

Abstract
A loop-mediated isothermal amplification (LAMP) method for the rapid detection of serotype 1 Marek's disease virus (MDV) was developed. The method used a set of three pairs of primers to amplify the MEQ gene for detecting serotype 1 MDV. The MDV LAMP method did not cross-react with serotype 2 and serotype 3, nor did the LAMP primers have binding sites for the common avian DNA viruses (reticuloendotheliosis virus, chicken anemia virus, subgroup J of the avian leukosis virus). Additionally, the assay could detect up to 10 copies of the MEQ gene in the MD viral genome, and it had 10 times higher sensitivity than the traditional PCR methods. The LAMP master mix was stable for 90 days at −20°C. Furthermore, the efficiency of LAMP for detection of serotype 1 MDV in clinical samples was comparable to those of PCR and viral isolation. The LAMP procedure is simple and does not rely on any special equipment. The detection of serotype 1 MDV by LAMP will be useful for detecting and controlling oncogenic Marek's disease.
INTRODUCTION
Marek's disease (MD) is a lymphoproliferative chicken disorder that virtually exists in all commercial chicken flocks (1, 4). The causative agent of MD is Marek's disease virus (MDV), a cell-associated virus that is a member of the Herpesviridae family, in the genus Mardivirus. There are three serotypes of MDV, differing in their virulence in chicken or their ability to induce T-cell lymphomas and antigenic properties. These serotypes are serotype 1 (Gallid herpesvirus 2), serotype 2 (Gallid herpesvirus 3), and serotype 3 (Meleagrid herpesvirus 1). Among these three serotypes, serotype 1 MDVs are oncogenic, and, based on their virulence, serotype 1 MDVs can be further classified as mild (m), virulent (v), very virulent (vv), and very virulent plus (vv+) strains (16, 17).
The MD virus initially replicates in a cell-associated form in the lymphoid tissues. Then, from 10 days postinfection onwards, MDV undergoes fully productive replication in the feather follicle epithelium. High levels of MDV antigens are expressed, and the cell-free virus is shed with the skin and feather debris throughout the life of an infected bird. This virus is the source of infection for other chickens, via a respiratory route (2). Several genes unique for MDVs have been identified. Among these genes, the MEQ gene is specific to serotype 1 MDVs.
MD is controlled by vaccination and good management practices (4). However, MD is still a problem throughout the world. Recently, the MDV-1 serotype has been reported in India (13). Several PCR and real-time-based techniques have been developed for detection, as well as quantification, of the MDV genome of field and vaccine strains (3, 8, 14, 20). These techniques use blood, organ samples, and feather tips for detection and quantification. However, PCR requires an expensive thermal cycler and operator skill, which are limited in the field. Additionally, virus isolation using chicken embryo fibroblast (CEF) cell culture takes more than 7 days for a definite diagnosis.
To overcome the difficulties of PCR-based techniques, Notomi et al. (10) developed a novel technique called loop-mediated isothermal amplification (LAMP). This technique requires four different primers, Bst DNA polymerase to displace DNA strands, and isothermal conditions for the amplification of target DNA. Since the technique was first described, it has been applied for the detection of several poultry pathogens (5, 6, 7, 18, 19). The advantages of this technique are that it requires only 30 to 60 min and can be performed at a single temperature ranging from 60°C to 65°C. The reaction can be carried out in a laboratory water bath or a dry heat block, and the results can be read with the naked eye.
Hence, the present study was carried out to develop a diagnostic method based on the LAMP reaction for rapid detection of the MD viral genome in feather samples. The sensitivity and specificity of LAMP were compared with those of conventional PCR. Further, to avoid procedural contamination, a LAMP master mix was prepared and tested for its stability and then used to detect MD viral genomes in field samples.
MATERIALS AND METHODS
Feather samples.
A total of 101 feather samples were collected from birds of 18 to 56 weeks of age suspected of having MD; the birds were from poultry farms in three Indian states (Tamil Nadu, Karnataka, and Andhra Pradesh). These samples were collected from 10 birds per poultry farm. Between 6 and 10 feathers from wings were collected and kept on ice for transportation. In the lab, feather samples were stored at −20°C until use.
Extraction of DNA from feather tips.
