

Abstract
E2F transcription can lead to cell proliferation or apoptosis, indicating that E2Fs control opposing functions. In a similar manner, DNA double-strand breaks can signal to induce cell cycle arrest or apoptosis. Specifically, pRB is activated following DNA damage, allowing it to bind to E2Fs and block transcription at cell cycle promoters; however, E2F1 is simultaneously activated, leading to transcription at proapoptotic promoters. We examined this paradoxical control of E2F transcription by studying how E2F1's interaction with pRB is regulated following DNA damage. Our work reveals that DNA damage signals create multiple forms of E2F1 that contain mutually exclusive posttranslational modifications. Specifically, E2F1 phospho-serine 364 is found only in complex with pRB, while E2F1 phosphorylation at serine 31 and acetylation function to create a pRB-free form of E2F1. Both pRB-bound and pRB-free modifications on E2F1 are essential for the activation of TA-p73 and the maximal induction of apoptosis. Chromatin immunoprecipitation demonstrated that E2F1 phosphorylated on serine 364 is also present at proapoptotic gene promoters during the induction of apoptosis. This indicates that distinct populations of E2F1 are organized in response to DNA damage signaling. Surprisingly, these complexes act in parallel to activate transcription of proapoptotic genes. Our data suggest that DNA damage signals alter pRB and E2F1 to engage them in functions leading to apoptotic induction that are distinct from pRB-E2F regulation in cell cycle control.
INTRODUCTION
The mammalian cell division cycle is intricately regulated by the retinoblastoma protein (pRB) and its associated E2F transcription factors. In the G1 phase, pRB interacts with E2F1 to -4 and negatively regulates transcription of cell cycle gene promoters that contain E2F binding sites (6). When cells are stimulated to proliferate, pRB is phosphorylated and E2Fs are released, activating expression of E2F target genes and stimulating the advancement of the cell cycle. While this simple model of cell cycle control has dominated our view of pRB and E2F function, more specialized aspects of their function are also beginning to emerge.
One function that has been suggested to set E2F1 apart from other E2F family proteins is its ability to induce apoptosis (13). In ectopic expression studies, E2F1 is the most potent E2F for inducing apoptosis (10). It has also been suggested that ectopic E2F1 may not induce cell cycle entry in primary cells unless accompanied by mutations that disrupt pRB or p53-p21 signaling (32). Furthermore, E2F1 may be required for apoptotic induction by other E2F family proteins (28). Gain-of-function studies have revealed a role for E2F1 in apoptosis, where it begins by escaping pRB transcriptional regulation (17, 26). E2F1's best known proapoptotic targets are the TA-p73 promoter and the INK4A promoter, which is specific for the ARF gene product (3, 20, 26). As a member of the p53 family, TA-p73 is capable of inducing expression of many proapoptotic genes to induce cell death (41). Similarly, increased expression of ARF blocks MDM2 from degrading p53 and also leads to cell death (13). In this way E2F1 can induce both p53-dependent and -independent pathways to apoptosis. E2F1's unique ability to activate transcription from these proapoptotic promoters relies on the specificity of recognition for the promoter, and it also has been shown that E2F1 can utilize the interactions through its marked box with specialized adapter proteins such as Jab1 to induce transcription of proapoptotic targets (14, 26, 40). These experiments have provided important insight into E2F1's mechanism of apoptotic induction; however, since they rely on overexpression, it is unclear how E2F1 can escape pRB regulation at endogenous levels. It is also unclear from this work what physiological circumstances activate E2F1 to use its proapoptotic activities.
A physiological stimulus that activates E2F1 independently of cell cycle control is DNA damage. DNA damage leads to the stabilization of E2F1 expression similarly to p53 (4, 16). This implies that DNA damage can activate E2F1 to participate in an apoptotic signaling pathway. In response to double-strand DNA breaks, E2F1 is phosphorylated on serine residues 31 and 364, acetylated on lysines 117, 120, and 125, and demethylated at lysine 185 (27, 29, 40, 43). Cells deficient for E2F1 are resistant to DNA damage-induced apoptosis, revealing that it has an essential role in this cell death pathway, although its importance may be greater in p53 mutant cells (21, 29, 33, 40, 47). A complicating factor in the interpretation of E2F1's role in DNA damage responsiveness is pRB. While DNA damage can induce cell death, it also leads to a pRB-dependent cell cycle arrest (5, 15). The RB protein is capable of responding to DNA damage and blocking the cell cycle in both G1 and S phases (2, 5, 15, 24, 25). This arrest includes the dephosphorylation of cyclin/cyclin-dependent kinase (CDK) sites on pRB, followed by the formation of repressive pRB-E2F complexes that block transcription of cell cycle-regulated genes (2, 5, 25). Intriguingly, pRB is also subject to posttranslational modifications in response to double-strand DNA breaks, as it is acetylated at lysine 874, methylated on lysines 810, 860, and 873, and phosphorylated at serine 612 (7, 19, 35, 38, 42). Thus, pRB and E2F1 are highly regulated as part of the cellular response to DNA damage, but it is difficult to understand how their opposing functions in cell cycle control and apoptosis can be managed simultaneously.
Studies that have evaluated posttranslational modifications on pRB and E2F1 in response to DNA breaks have drawn little consensus. It has been suggested that acetylation of pRB releases E2F1 to induce cell death, leaving in question how pRB interacts with other E2Fs under these circumstances (35). Conversely, it has been reported that phosphorylation of pRB at serine 612 induces complex formation with E2F1 to maintain cell viability (19). Lastly, it has also been proposed that pRB and E2F1 form a complex in response to DNA damage to activate transcription of proapoptotic target genes (18). Independent of its response to DNA damage, pRB is also capable of forming two distinct types of interaction with E2F1, one that is disrupted by cyclin-dependent kinase phosphorylation and one that is not, indicating that regulation of their interaction is likely more complex than for other E2Fs (8, 9, 11, 23). Taking these findings together, there are many possibilities for how pRB and E2F1 can interact and function in the DNA damage response, but a cohesive mechanism has yet to emerge.
We have studied pRB-E2F1 interactions following DNA damage and determined that distinct pRB-“free” E2F1 and pRB-E2F1 complexes form in response to DNA double-strand breaks. Using a combination of mutational analysis to block addition of posttranslational modifications to E2F1, as well as modification specific antibodies, we have determined that serine 31 phosphorylation and lysine 117, 120, and 125 acetylation on E2F1 contribute to the formation of a “free” form of E2F1 that does not bind pRB. Furthermore, we demonstrate that serine 364 phosphorylation of E2F1 is a component of a unique complex of hyperphosphorylated pRB and E2F1 in response to DNA damage. Reconstitution of E2f1−/− 3T3 cells with human E2F1 mutants reveals that posttranslational modifications that characterize separate populations of E2F1 are necessary for maximal activation of transcription at proapoptotic target genes and for the induction of apoptosis in response to DNA damage. Despite its critical role in DNA damage-dependent apoptosis, serine 364 on E2F1 is found only in primates, indicating that this aspect of E2F1 function is unique to their DNA damage response.
MATERIALS AND METHODS
Cell culture.
Human U2OS cells and primary IMR90 fibroblasts were obtained from the American Type Culture Collection. Phoenix-Eco cells were obtained from the National Gene Vector Laboratory Biorepository (Stanford University Medical Center). E2f1−/− 3T3 cells were generated using standard methods (46). Briefly, E2f1−/− fibroblasts were plated at a density of 1 × 106 cells per 10-cm dish and subcultured at the same density every 3 days. Immortal E2f1−/− 3T3 cells were used between passages 20 and 25. All cells were cultured in growth medium that contained Dulbecco modified Eagle medium (DMEM) supplemented with l-glutamine, streptomycin, penicillin, and 10% fetal bovine serum. Cells were maintained in a humidified chamber at 37°C with 5% CO2. Cells were treated with etoposide at the concentrations and times described in the figure legends. All untreated controls received dimethyl sulfoxide (DMSO) vehicle only, for the same time period.
