Noisy-le-grand) 57:40–46 [PMC free article] [PubMed] [Google Scholar]
- 16. Koczor CA, Lewis W. 2010. Nucleoside reverse transcriptase inhibitor toxicity and mitochondrial DNA. Expert Opin. Drug Metab. Toxicol. 6:1493–1504 [DOI] [PubMed] [Google Scholar]
- 17. Kodama EI, et al. 2001. 4′-Ethynyl nucleoside analogs: potent inhibitors of multidrug-resistant human immunodeficiency virus variants in vitro. Antimicrob. Agents Chemother. 45:1539–1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kohler JJ, Lewis W. 2007. A brief overview of mechanisms of mitochondrial toxicity from NRTIs. Environ. Mol. Mutagen. 48:166–172 [DOI] [PubMed] [Google Scholar]
- 19. Lee YS, Kennedy WD, Yin YW. 2009. Structural insight into processive human mitochondrial DNA synthesis and disease-related polymerase mutations. Cell 139:312–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lewis W, Day BJ, Copeland WC. 2003. Mitochondrial toxicity of NRTI antiviral drugs: an integrated cellular perspective. Nat. Rev. Drug Discov. 2:812–822 [DOI] [PubMed] [Google Scholar]
- 21. Li Y, Kong Y, Korolev S, Waksman G. 1998. Crystal structures of the Klenow fragment of Thermus aquaticus DNA polymerase I complexed with deoxyribonucleoside triphosphates. Protein Sci. 7:1116–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lim SE, Longley MJ, Copeland WC. 1999. The mitochondrial p55 accessory subunit of human DNA polymerase gamma enhances DNA binding, promotes processive DNA synthesis, and confers N-ethylmaleimide resistance. J. Biol. Chem. 274:38197–38203 [DOI] [PubMed] [Google Scholar]
- 23. Longley MJ, Ropp PA, Lim SE, Copeland WC. 1998. Characterization of the native and recombinant catalytic subunit of human DNA polymerase gamma: identification of residues critical for exonuclease activity and dideoxynucleotide sensitivity. Biochemistry 37:10529–10539 [DOI] [PubMed] [Google Scholar]
- 24. Michailidis E, et al. 2009. Mechanism of inhibition of HIV-1 reverse transcriptase by 4′-ethynyl-2-fluoro-2′-deoxyadenosine triphosphate, a translocation-defective reverse transcriptase inhibitor. J. Biol. Chem. 284:35681–35691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Murakami E, Basavapathruni A, Bradley WD, Anderson KS. 2005. Mechanism of action of a novel viral mutagenic covert nucleotide: molecular interactions with HIV-1 reverse transcriptase and host cell DNA polymerases. Antiviral Res. 67:10–17 [DOI] [PubMed] [Google Scholar]
- 26. Nakata H, et al. 2007. Activity against human immunodeficiency virus type 1, intracellular metabolism, and effects on human DNA polymerases of 4′-ethynyl-2-fluoro-2′-deoxyadenosine. Antimicrob. Agents Chemother. 51:2701–2708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ohrui H, et al. 2007. 2′-Deoxy-4′-C-ethynyl-2-fluoroadenosine: a nucleoside reverse transcriptase inhibitor with highly potent activity against wide spectrum of HIV-1 strains, favorable toxic profiles, and stability in plasma. Nucleosides Nucleotides Nucleic Acids 26:1543–1546 [DOI] [PubMed] [Google Scholar]
- 28. Ray AS, et al. 2007. Interaction of 2′-deoxyguanosine triphosphate analogue inhibitors of HIV reverse transcriptase with human mitochondrial DNA polymerase gamma. Antivir. Chem. Chemother. 18:25–33 [DOI] [PubMed] [Google Scholar]
- 29. Yakubovskaya E, Chen Z, Carrodeguas JA, Kisker C, Bogenhagen DF. 2006. Functional human mitochondrial DNA polymerase gamma forms a heterotrimer. J. Biol. Chem. 281:374–382 [DOI] [PubMed] [Google Scholar]