The tips of the feathers (approximately 10 mm) with feather pulp were cut with sterile scissors and placed into a sterile 1.5-ml microcentrifuge tube (Eppendorf, Germany). The total DNA was extracted with a Charge switch DNA extraction kit (Invitrogen, USA), according to the manufacturer's instructions. The purity and concentration of the extracted DNAs were estimated with Biospectrometer (Eppendorf, Germany). The DNAs were stored at −20°C until use.
LAMP for MD viral genome.
The MEQ gene was selected as a target gene, as it is present only in the stereotype 1 of the MDV. LAMP primers were designed using Primer Explorer V4 software (Eiken Chemical Co., Ltd., Japan) and synthesized by Sigma-Aldrich India Pvt., Ltd., India. The six primers consisted of one pair each of the outer primers (F3 and B3), inner primers (FIP and BIP), and loop primers (LF and RF) (Fig. 1). The LAMP reactions (25 μl) were carried out with primers at concentrations of 10 pmol each of F3 and B3, 40 pmol each of FIP and BIP, and 20 pmol each of LF and RF. The mixture also contained a 10 mM final concentration of deoxynucleoside triphosphates (dNTPs), 8 mM MgSO4, 0.8 M betaine, 20 mM Tris-HCl, 10 mM (NH4)2SO4, 6 mM MgSO4, 0.1% Tween 20, 8 U of Bst DNA polymerase (New England BioLabs) and 2 μl template DNA, with nuclease-free water to make up the volume to 25 μl. The mixture was incubated at 60°C for 60 min in a water bath, and the reaction was terminated by incubating the tubes at 800°C for 5 min. The LAMP products were analyzed by 2% agarose gel electrophoresis with UV light transillumination and naked-eye observation of a color change after the addition of 2 μl of SYBR green I dye (1:1,000) (Invitrogen). The amplified products were also digested with MboI restriction enzyme (New England BioLabs). The digested LAMP products produced 110- and 70-bp fragments by electrophoresis on a 3% agarose gel.
Fig 1.

LAMP primer designs for the detection of serotype 1 MDV. (A) Locations of LAMP primers in relation to the defined regions of the target sequence. The outer primers (F3 and B3), inner primers (FIP and BIP), and loop primers (LF and LR) are complementary to the indicated regions of the MEQ gene. FIP and BIP contain two distinct sequences (F1c + F2 and B1c + B2, respectively). (B) The positions of the LAMP primers are shown relative to the serotype 1 MDV isolate (accession no. FJ620900).
Standard plasmid preparation.
A fragment (180 bp) of the MEQ gene of the MDV-1 field isolate (Ind-KA01-06) was amplified by PCR using the LAMP F3 and B3 primers and cloned into TA easy vectors (RBC, USA) (2,748 bp). The recombinant Escherichia coli strain carrying the recombinant plasmids was inoculated into Luria-Bertani broth (10 ml) and incubated at 37°C overnight with horizontal shaking. Plasmid DNAs were then extracted from the culture with a Spin miniprep kit (RBC, USA) and checked by sequencing. The recombinant plasmid (pMDV-1) was used as a standard for determining assay sensitivity.
PCR.
PCR amplification of the MEQ gene of serotype 1 MDV was performed with the LAMP F3 and B3 primers, and it would produce a 180-bp PCR amplicon in gel electrophoresis. The 20-μl reaction mixtures contained 10 μl of PCR master mix (Genei, Bangalore, India), 10 pmol of each primer, approximately 100 ng of template DNA, and nuclease-free water to make the final volume 20 μl. The PCR was as follows: initial denaturation at 94°C for 5 min, 30 cycles of 94°C for 30 s, 60°C for 45 s, and 72°C for 30 s, and a final extension at 72°C for 7 min. The PCRs were carried out using the PTC-200 (MJ Research), and the products were confirmed by 2% agarose gel electrophoresis.
Stability of the LAMP master mix.
The LAMP master mix was prepared by adding all the materials, including the buffer, dNTPs, primers, MgSO4, betaine, and Bst polymerase, and then storing the mix at −20°C. At 15-day intervals, the master mix was removed from the −20°C deep freezer and used for LAMP analysis, to detect the MEQ gene in the MD viral genome by adding the template DNA (pMDV-1).
RESULTS
Assay design.