Plasmid constructs.
CMV-HA-E2F1, CMV-HA-DP1, and CMV-β-Gal have been described previously (11). CMV-HA-E2F1 constructs with K117R, K120R, K125R (KR), and S31A substitutions and the -DM combination were gifts of A. Gulino and M. Levrero (University of Rome) (40).
The E2F1 S364A allele was constructed by PCR mutagenesis using previously described methods (12). Inserting the BglII-NotI S364A mutant fragment into the same sites of CMV-HA-E2F1-DM created the E2F1-TM mutant. pBABE-HA-E2F1 and corresponding mutants were constructed by transferring the respective HA-E2F1 cDNAs into the pBABE-puro vector. The pMOV-ψ plasmid carrying the Gag, Pol, and Env genes was described by Mann et al. and was a gift from M. Golding (Texas A&M) (34).
Recombinant proteins.
Previously reported glutathione S-transferase (GST)-tagged wild-type and mutant pRB large-pocket (RBLP) fragments were inserted into the pSCodon1 plasmid, and proteins were expressed using the Staby Codon T7 system (Delphi Genetics, Belgium) (23). LB-Amp cultures were grown at room temperature and induced, and proteins were purified from extracts using standard procedures. All proteins were eluted with glutathione and dialyzed overnight against a 1,000× excess of buffer, and the final protein was subjected to SDS-PAGE to determine quantity and purity.
Transfections and infections.
U2OS cells were plated at 1 million to 2 million cells per 10-cm dish at 24 h prior to transfection and refed fresh growth medium at 3 h prior to transfection by calcium phosphate precipitation. Cells were transfected with 1 μg of CMV-HA-E2F1, 1 μg of CMV-HA-DP1, and 20 μg of CMV-β-Gal plasmid for 16 h. The following day, cells were refed with fresh medium and collected at 48 h posttransfection.
E2f1−/− 3T3 cells were retrovirally transduced at passage 21. Briefly, Phoenix-Eco retroviral packaging cells were plated at a density of 11 million cells per 15-cm dish in 20 ml of medium 1 day before transfection. At 4 h pretransfection, cells were refed with 20 ml of fresh medium. Phoenix-Eco cells were transfected with 45 μg of pBABE-puro vectors along with 30 μg of pMOV-ψ. At 12 h posttransfection cells were refed with fresh medium, and viral supernatants were collected at 48 and 60 h posttransfection. Viral supernatants were filtered, supplemented with Polybrene, and added to E2f1−/− 3T3 cells. Each infection was allowed to take place for 12 h. After two rounds of viral infection, fresh medium containing 4 μg/ml puromycin was added and left for 4 days to select infected cells. Cells were then passaged once, after which they were plated and used for functional assays.
Protein binding assays.
For GST pulldown assays, either transiently transfected or untransfected cells were harvested and nuclear extracts were prepared (8). Extracts were diluted in low-salt GSE buffer (20 mM Tris [pH 7.5], 200 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 1 mM dithiothreitol [DTT], and 0.1% NP-40) and incubated with GST or GST-RBLP proteins. Protein complexes were collected with 20 μl of bead slurry for 1 h and eluted in 1× Laemmli buffer.
Quantitative immunoprecipitation (IP) experiments were performed on 5 mg of U2OS nuclear extract in 1.5 ml. Extracts were precleared with 50 μl of protein G Dynabeads prebound to 4 μg of nonspecific rabbit IgG. The cleared supernatant was incubated with 200 μl of protein G Dynabeads prebound to 1 mg/ml of purified anti-pRB (C15) rabbit polyclonal antibody overnight at 4°C. The next morning the supernatant was removed and reincubated with 80 μl of protein G Dynabeads prebound to 1 mg/ml of purified anti-pRB (C15) rabbit polyclonal antibody for 2 h at 4°C, and the initial 200 μl of antibody-bead complexes was collected and stored on ice. After the second depletion of pRB complexes, the supernatant was removed and incubated with 200 μl of protein G Dynabeads prebound to 1 mg/ml of anti-E2F1 (C20) rabbit polyclonal antibody (Santa Cruz) for 8 h at 4°C. Finally, all antibody-bead complexes were collected, washed in low-salt GSE buffer, and eluted in 1× Laemmli buffer. Supernatant fractions were mixed with 5× Laemmli buffer so that equivalent total volumes could be analyzed side by side using SDS-PAGE and Western blotting.
Nonquantitative immunoprecipitations were performed as described above but with the following modifications: 2 mg of nuclear extract was used, 2 μg of antibody was prebound to 50 μl of protein G Dynabeads, immunoprecipitations were performed overnight at 4°C, and elution was in 50 μl of sample buffer. A nonspecific rabbit IgG immunoprecipitation was used in parallel in equal quantities as a negative control for all immunoprecipitation experiments.
Protein detection.
Western blotting was carried out using standard methods. All antibodies were used at a dilution of 1/1,000 of the stock in blocking buffer (1% nonfat milk or 5% bovine serum albumin [BSA] in the case of anti-acetyl-lysine and anti-E2F1 pSer364) for Western blots. The antibodies used for immunoprecipitations were rabbit anti-pRB (C15) produced and purified against a C-terminal 15-amino-acid peptide from human pRB, rabbit anti-E2F1 pSer364 (Rockland 18434), and rabbit anti-E2F1 (C20). Antibodies used for Western blots were mouse anti-E2F1 KH20 (Santa Cruz), high-affinity rat antihemagglutinin (anti-HA) (Roche 11867423001), mouse anti-pRB G3-245 (BD Pharmingen), rabbit anti-p53 pSer15 (Cell Signaling 9284), rabbit anti-E2F1 pSer364 (Rockland 18434), mouse anti-acetyl-lysine (Millipore 05515), mouse anti-pRB pSer612 clone 4E4 (CY-M1012; Cyclex), mouse anti-p53 monoclonal antibody (MAb) (Ab1; Oncogene Research Products), mouse anti-TopBP1 (BD Transduction Laboratories 611874), rabbit anti-β-actin (Sigma A2066), and mouse anti-lamin A/C (Chemicon International 3211).
Chromatin immunoprecipitation (ChIP) and real-time PCR.
For expression analysis, cells were harvested following treatment, and RNA was extracted using TRIzol reagent as per the manufacturer's recommended protocol (Invitrogen). Quantitative real-time PCR was performed using the QuantiFast SYBR green reverse transcription-PCR (RT-PCR) kit as per the manufacturer's instructions (Qiagen). Relative expression was normalized to a β-actin control and is the average from three independent experiments. RNA was amplified by PCR using primers specific to the mRNA sequences of mouse cyclin A2 (GAGAATGTCAACCCCGAAAA and GCAGTGACATGCTCATCGTT), cyclin E1 (CCTCCAAAGTTGCACCAGTT and CACCCGTGTCGTTGACATAG), TA-p73 (CTTCGAGCACCTGTGGAGTT and TGCTGAGCAAATTGAACTGG), caspase-7 (TTTGCTTACTCCACGGTTCC and GAGCATGGACACCATACACG), and β-actin (CTGTCGAGTCGCGTCCACCC and ACATGCCGGAGCCGTTGTCG).
ChIP assays were performed as described previously (1, 44), using rabbit anti-E2F1 pSer364 and rabbit anti-E2F1 (C20) antibodies and 3 × 107 cells per immunoprecipitation. DNA from precipitated complexes was amplified by quantitative PCR (qPCR) using iQ SYBR green master mix (Bio-Rad) and primers specific to the promoter regions of human TA-p73 (TGAGCCATGAAGATGTGCGAG and GCTGCTTATGGTCTGATGCTTATG) (49) and human GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (GATGGTGATGGGATTTCC and TCTGGTAAAGTGGATATTGTT) (45).
Cell cycle, growth, and cell viability assays.
Bromodeoxyuridine (BrdU) incorporation and cell cycle analysis were performed using standard techniques. Briefly, cells were pulse-labeled with BrdU, fixed, and stained for BrdU incorporation and with propidium iodide (PI). The proportion of cells in each phase of the cell cycle was determined by flow cytometry on a Beckman-Coulter Epics XL-MCL instrument (22). For growth curves, cells were plated in triplicate at a density of 5 × 104 cells per well in 6-well tissue culture dishes. Every 24 h the cells were trypsinized, resuspended in 1× phosphate-buffered saline (PBS), and counted using a hemocytometer. Cell viability was quantitatively measured using the alamarBlue cell viability reagent as per the manufacturer's instructions (Invitrogen). Cells were plated at a density of 5,000 cells in 100 μl medium per well of a 96-well dish. One day later, cells were treated with DMSO or etoposide. At 20 h posttreatment, alamarBlue was added to each well, and after a 6-h incubation, fluorescence was measured at 570 nm using a Wallac Victor2 1420 Multilabel counter. Cell viability measurements were in comparison to an untreated control and were represented as the average from three independent experiments. Apoptosis was quantitatively measured using the Apo-ONE homogeneous caspase-3/7 assay (Promega). Cells were plated and treated as described above. At 24 h posttreatment, cells were lysed using the 2× Apo-ONE caspase-3/7 reagent and agitated on a shaker for 1 h. Supernatants were transferred to a white 96-well plate, and the cleavage of the profluorescent caspase-3/7 substrate Z-DEVD-R110 was quantitated using an excitation wavelength of 499 nm and measuring emittance at 521 nm using a fluorescent plate reader (Biotek Synergy H4 hybrid reader). Fluorescence of the caspase-3/7 substrate was represented as the average from three independent experiments.
RESULTS
Distinct E2F1 and pRB-E2F1 complexes form following DNA double-strand breaks.
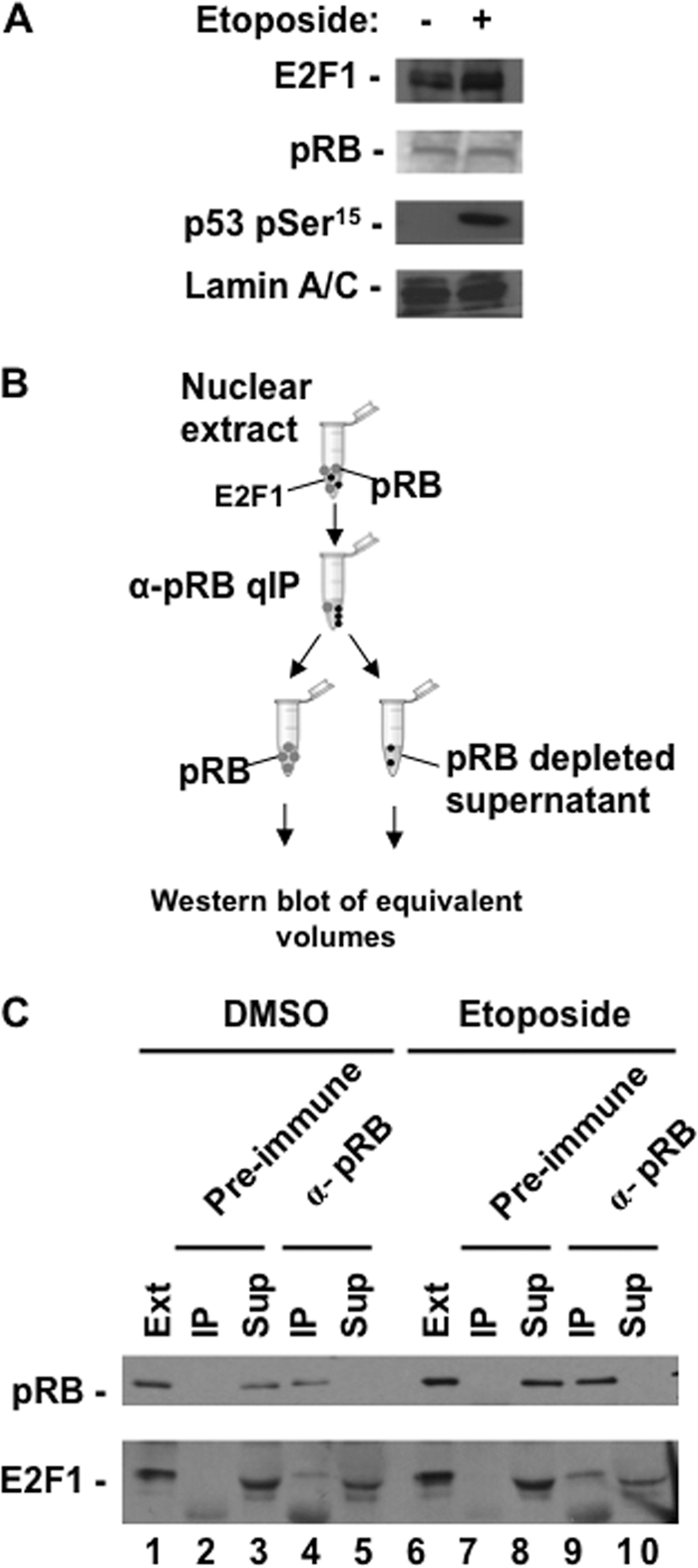
The complexity of possible interactions and functions for pRB and E2F1 following DNA double-strand breaks motivated us to investigate their interactions in response to this stimulus. For these experiments we utilized the U2OS cell line, which is p53 positive and is known to induce higher levels of E2F1 in response to DNA double-strand breaks. We chose a topoisomerase II inhibitor, etoposide, to induce double-strand DNA breaks in our experiments, and we confirmed its effects by Western blotting for the appearance of p53 phosphoserine 15 (pSer15). Figure 1A shows expression levels of E2F1 and pRB in control and etoposide-treated cultures, demonstrating elevated expression of E2F1. We next performed immunoprecipitations to fractionate pRB-E2F1 complexes from unbound E2F1 following DNA damage. In this experiment, pRB was quantitatively precipitated from nuclear extracts and E2F1 was in turn quantitatively precipitated from the supernatant (Fig. 1B). Pellets were resuspended in an equal volume of buffer as the supernatant, allowing equivalent amounts of all fractions to be analyzed by Western blotting for E2F1 and pRB levels (Fig. 1C). This experiment indicates that in response to DNA double-strand breaks, the abundance of E2F1 in pRB-bound complexes increases (Fig. 1C, compare E2F1 in lanes 3 and 6), but a free form of E2F1 also remains (Fig. 1C, lane 7). Importantly, we have accounted for all pRB and E2F1 in these fractions because a control nuclear protein, p53 pSer15, is readily detected in the supernatant when pRB and E2F1 are not (Fig. 1C, lane 8). Using GST-E2F1 as a standard, we estimated that the abundance of pRB-bound E2F1 following DNA damage was approximately 5 times that of free E2F1 (Fig. 1D, compare E2F1 in lanes 2 and 3). To rule out “free” E2F1 as an artifact of purification, we also utilized it in binding assays with GST-RB large pocket (GST-RBLP). Figure 1E reveals that “free” E2F1 in control extracts is capable of binding pRB, whereas the “free” form following DNA damage is resistant to this interaction (Fig. 1E, compare lanes 4 and 6). Taken together, these experiments indicate that pRB-E2F1 complexes become more abundant in response to DNA damage and a separate population of E2F1 becomes resistant to pRB binding. Thus, distinct populations of E2F1 are created following DNA damage. We refer to them as pRB-E2F1 complexes and pRB-free E2F1 throughout this report.