The MEQ gene was selected as the target gene because it is unique to MDV-1 and present in all pathotypes of MDV-1. LAMP primers were designed, and the nucleotide sequences were screened with the BLAST program (www.ncbi.nlm.nih.gov) to confirm the presence of these sequences in all of the pathotypes of serotype 1 MDV. The sequences and their locations are shown in Fig. 1. Feather samples were chosen as a target tissue because they are easy to collect and a noninvasive method to detect the MD viral genome. The LAMP reaction was optimized by varying the ratio of inner (40 pmol each), outer (10 pmol each), and loop primers (20 pmol each), the concentrations of MgSO4 (8 mM) and dNTPs (10 mM), the amplification temperature (60°C), and the reaction time (60 min). A LAMP-positive reaction showed ladder like DNA bands of different sizes following 2% agarose gel electrophoresis.
Use of analytical sensitivity of LAMP to detect serotype 1 MD genome.
The recombinant plasmid containing the 180-bp MEQ gene fragment was used to compare the sensitivity of the LAMP assay with that of conventional PCR. Copy numbers of the plasmid DNA were calculated, and then it was serially diluted. Each dilution was used as a template for both LAMP and PCR methods. The results are shown in Fig. 2. The detection limit of the LAMP assay was 10 copies per reaction during a 60-min reaction, whereas the sensitivity of the conventional PCR was 100 copies per tube. Thus, the LAMP was 10-fold more sensitive than the conventional PCR.
Fig 2.

Comparison of the sensitivities of LAMP and PCR methods for the detection of the serotype 1 MD viral genome by 2% agarose gel electrophoresis. The plasmid containing the 180-bp MEQ gene fragment of the Ind/KA01/06 isolate was serially diluted. Each plasmid concentration (copies/tube) was subjected to both the LAMP and PCR assays. (A) LAMP assay specific to serotype 1 MDV. (B) PCR amplification of the 180-bp fragment of the MEQ gene from serotype 1 MDV. Lanes: M, 100-bp DNA marker (Genei, Bangalore, India); PC, positive control; NC, negative control.
Analytical specificity of LAMP.
To confirm the amplification of specific LAMP products, the LAMP product from the Ind/KA01/06 isolate was digested with MboI. It showed the specific products of 110-bp and 70-bp bands on agarose gel (Fig. 3). DNA from the SB1 vaccine (MDV serotype 2) and the HVT vaccine (MDV serotype 3) were not amplified by the LAMP primers (Fig. 3). DNA from healthy chicken feathers and nuclease-free water also was not amplified (data not shown).
Fig 3.
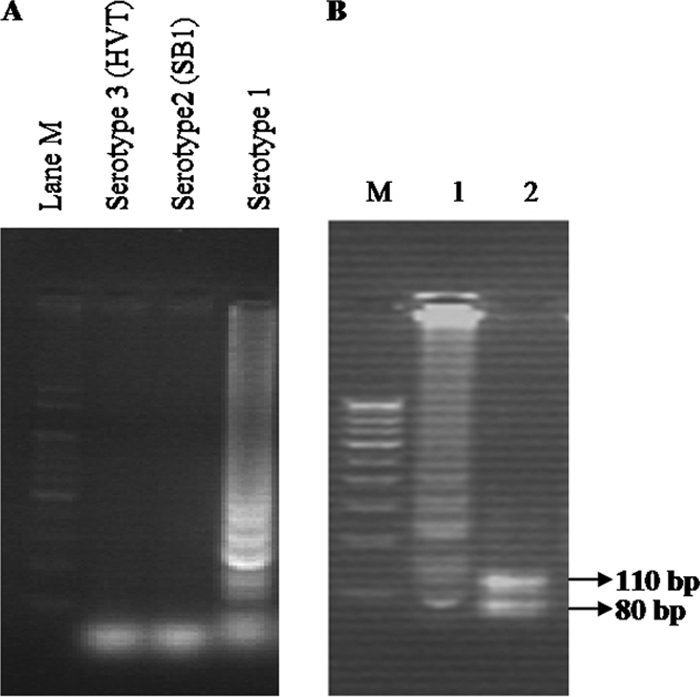
Specificity of the LAMP assay in the detection of serotype 1 MDV. (A) Agarose gel electrophoresis of different serotypes of MDV. Lanes: M, 100-bp DNA marker; Serotype 3 (HVT), DNA from HVT vaccine; Serotype 2 (SB1), DNA from SB1 vaccine; Serotype 1, DNA from Ind/KA01/06 isolate. (B) Restriction enzyme analysis of the LAMP products on 3% agarose gel electrophoresis. Lanes: M, 100-bp DNA marker; 1, undigested LAMP product; 2, MboI-digested LAMP product from the Ind/KA01/06 isolate.