Fig 1.

DNA damage signaling generates two distinct populations of E2F1. (A) U2OS cells were treated with 100 μM etoposide or DMSO for 8 h. Relative expression levels of pRB, E2F1, p53 pSer15, and lamin A/C in U2OS nuclear extracts were determined by Western blotting. (B) Schematic diagram of quantitative immunoprecipitation (qIP) method. All pRB-containing complexes are first removed from nuclear extracts by immunoprecipitation, followed by all unbound E2F1, and lastly depleted supernatant is retained as a control for the quantitative nature of the IP. (C) Nuclear extracts were successively precipitated with antibodies against pRB and E2F1 as shown in panel B. Fractions were assayed for pRB, E2F1, and p53 pSer15 levels by Western blotting. (D) Quantitative immunoprecipitation fractions from the DNA-damaged nuclear extracts in panel C were analyzed by SDS-PAGE alongside a range of recombinant GST-E2F1 protein standards. E2F1 Western blot analysis was used to compare pRB-free E2F1 and pRB-E2F1 protein quantities. The asterisk indicates a nonspecific band. (E) pRB was quantitatively immunoprecipitated from untreated and etoposide-treated extracts. Fractions were Western blotted for pRB levels to ensure its depletion. Untreated and treated supernatants were normalized for pRB-free E2F1 levels and used in GST pulldown experiments. Input levels of E2F1, as well as E2F1 from GST-RBLP and GST-only pulldowns, were detected by Western blotting.
We also investigated the organization of pRB and E2F1 interactions following etoposide-induced DNA damage in IMR90 cells. We observed a similar increase in E2F1 expression (Fig. 2A) and utilized a similar quantitative immunoprecipitation experiment to assess pRB-E2F1 interactions following DNA damage (Fig. 2B). As before, the abundance of E2F1 found in complex with pRB increases following DNA damage (Fig. 2C, compare E2F1 in lanes 4 and 9), but a population of pRB-free E2F1 is also evident in IMR90 nuclear extract (Fig. 2C, lane 10). These analyses of pRB-E2F1 complexes following DNA damage from different cell lines suggest that multiple forms of E2F1 organize in response to double-strand DNA breaks.
Fig 2.
pRB-E2F1 interactions in IMR90 cells following DNA damage. (A) IMR90 cells were treated with 100 μM etoposide for 8 h, and nuclear extracts were prepared. Western blots display the levels of E2F1 and pRB in response to DNA damage. Blots of p53 pSer15 and lamin A/C control for DNA damage treatment and loading, respectively. (B) Schematic diagram of quantitative immunoprecipitation (qIP) method. All pRB-containing complexes are first removed from nuclear extracts by immunoprecipitation, and depleted supernatant is retained as a control for the quantitative nature of the IP. The immunoprecipitated fraction is resuspended in an volume equivalent to that of the supernatant and used in Western blot analysis. (C) Anti-pRB and anti-E2F1 Western blots demonstrate their respective levels in each fraction from quantitative immunoprecipitations of untreated and etoposide-treated cells.
Serine 31 phosphorylation and lysine acetylation are required to form a pRB-free fraction of E2F1.
The data in Fig. 1E indicate that a portion of E2F1 is modified by DNA damage signaling to be resistant to pRB binding. To understand the molecular basis for this change, we developed an assay for E2F1 binding following DNA damage (Fig. 3). GST-RBLP was used to precipitate endogenous E2F1 from whole-cell lysates derived from DNA-damaged cells or untreated controls. These experiments revealed that elevated E2F1, expressed in response to DNA damage (Fig. 3A, lane 6), was largely unable to bind to GST-RBLP, but a basal level of E2F1 binding to GST-RBLP was evident (Fig. 3A, lanes 8 and 9). In this regard it recapitulated the endogenous behavior of pRB and E2F1 following DNA damage, in which some E2F1 binds pRB while a portion becomes resistant. Similarly, normalizing the levels of input E2F1 between untreated and etoposide-treated extracts (Fig. 3B, lanes 1 and 2) allowed for a simpler comparison of GST-RBLP binding by E2F1 in this assay (Fig. 3B, compare lanes 4, 5, and 7 with lanes 9, 10, and 12). Furthermore, since pRB is capable of interacting with E2F1 through either of two distinct configurations, called the “general” and “specific” interactions, in our experiments we used mutant GST-RBLP proteins that would allow us to distinguish whether E2F1's affinity was altered toward either interaction (23). In this way, not only can we detect if E2F1 can bind wild-type pRB following DNA damage (Fig. 3B, compare lanes 7 and 12), but we can also determine if there is an effect on the general interaction (Fig. 3B, compare lanes 5 and 10) or the specific interaction (Fig. 3B, compare lanes 4 and 9) in isolation.
Fig 3.

Development of GST-RBLP binding assays to investigate pRB-free E2F1 following DNA damage. (A) U2OS cells were treated with 100 μM etoposide for 8 h, and whole-cell extracts were prepared. Equal quantities of extract were mixed with the indicated GST-RBLP fusion proteins before they were precipitated on beads and analyzed by SDS-PAGE and Western blotting. Input levels of the various GST proteins are shown by SDS-PAGE and Coomassie blue staining. (B) Extracts were prepared as described above, but this time the input amount used in GST pulldown experiments was normalized to E2F1. For simplicity, this type of experiment was used for Fig. 4.
We used this assay to investigate the effects of known modifications that are added to E2F1 in response to double-strand breaks. Figure 4A shows a schematic diagram of the mutant E2F1 constructs used in this study that eliminate sites of modification individually or in groups. To investigate their effects on DNA damage signaling and pRB-E2F1 interactions, U2OS cells were cotransfected with expression vectors for HA-E2F1 and HA-DP1 or for HA-E2F1-TM and HA-DP1. Subsequently, transfected cell cultures were equally divided and either treated with etoposide as before or processed in parallel as an untreated control. Whole-cell extracts were prepared and analyzed by SDS-PAGE and Western blotting (Fig. 4B). Samples were analyzed for both exogenous HA-tagged E2Fs and total E2F1 expression levels. Blots probed with E2F1 antibodies reveal that the exogenously introduced HA-E2F1 proteins provide a modest increase in total E2F1 levels (Fig. 4B, compare E2F1 in lanes 1 and 3). Because exogenously introduced wild-type HA-E2F1 is stabilized by DNA damage (Fig. 4B, compare HA-E2F1 in lanes 3 and 4), this suggests that this transfection system allows us to modify exogenous HA-E2F1 proteins expressed at this level or lower levels and study their effects on pRB interaction.
Fig 4.

Serine 31 phosphorylation and lysine acetylation of E2F1 are necessary to form a pRB-free fraction of E2F1. (A) Schematic diagram of HA-E2F1 expression vectors used in this study. Substitutions of serine to alanine and/or lysine to arginine found in each construct are shown. The corresponding name for each mutant is also shown to the left. (B) U2OS cells were cotransfected with expression vectors for HA-E2F1 and HA-DP1 or for HA-E2F1-TM and HA-DP1. Cultures were equally divided, and one was treated with 100 μM etoposide for 8 h and the other with DMSO as a control. Relative levels of exogenous HA-E2F1, HA-DP1, total E2F1, p53 pSer15, and β-actin were determined by Western blotting. (C) Whole-cell extracts from treated and untreated controls were normalized for E2F1 levels. Normalized extracts were mixed with the indicated GST fusion proteins, coprecipitated on beads, and detected by Western blotting for the HA epitope on exogenous HA-E2F1 and HA-E2F1-TM. (D) U2OS cells were transfected with HA-E2F1, HA-E2F1-TM, HA-E2F1-KR, HA-E2F1-S31A, or HA-E2F1-S364A expression vectors along with HA-DP1. Cultures were split and each half treated with DMSO or 100 μM etoposide for 8 h. Relative expression levels of HA-E2F1 proteins, p53 pSer15, and β-actin from whole-cell lysates were determined by Western blotting. (E) Whole-cell extracts from panel D were normalized for E2F1 levels, mixed with the indicated GST fusion proteins, and coprecipitated on beads, and E2F1 was detected by Western blotting for HA. (F) Etoposide-treated extracts and GST-RBLP-bound E2F1 were Western blotted to detect total levels of E2F1 and E2F1 pSer364.
Figure 4C shows a GST pulldown experiment using U2OS cell extracts that were normalized for HA-E2F1 levels from control and etoposide-treated cells (Fig. 4C, lanes 1 and 2). This revealed loss of interaction between wild-type HA-E2F1 and the different GST-RB constructs in response to DNA damage, as E2F1 that is capable of binding GST-RBLP is not visible at this level of film exposure. In contrast all HA-E2F1-TM remained capable of binding pRB following DNA damage. This implicates known posttranslational modifications on E2F1 in regulating its ability to contact pRB at either the “general” or “specific” binding sites in response to double-strand DNA breaks.
To identify individual sites of modification on E2F1 responsible for blocking pRB-E2F1 interactions, we repeated this assay using HA-E2F1 mutants that individually eliminate posttranslational modifications. Figure 4D demonstrates the expression levels of each mutant relative to the wild type in the input extracts and reveals that they were lower than that of the induced wild-type control (Fig. 4D, compare HA-E2F1 in lane 2 with lanes 3 to 10). GST-RBLP pulldown experiments using these extracts showed that mutation of either S31 or K117, 120, and 125 allowed E2F1 to bind to GST-RBLP following DNA damage (Fig. 4E). Conversely, HA-E2F1-S364A showed loss of interaction with pRB following DNA damage like the wild type, suggesting that phosphorylation at serine 364 is not involved in disrupting E2F1-pRB interactions. In addition, probing blots of E2F1 precipitated from etoposide-treated cells indicates that E2F1 phosphoserine 364 is actually enriched in the low levels of E2F1 that bind GST-RBLP following DNA damage (Fig. 4F).
Taken together, these experiments suggested that no individual modification on E2F1 affects “general” or “specific” pRB-E2F1 interactions in isolation. Instead, S31 and K117, 120, and 125 are involved in modifications that contribute to creating a pRB-free form of E2F1. A mechanism by which these modifications release a portion of the E2F1 population from pRB control is proposed in Discussion.
Serine 364 phosphorylation is associated exclusively with pRB-E2F1 complexes.
In order to better understand the role of posttranslational modifications on pRB and E2F1 in regulating their interactions, we also performed IP and Western blot analysis to detect phosphoserine 364 on E2F1 (henceforth referred to as E2F1 pSer364) and other posttranslational modifications on pRB and E2F1. For Fig. 5A we investigated the abundance of E2F1 pSer364 in response to DNA damage. We normalized E2F1 levels in nuclear extracts from untreated and etoposide-treated cells, precipitated E2F1 pSer364, and Western blotted for E2F1 (Fig. 5A). This experiment confirmed that this modification is induced by DNA double-strand breaks, as previously reported (43). We then normalized pRB-E2F1 and pRB-free E2F1 fractions for E2F1 levels and blotted for E2F1 pSer364. Strikingly, this modification is found exclusively with pRB-E2F1 complexes following DNA damage (Fig. 5B). This is consistent with experiments shown in Fig. 4F, as low levels of E2F1 bound to GST-RBLP following DNA damage were also highly enriched for E2F1 pSer364.
Fig 5.

Mutually exclusive posttranslational modifications on E2F1 complexes following DNA damage. (A) U2OS cells were treated with 100 μM etoposide for 8 h, after which nuclear extracts were prepared and normalized to E2F1 levels (shown in the upper panel). E2F1 was immunoprecipitated with anti-E2F1 pSer364 antibodies and detected by Western blotting. (B) Nuclear extracts from etoposide-treated cells were quantitatively immunoprecipitated to produce pRB-E2F1 and pRB-free E2F1 fractions. IP samples were normalized with respect to E2F1 levels and Western blotted for pRB, E2F1 pSer364, and total E2F1. (C) Nuclear extracts from etoposide-treated cells were immunoprecipitated with the indicated antibodies, and samples were normalized with respect to E2F1 levels. Western blots for pRB, E2F1 pSer364, and total E2F1 are shown. (D) Nuclear extracts from etoposide-treated cells were immunoprecipitated with the indicated antibodies and Western blotted using an anti-pRB pSer612-specific antibody and an anti-E2F1 pSer364 antibody. (E) Nuclear extracts from etoposide-treated cells were immunoprecipitated with the indicated antibodies, and IP samples were normalized with respect to total E2F1. Western blots for pRB, acetylated lysine, E2F1 pSer364, and total E2F1 were used to assay the posttranslational modifications present on both pRB and E2F1 in response to DNA damage. ppRB represents hyperphosphorylated pRB, and pRB represents hypophosphorylated pRB; they were determined by relative migration in SDS-PAGE. (F) Extracts from etoposide-treated cells were quantitatively precipitated with anti-pRB antibodies, and the precipitate and supernatant were blotted for TopBP1, pRB, and E2F1 to determine their presence in these fractions. (G) Model summarizing posttranslational modifications implicated in the distinct pRB-E2F1 complex characterized in this figure.
To better determine the abundance of complexes containing E2F1 pSer364 and pRB, we selectively immunoprecipitated E2F1 pSer364 and total E2F1 and compared the levels of pRB found in each (Fig. 5C). This reveals that the most prevalent form of pRB-E2F1 complex that is assembled in response to DNA damage contains E2F1 pSer364. When considered together with the abundance of pRB-E2F1 versus pRB-free E2F1 determined in Fig. 1D, this indicates that pRB-E2F1 complexes containing E2F1 pSer364 account for a substantial proportion of E2F1 following DNA damage. This modification is added to E2F1 by Chk2, and it is also known that pRB is phosphorylated by Chk2 in response to DNA damage (19). For this reason, we sought to investigate if these Chk2 phosphorylation sites are coincidentally modified and if this could explain the exclusivity of E2F1 pSer364 with pRB. Comparison of phosphoserine 612 on pRB (henceforth called pRB pSer612) in anti-E2F1 and anti-E2F1 pSer364 immunoprecipitates demonstrates that pRB pSer612 is largely excluded from interactions with E2F1 pSer364 but is readily detected in immunoprecipitates of total E2F1 (Fig. 5D, compare lanes 2 and 3). Similarly, immunoprecipitation of pRB pSer612 does not yield E2F1 pSer364 by Western blotting (Fig. 5D, compare lanes 3 and 4). This is highly suggestive that phosphorylated serine 364 is an important component of pRB-E2F1 complexes following DNA damage and that its abundance and exclusive nature are unlikely to be a coincidence.
We further compared equal quantities of total E2F1 and E2F1 pSer364 obtained from immunoprecipitation (Fig. 5E). Western blot analysis of these fractions revealed that hyperphosphorylated pRB predominates in E2F1 pSer364-containing complexes (Fig. 5E, pRB blot, lane 2). This is striking because pRB in total E2F1 IPs was largely dephosphorylated at this stage following DNA damage (Fig. 5E, pRB blot, lane 3). This suggests that hyperphosphorylated pRB-E2F1 pSer364 complexes exist through the specific interaction configuration, as this is the only way for E2F1 to bind to hyperphosphorylated pRB (8). These experiments also revealed that acetylation on pRB is excluded from E2F1 pSer364-containing complexes, further emphasizing the distinct nature of pRB-free and pRB-E2F1 complexes following DNA damage. Lastly, acetylated E2F1 is also excluded from pRB-E2F1 pSer364, and this also highlights the unique properties of this complex (Fig. 5E, compare AcK blots for lanes 2 and 3).