Visual detection of LAMP products.
Apart from the gel electrophoresis, visual detection of LAMP products was also performed by adding SYBR green I dye to the terminated LAMP reaction. The LAMP reaction for positive field isolates fluoresced apple green, whereas the negative reactions for SB1 DNA, HVT DNA template, and healthy chicken DNA looked brown when visualized by the naked eye (Fig. 4).
Fig 4.

Visual detection of LAMP products for the detection of serotype 1 MDV. SYBR green I dye (1:1,000) (2 μl) was added to all the tubes after termination of the LAMP reaction. LAMP-positive reactions turned to apple green, while negative reactions remained brown. Tube 1, DNA from Ind/KA01/06 serotype 1 MDV isolate; tube 2, DNA from serotype 2 SB-1 vaccine; tube 3, DNA from serotype 3 HVT vaccine; tube 4, DNA from healthy chicken feather.
Stability of the LAMP master mix.
The stability of the LAMP master mix at −20°C was studied for 120 days. The results showed that the LAMP master mix could be stable for 90 days without any change in the ability to amplify the template DNA. At 120 days, there was a reduction in the intensity of the higher-molecular-weight band. Hence, the template DNA could be amplified until 90 days when stored at −20°C.
Detection of the MEQ gene of the MD viral genome from field samples.
Out of 101 samples screened, 15 samples analyzed for the presence of the MEQ gene of the MDV-1 viral genome were positive with LAMP. The LAMP results were compared to those from conventional PCR and virus isolation for selected positive samples. All the samples positive with LAMP were also positive with PCR, and MD-specific virus plaques could be identified in the field isolates Ind/KA01/06, Ind/AP02/09, Ind/TN01/06, Ind/TN11/07, Ind/TN14/08, and Ind/TN21/10.
DISCUSSION
Serotype 1 MDV infections pose a serious threat to the poultry industry and are responsible for significant financial losses throughout the world (13, 17). Thus, it is important to establish simple, rapid, and reliable methods for the diagnosis of serotype 1 MDV to prevent further disease transmission or outbreaks. At present, PCR is the diagnostic method used most widely. However, it requires the use of expensive equipment and costly consumables. The LAMP reaction is performed under isothermal conditions and does not require expensive equipment or costly kits. LAMP has been used widely for the detection of bacterial, viral, fungal, and parasitic pathogens of animals, plants, and humans. In poultry, LAMP has been developed for the detection of various viral pathogens, such as the reticuloendotheliosis virus (7), infectious bronchitis virus (5), infectious laryngotracheitis virus (18), infectious bursal disease virus (19), and avian influenza virus (6). Bst DNA polymerase has an optimal activity at 65°C. However, several reports have shown that this enzyme can amplify the target DNA template at lower temperatures in the LAMP reaction (11, 12). To use LAMP as a field test, the Bst polymerase should show equal amplification at a range of temperatures from 60 to 64°C, due to the variations in the precision of a water bath or dry heating blocks that are used to conduct the LAMP test.
In this study, the LAMP products could be detected at reaction times of 45, 60, and 90 min with the loop primers, whereas without loop primers it took 90 min to amplify the target template. Loop primers could accelerate the reaction because they hybridize to the stem-loops, except for those loops that are hybridized by the inner primers and prime strand displacement DNA synthesis (9). The loop primers could also provide higher specificity, as the six primers could recognize the eight distinct regions of the target DNA (11, 15).
The high sensitivity of the LAMP system makes it susceptible to false-positive results due to carryover or cross-contamination. Amplification and detection should therefore be carried out in separate working areas. In this study, to avoid carryover contamination, the LAMP master mix was prepared and showed that the LAMP master mix was stable for 90 days at −20°C. The LAMP master mix was tested with the field samples. Comparison of results showed that there was no false-positive reaction.