Given the similar role for Ser31 phosphorylation and lysine acetylation in creating a pRB-free population of E2F1, we were also curious to see if Ser31 phosphorylation of E2F1 is excluded from pRB-E2F1 complexes. Phosphorylation-specific antibodies to this site are unavailable, so we used a surrogate marker for its presence on E2F1. TopBP1 has been shown to bind to E2F1 in a Ser31 phosphorylation-dependent manner (30); consequently, we blotted for it in fractions of pRB-E2F1 and pRB-free E2F1. This analysis revealed that TopBP1 is found exclusively in the pRB-free fraction (Fig. 5F, compare TopBP1 blots for lanes 2 and 3), suggesting that Ser31 phosphorylation of E2F1 is also largely separate from Ser364 phosphorylation in populations of E2F1 following DNA damage.
These experiments demonstrate a remarkable contrast between pRB-E2F1 complexes and pRB-free E2F1 that are established in response to DNA damage. Figure 5G shows a diagram summarizing the posttranslational modifications that characterize and are excluded from the pRB-E2F1 pSer364 complex. Based on our experiments studying posttranslational modifications and associated proteins, DNA damage induces the formation of at least two distinct molecular species containing E2F1 that either contain pRB or exclude it.
DNA damage-induced posttranslational modifications on E2F1 are dispensable for the induction of proliferation.
In order to determine the roles played by each species of E2F1 following DNA damage, we generated an E2f1−/− 3T3 cell line. These cells are deficient for p53 function, as determined by the inability of DNA damage signaling to stabilize and elevate its expression or to induce subsequent p21 expression (Fig. 6A). We used retroviral expression of HA-E2F1 or mutants that are defective for DNA damage-dependent posttranslational modifications to reconstitute E2F1 expression. Western blots revealed uniform expression of all HA-E2F1 proteins in asynchronous cultures (Fig. 6B) and modest elevation of wild-type HA-E2F1 in response to DNA damage (Fig. 6B, E2F1 blot, lane 4). Importantly, DNA damage signaling led to phosphorylation of HA-E2F1 at serine 364 as shown by IP-Western blot experiments using anti-E2F1 pSer364 antibodies (Fig. 6C).
Fig 6.

E2F1 mutants complement cell cycle and transcription defects in E2f1−/− cells. (A) Wild-type mouse embryonic fibroblasts (wt MEF) and E2f1−/− 3T3 cells were treated with DMSO or 100 μM etoposide for 8 h, after which extracts were analyzed by Western blotting. Relative expression levels of p53 and p21 are shown. β-Actin serves as a loading control. (B) The indicated empty vector or E2F1-reconstituted (-WT, -TM, -DM, or -S364A) E2f1−/− 3T3 cells were treated as for panel A, after which whole-cell extracts were analyzed by Western blotting for E2F1 and β-actin. (C) The indicated E2F1-reconstituted E2f1−/− 3T3 cells were treated with 100 μM etoposide for 8 h. Nuclear extracts were prepared and immunoprecipitated with the indicated antibodies. Immunoprecipitated E2F1 was detected by Western blot analysis. (D) Growth curves for the indicated E2F1-reconstituted 3T3 lines. The total cell number was counted every 24 h following plating for 6 days. Error bars indicate one standard deviation from the mean (n = 3). (E) Asynchronously growing cultures of the indicated E2F1-reconstituted E2f1−/− 3T3 cells were pulse-labeled with BrdU for 1 h, processed for BrdU and PI staining, and analyzed by flow cytometry. Results are graphed as the percentage of total cells in G0/G1, S, or G2/M. Error bars indicate one standard deviation from the mean (n = 3). (F) RNA was extracted from reconstituted E2f1−/− 3T3 cells, and cyclin A2 and cyclin E1 expression was assayed by real-time RT-PCR. Results are normalized to the expression of β-actin and shown relative to the levels observed in empty-vector-transduced cells (set to 1). Error bars indicate one standard deviation from the mean (n = 3).
E2f1−/− 3T3 cells proliferated slowly compared to wild-type 3T3 cells. We investigated whether reexpression of HA-E2F1 in these cells could complement this slow-growth phenotype. Each mutant restored proliferation to the same level as for wild-type HA-E2F1 in a 6-day growth curve experiment (Fig. 6D). Likewise, the distributions of cells in each phase of the cell cycle were similar among HA-E2F1 mutant-expressing cultures, and these were collectively distinct from that of vector-only controls that contained elevated levels of G1, consistent with slower proliferation (Fig. 6E). Lastly, two key E2F target genes that induce proliferation, cyclin A2 and cyclin E1, were assayed using real time RT-PCR and found to be elevated in response to reexpression of wild-type and mutant HA-E2F1 proteins (Fig. 6F).
These analyses establish a number of key baseline parameters of our cell culture system that are critical to studying DNA damage signaling through E2F1. First, this system allows us to investigate apoptotic signaling in the absence of cell cycle control because the p53-p21 cell cycle arrest axis is deficient. Defective cell cycle regulation also reduces the effect of arrest-dependent DNA repair pathways on our analysis. In addition, expression of the different mutants has relatively uniform effects on proliferation and E2F transcription of cell cycle genes. Lastly, exogenously introduced E2F1 proteins can be modified in response to double-strand DNA breaks. Taken together, these factors ensure that the comparisons of DNA damage responsiveness in apoptosis described below are unlikely to be affected by differences in cell cycle regulation.
Both pRB-E2F1 and pRB-free E2F1 complexes are required for maximal induction of apoptotic target gene expression and cell death.
As shown in our previous experiments, phosphorylation of serine 31 and acetylation of lysine residues 117, 120, and 125 are essential to the formation of the pRB-free form of E2F1. Conversely, phosphorylation of serine 364 by Chk2 is a component of a distinct pRB-E2F1 pSer364 complex following DNA damage. For this reason we compared a double mutant, HA-E2F1 (which contains S31A and K117, 120, and 125R substitutions), and an S364A mutant (which blocks the defining feature of pRB-E2F1 complexes following DNA damage) in DNA damage-dependent apoptosis assays. In addition, we utilized wild-type HA-E2F1 and a triple mutant missing all three sites of posttranslational modification as a further comparison in these experiments.
Figure 7A shows real-time RT-PCR analyses of TA-p73 and caspase-7 expression in response to DNA damage in E2f1−/− 3T3 cells expressing our mutant forms of HA-E2F1. This revealed that wild-type HA-E2F1 induced the highest levels of expression in response to DNA damage. Furthermore, each mutant was defective for activation. This suggests that eliminating modifications on E2F1 that are key to forming either the pRB-free E2F1 population or pRB-E2F1 complexes is disruptive to the activation of transcription. Importantly, none of these mutants has a meaningful effect on transcription of cyclin A2 or cyclin E1, as their expression either dropped slightly or remained constant following DNA damage (Fig. 7B). We investigated the effects of diminished transcriptional activation of proapoptotic target genes by two different means. First, using alamarBlue viability dye, we demonstrated that both E2F1-DM and -S364A mutants had intermediate effects on cell viability following etoposide treatment (Fig. 7C). As expected, wild-type E2F1 reduced viability the most and HA-E2F1-TM the least in response to DNA damage. Second, using an assay to measure caspase-3 and -7 catalytic activity directly, we demonstrated that both HA-E2F1-DM and -S364A reduced caspase activation relative to that with wild-type HA-E2F1 (Fig. 7D). The presence of E2F1 pSer364 exclusively with pRB following DNA double-strand breaks suggests that this modification may mediate this interaction. We investigated this further using our reconstituted cell lines by immunoprecipitating HA-E2F1 and Western blotting for pRB. As shown in Fig. 7E, binding between pRB and HA-E2F1-S364A was indistinguishable from that of the wild-type control either before or after DNA damage. The implications for how Ser364 phosphorylation affects E2F1 function in transcriptional activation and apoptosis will be further explored in Discussion.