The detection limit of the MDV LAMP assay was determined by amplification of recombinant plasmid DNA, which carries the MEQ gene of serotype 1 MDV. The LAMP assay had a detection limit of 10 copies of the MEQ gene of serotype 1 MDV, whereas the PCR assay was able to detect 100 copies of the MEQ gene of MDV. The detection limit of the LAMP assay was 10-fold higher than that of conventional PCR. LAMP is also considered superior because it is a comparatively simple technique that can be carried out in most situations where rapid diagnosis is required, like in field conditions. A water bath or block heater is sufficient for DNA amplification because the method requires isothermal conditions. The high specificity and amplification capability of the LAMP method also allow for the easy and rapid visualization of the amplified products without the need for gel electrophoresis; this makes it a very simple and cheap diagnostic tool.
The MEQ gene was used as the target gene in the serotype 1 MDV LAMP assay because the MEQ gene is unique to the oncogenic strains of MDV located within the BamHI-I2 and L fragments. As a protein highly expressed in MDV-induced tumors and lymphoblastoid cells and present uniquely in oncogenic strains, MEQ is considered an important candidate gene for the diagnosis of serotype 1 MDV virus. The MDV LAMP assay described above did not amplify other serotypes of MDV, including SB-1 (serotype 2) and HVT (serotype 3), nor did the assay amplify the DNA of normal, healthy chicken. The MDV LAMP primer sequences were screened using BLAST (National Centre for Biotechnology Information [NCBI]), and it was observed that the primers could detect all the strains belonging to serotype 1, including the following: the mMDV CU2 strain (GenBank accession no. AY362708), the vMDV strains CVI988 and 567 (GenBank accession no. AF493555 and AY362709), the vvMDV strains 549 and RB1B (GenBank accession no. AY362714 and HM488349), and the VV+MDV strains 648a and L (GenBank accession no. AY362725 and AY362717). Moreover, these LAMP primers were also BLASTed against the complete genomes of other avian oncogenic and related viruses, which included reticuloendotheliosis virus (GenBank accession no. GQ375848), chicken anemia virus (GenBank accession no. AF311900), and avian leukosis-J virus (GenBank accession no. HM776937). These LAMP primer sequences were absent from these other viruses. Thus, this LAMP technique could be useful for the detection of serotype 1 MDVs.
The practical application of the LAMP assay was evaluated using 101 samples from three South Indian states. Both LAMP and PCR methods detected serotype 1 MDV in 15 samples, which was further confirmed by the appearance of typical plaques in CEF cells.
A major challenge is differentiating CVI988 and field MD virus/other virulent viruses in field samples because of the close sequence similarities between the two viruses. Although there is some experimental evidence to suggest that a single point mutation in the pp38 gene can be used for such differentiation, this approach has not been evaluated under field conditions. There are no methods available that can differentiate between these viruses in the field. In this paper, we have targeted the MEQ gene, which has a 178-bp insertion in CVI988. However, as it is only a tandem repeat of an existing sequence, it will not be a suitable target for PCR-based differentiation. Hence, the LAMP method described here will not differentiate CVI988 strain and field MD virus/virulent viruses. Nevertheless, we would expect that the titers of CVI988 in the feather are likely to be very low in a vaccinated bird compared to those of the field viruses. Additionally, any strongly positive sample with the LAMP assay is most likely to be one of the field viruses. However, the contribution of CVI988 to this result cannot be ruled out.
Considering the prevalence and economic impact of MD, the development of a simple, cost-effective, sensitive, and rapid diagnostic technique is important. The LAMP protocol described in this study represents a technique with these attributes that can be used under field conditions for the diagnosis or surveillance of serotype 1 MDV infection. This technique has considerable potential for routine diagnosis in less-equipped laboratories, as well as under field conditions.
ACKNOWLEDGMENTS
This work was supported by a grant received from the Department of Biotechnology, New Delhi, India (BT/PR9481/AAQ/01/344/2007).
We thank the Tamil Nadu Veterinary and Animal Sciences University in Chennai, India, for providing the facilities to carry out this work. We also thank Venugopal Nair (IAH, Compton, United Kingdom) for his valuable suggestions.