Fig 7.

Multiple E2F1 complexes are required for transcription of proapoptotic genes and induction of apoptosis. (A) Reconstituted E2f1−/− 3T3 cells were treated with 100 μM etoposide or DMSO for 6 h. RNA was extracted, and TA-p73 and caspase-7 induction was assayed by real-time RT-PCR. Results are normalized to the expression of β-actin and shown relative to the levels observed in untreated cells (set to 1). Error bars indicate one standard deviation from the mean (n = 3). Means were compared by the t test, and the resulting P values are indicated. (B) Cyclin A2 and cyclin E1 induction was assayed by real-time RT-PCR as for panel A. Fold induction is shown relative to the levels observed in untreated cells (set to 1). (C) Reconstituted E2f1−/− 3T3 cells were treated with 10 μM etoposide for 24 h. Cell viability was determined by using the alamarBlue cell viability reagent and measuring fluorescence at 570 nm. Percent viability is represented as relative to the levels observed in empty-vector-transduced cells. Error bars indicate one standard deviation from the mean (n = 3). Means were compared by the t test, and the resulting P values are indicated. (D) E2f1−/− 3T3 cells were treated with DMSO or 10 μM or 25 μM etoposide for 24 h. Apoptosis was quantitated by fluorescence emission created by cleavage of a rhodamine-linked caspase-3/7 substrate. Error bars indicate one standard deviation from the mean (n = 3). Means were compared by the t test, and the resulting P values are indicated. (E) HA-E2F1 was immunoprecipitated from extracts of untreated and treated 3T3 cells containing empty vector, HA-E2F1, or HA-E2F1-S364A. The levels of precipitated HA-E2F1 and pRB were determined by Western blotting.
These experiments reveal that DNA damage signaling pathways liberate a portion of E2F1 from pRB regulation. These same signals also organize a distinct pRB-E2F1 complex containing E2F1 pSer364. Surprisingly, both forms of E2F1 are required for transcriptional activation of the same target genes and, ultimately, the induction of apoptosis.
E2F1 pSer364 is present at the TA-p73 promoter following DNA double-strand breaks.
Previous analysis of the TA-p73 promoter has suggested that multiple E2F transcription factor binding sites at the TA-p73 transcriptional start site are inducibly occupied by E2F1 in an S31 phosphorylation- and lysine acetylation-dependent manner (40). In order to determine how directly the pRB-E2F1 pSer364 complex affects transcriptional control, we sought to identify it at the same region of this promoter by ChIP analysis. Using a modification-independent antibody to E2F1, we detected an increase in its occupancy of the TA-p73 promoter that is similar to that described in previous reports (Fig. 8). We also precipitated E2F1 pSer364 using modification-specific antibodies. This revealed that E2F1 pSer364 is detectable in the same region of TA-p73 following DNA damage (Fig. 8). This analysis, coupled with previously published work on Ser31 phosphorylation and lysine acetylation, suggests that differentially modified forms of E2F1 impinge on TA-p73 transcriptional regulation at a small region of its promoter that contains multiple E2F binding sites.
Fig 8.

E2F1 localization to the TA-p73 promoter in response to DNA damage. A graph of real-time PCR data from E2F1 and E2F1 pSer364 ChIP experiments is shown. A region proximal to the TA-p73 start site that is surrounded by multiple E2F binding sites was amplified along with a region of the GAPDH promoter as a negative control. Error bars indicate one standard deviation from the mean (n = 3). Means were compared by the t test, and the resulting P values are indicated.
DISCUSSION
This paper examines how signaling from DNA double-strand breaks impinges on the interactions of pRB and E2F1, two proteins that are known to interact but are capable of inducing different cellular outcomes in response to this stimulus. Our data indicate that serine 31 phosphorylation and lysine 117, 120, and 125 acetylation modifications on E2F1 are essential to create a pRB-free form of E2F1. Furthermore, a distinct pRB-E2F1 complex containing E2F1 pSer364 is also organized in response to etoposide-induced DNA damage. Strikingly, both forms of E2F1 are essential for maximal induction of transcription of E2F1 proapoptotic target genes and to induce apoptosis. Thus, our work reveals previously unappreciated mechanisms that govern pRB-E2F1 interactions that allow a distinct E2F1 transcriptional program to activate transcription under circumstances where pRB is normally dephosphorylated to repress E2F transcription in cell cycle control.
The dual requirement for serine 31 phosphorylation and acetylation of lysine 117, 120, and 125 in the creation of pRB-free E2F1 raises the question of how these modifications control this interaction and affect apoptotic signaling. Since there are no modification-specific antibodies available to either, we and others who have studied this question have relied on loss-of-function genetic analysis to guide the investigation of these modifications. Until reagents that allow us to positively identify E2F1 containing either serine 31 phosphorylation or acetylation of lysine 117, 120, and 125 become available, we think that the following model offers the most appropriate interpretation of the available data on the effect of these modification sites. Our experiments indicate that neither modification alone can affect either of the “specific” or “general” interaction configurations between E2F1 and pRB, and this suggests that these modifications contribute to a greater change in E2F1 that prevents its interaction with pRB. One explanation is that these modifications participate in the recruitment of new partners for binding to E2F1, such as 14-3-3τ or TopBP1, which have been shown to require S31 on E2F1 for DNA damage-induced interactions (30, 48). Furthermore, P/CAF, MCPH1/BRIT1, and SirT1 are reported to bind to E2F1 following DNA damage, suggesting that other proteins may be part of the pRB-free E2F1 complex described in this report (39, 40, 49). In this way E2F1's interacting partners would provide steric interference and prevent binding to pRB. Alternatively, the modifications could alter the conformation of E2F1 so that it is unable to interact with pRB. This type of change is consistent with the observation that acetylation of E2F1's DNA binding domain increases its affinity for DNA (36, 37), and K117, 120, and 125 as well as S31 are required for recruitment of E2F1 to the TA-p73 promoter in response to DNA damage (40). It is also possible that pRB-free E2F1 is unable to bind to pRB because of a combination of these two scenarios. We expect that phosphorylated serine 31 and acetylated lysines are found together on E2F1, because mutation of either site alone has similar effects on pRB binding following DNA damage (as shown here), as well as on recruitment to the TA-p73 promoter (40); however, we cannot rule out individual roles for them. We propose that pRB-free E2F1 contains these modifications and additional cofactors and localizes directly to promoters of apoptotic transcriptional targets (Fig. 9A).
Fig 9.

DNA damage induces the proapoptotic activity of E2F1 through multiple signaling pathways. (A) DNA damage leads to the formation of a hyperphosphorylated pRB-E2F1 complex. This complex requires phosphorylation of serine 364 of E2F1 to support maximal activation of proapoptotic target genes such as that for TA-p73, and it is physically present at the TA-p73 promoter. In addition, a pRB-free fraction of E2F1 is generated through phosphorylation of serine 31 and acetylation of lysine residues. Phosphorylation of S31 is known to recruit 14-3-3τ or TopBP1, and other factors may also participate in this complex. Serine 31 phosphorylation and lysine acetylation of E2F1 are also required for the maximal induction of TA-p73 and caspase-7 transcription, and these modifications have been shown to recruit E2F1 to the TA-p73 promoter. Therefore, multiple distinct molecular complexes containing E2F1 are utilized in the DNA damage response to induce transcription and apoptosis. (B) Conservation of amino acid identity in mammalian E2F1 proteins surrounding the pSer364 site. Sequences from human, chimpanzee, rhesus monkey, cow, mouse, and rat are shown. The consensus Chk2 phosphorylation site is shown at the bottom.