Footnotes
Published ahead of print 14 December 2011
REFERENCES
- 1. Baaten BJ, Butter C, Davison TF. 2004. Study of host-pathogen interactions to identify sustainable vaccine strategies to Marek's disease. Vet. Immunol. Immunopathol. 100:165–177 [DOI] [PubMed] [Google Scholar]
- 2. Baigent S, Davison F. 2004. Marek's disease virus: biology and life cycle, p 62–77 In Davison F, Nair V. (ed), Marek's disease, an evolving problem, 1st ed Elsevier Academic Press, Oxford, United Kingdom [Google Scholar]
- 3. Becker Y, et al. 1992. PCR detection of amplified 132 bp repeats in Marek's disease virus type 1 (MDV-1) DNA can serve as an indicator for critical genomic rearrangement leading to the attenuation of virus virulence. Virus Genes 3:277–287 [DOI] [PubMed] [Google Scholar]
- 4. Calnek BW. 2001. Pathogenesis of Marek's disease virus infection. Curr. Top. Microbiol. Immunol. 255:25–55 [DOI] [PubMed] [Google Scholar]
- 5. Chen HT, et al. 2010. Reverse transcription loop-mediated isothermal amplification for the rapid detection of infectious bronchitis virus in infected chicken tissues. Mol. Cell Probes 24:104–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen HT, et al. 2008. Development of reverse transcription loop-mediated isothermal amplification for rapid detection of H9 avian influenza virus. J. Virol. Methods 151:200–203 [DOI] [PubMed] [Google Scholar]
- 7. Deng X, et al. 2010. Development of reverse transcription loop-mediated isothermal amplification for rapid detection of reticuloendotheliosis virus. J. Virol. Methods 168:82–86 [DOI] [PubMed] [Google Scholar]
- 8. Handberg KJ, Nielsen OL, Jorgensen PH. 2001. The use of serotype 1- and serotype 3-specific polymerase chain reaction for the detection of Marek's disease virus in chickens. Avian Pathol. 30:243–249 [DOI] [PubMed] [Google Scholar]
- 9. Nagamine K, Hase T, Notomi T. 2002. Accelerated reaction by loop-mediated isothermal amplification using loop primers. Mol. Cell Probes 16:223–229 [DOI] [PubMed] [Google Scholar]
- 10. Notomi T, et al. 2000. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 28:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Parida M, et al. 2004. Real-time reverse transcription loop-mediated isothermal amplification for rapid detection of West Nile virus. J. Clin. Microbiol. 42:257–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Poon LLM, et al. 2004. Rapid detection of the severe acute respiratory syndrome (SARS) coronavirus by a loop-mediated isothermal amplification assay. Clin. Chem. 50:1050–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Raja A, et al. 2009. Detection of virulent Marek's disease virus in poultry in India. Acta Virol. 53:255–260 [DOI] [PubMed] [Google Scholar]
- 14. Silva RF. 1992. Differentiation of pathogenic and non-pathogenic serotype 1 Marek's disease viruses (MDVs) by the polymerase chain reaction amplification of the tandem direct repeats within the MDV genome. Avian Dis. 36:521–528 [PubMed] [Google Scholar]
- 15. Soliman H, El-Matbouli M. 2006. Reverse transcription loop-mediated isothermal amplification (RT-LAMP) for rapid detection of viral hemorrhagic septicaemia virus (VHS). Vet. Microbiol. 114:205–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. van Regenmortel MHV, et al. (ed). 1999. Virus taxonomy. Seventh report of the International Committee on Taxonomy of Viruses Academic Press, New York, NY [Google Scholar]
- 17. Venugopal N. 2005. Evolution of Marek's disease—a paradigm for incessant race between the pathogen and the host. Vet. J. 170:175–183 [DOI] [PubMed] [Google Scholar]
- 18. Xie QM, et al. 2010. Rapid detection of infectious laryngotracheitis virus isolates by loop mediated isothermal amplification. J. Virol. Methods 165:71–75 [DOI] [PubMed] [Google Scholar]
- 19. Xu J, et al. 2009. Development of reverse-transcription loop-mediated isothermal amplification for detection of infectious bursal disease virus. J. Virol. Methods 162:267–271 [DOI] [PubMed] [Google Scholar]
- 20. Zhu G-S, et al. 1992. Differentiation of oncogenic and nononcogenic strains of Marek's disease virus type 1 with polymerase chain reaction DNA amplification. Avian Dis. 36:637–645 [PubMed] [Google Scholar]