A number of recent reports have suggested ways that DNA damage signaling can modify pRB-E2F1 interactions to either induce cell death or preserve viability. Markham et al. have suggested that acetylation of pRB at lysine 873 and 874 will release E2F1 because these amino acids have previously been shown to form part of the “general” E2F interaction site on pRB (35). They demonstrate that a C-terminal fragment of pRB is unable to bind to E2F1 when these amino acids are changed to glutamine to mimic lysine acetylation, and since DNA damage induces acetylation of pRB, this likely causes E2F1 to be released. This observation is seemingly contradicted by two other reports that indicate that DNA damage induces formation of a complex between pRB and E2F1. In particular, Inoue et al. described an ATM- and Chk2-dependent phosphorylation event on pRB at serine 612 that is enriched in pRB-E2F1 complexes following DNA damage (19). Given the complexity of pRB-E2F1 interactions described in this report, these seemingly contradictory results can be accommodated by the fact they likely describe different populations of pRB-E2F1 complexes. For example, pRB pSer612 bound to E2F1 is clearly distinct from the pRB-E2F1 species containing E2F1 pSer364 described here. Furthermore, acetylation of pRB (at K873 and K874) is excluded from pRB when in complex with E2F1 pSer364, suggesting that the apparently contradictory results can be reconciled by the fact that they likely describe events that take place in different populations of pRB and E2F1 that are created in response to DNA damage. This raises the question of how many distinct forms of E2F1 exist in response to DNA damage. We are able to describe two separate signaling pathways that involve differentially modified E2F1 in transcriptional activation and apoptotic induction. Previous work on pRB pSer612 bound to E2F1 and E2F1 interactions with TopBP1 suggests that transcriptionally inactive complexes also exist and contribute to viability and repair functions, suggesting that there are more (19, 31). Future work to fully understand E2F1 in the DNA damage response will need to encompass many different forms of pRB and E2F1 beyond those examined in this study.
It has also been reported that pRB-E2F1 complexes activate transcription at proapoptotic promoters such as TA-p73 in response to DNA damage and that this leads to apoptosis (18). In agreement with the report by Ianari et al. (18) where they describe CDK-phosphorylated pRB in complex with E2F1, we find hyperphosphorylated pRB in complex with E2F1 pSer364. Our observation that E2F1 pSer364 is required for maximal transcriptional activation of proapoptotic promoters is consistent with this model. However, in our work we are unable to rule out the possibility that phosphorylation of serine 364 on E2F1 serves to inactivate a repressor function, and thus we are less confident that pRB-E2F1 is truly a transcriptional activator. We think it is unlikely that phosphoserine 364 on E2F1 alters its affinity for pRB, and instead we propose that a unique complex of hyperphosphorylated pRB and E2F1 pSer364 localizes directly to proapoptotic target genes of E2F1 and has a net positive impact on transcription (Fig. 9A). In this way, two distinct signaling pathways, both of which include E2F1, participate in transcriptional activation of proapoptotic target genes such as that for TA-p73.
Perhaps the most intriguing aspect of E2F1 pSer364 in DNA damage signaling is that this modification site appears to be restricted to primate E2F1 proteins (Fig. 9B). For this reason, the distinct pRB-E2F1 complex that contains E2F1 pSer364 is truly a primate-specific mechanism in the DNA damage response. The fact that we can detect a role for E2F1 pSer364 in mouse cells suggests that phosphorylation of this site creates a change in pRB-E2F1 structure or function that is intrinsic to the complex. It is not clear what new function exists for this modification, but our work offers a brief glimpse into the creation of a truly distinct signaling mechanism. The exclusion of other known DNA damage-dependent modifications on E2F1 emphasizes that phosphoserine 364 offers a new function beyond what is available in nonprimates.
ACKNOWLEDGMENTS
We are indebted to A. Gulino and M. Levrero (University of Rome) for plasmids used in this study. Primer design and assistance with real-time PCR from S. Cregan (UWO) are gratefully acknowledged. We thank many members of the London Regional Cancer Program and Children's Health Research Institute for advice during the course of this work, in particular Matt Cecchini for help with plasmid constructions.
O.P. was partially supported by an ERA award. J.C. and L.A.S. received stipend support from the Strategic Training Program in Cancer Research (CaRTT), and L.A.S. also received support from OGSST. This work was funded by grant MOP-89765 from the CIHR to F.A.D.
Footnotes
Published ahead of print 19 December 2011
REFERENCES
- 1. Aparicio O, et al. 2005. Chromatin immunoprecipitation for determining the association of proteins with specific genomic sequences in vivo, p 21.23.21–21.23.33 In Ausubel FM, Brent R, Kington RE, Moore DD, Seidman JG, Smith JA, Struhl KE. (ed), Current protocols in molecular biology. Greene Publishing Associates, New York, NY: [DOI] [PubMed] [Google Scholar]
- 2. Avni D, et al. 2003. Active localization of the retinoblastoma protein in chromatin and its response to S phase DNA damage. Mol. Cell 12:735–746 [DOI] [PubMed] [Google Scholar]
- 3. Bates S, et al. 1998. p14ARF links the tumour suppressors RB and p53. Nature 395:124–125 [DOI] [PubMed] [Google Scholar]
- 4. Blattner C, Sparks A, Lane D. 1999. Transcription factor E2F-1 is upregulated in response to DNA damage in a manner analogous to that of p53. Mol. Cell. Biol. 19:3704–3713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brugarolas J, et al. 1999. Inhibition of cyclin-dependent kinase 2 by p21 is necessary for retinoblastoma protein-mediated G1 arrest after gamma-irradiation. Proc. Natl. Acad. Sci. U. S. A. 96:1002–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Burkhart DL, Sage J. 2008. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat. Rev. Cancer 8:671–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Carr SM, Munro S, Kessler B, Oppermann U, La Thangue NB. 2011. Interplay between lysine methylation and Cdk phosphorylation in growth control by the retinoblastoma protein. EMBO J. 30:317–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cecchini MJ, Dick FA. 2011. The biochemical basis of CDK phosphorylation-independent regulation of E2F1 by the retinoblastoma protein. Biochem. J. 434:297–308 [DOI] [PubMed] [Google Scholar]
- 9. Chau BN, Pan CW, Wang JY. 2006. Separation of anti-proliferation and anti-apoptotic functions of retinoblastoma protein through targeted mutations of its A/B domain. PLoS One 1:e82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. DeGregori J, Leone G, Miron A, Jakoi L, Nevins JR. 1997. Distinct roles for E2F proteins in cell growth control and apoptosis. Proc. Natl. Acad. Sci. U. S. A. 94:7245–7250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dick FA, Dyson N. 2003. pRB contains an E2F1-specific binding domain that allows E2F1-induced apoptosis to be regulated separately from other E2F activities. Mol. Cell 12:639–649 [DOI] [PubMed] [Google Scholar]
- 12. Dick FA, Sailhamer E, Dyson NJ. 2000. Mutagenesis of the pRB pocket reveals that cell cycle arrest functions are separable from binding to viral oncoproteins. Mol. Cell. Biol. 20:3715–3727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ginsberg D. 2002. E2F1 pathways to apoptosis. FEBS Lett. 529:122–125 [DOI] [PubMed] [Google Scholar]
- 14. Hallstrom TC, Nevins JR. 2006. Jab1 is a specificity factor for E2F1-induced apoptosis. Genes Dev. 20:613–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Harrington EA, Bruce JL, Harlow E, Dyson N. 1998. pRB plays an essential role in cell cycle arrest induced by DNA damage. Proc. Natl. Acad. Sci. U. S. A. 95:11945–11950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hofferer M, Wirbelauer C, Humar B, Krek W. 1999. Increased levels of E2F-1-dependent DNA binding activity after UV- or gamma-irradiation. Nucleic Acids Res. 27:491–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hsieh J-K, Fredersdorf S, Kouzarides T, Martin K, Lu X. 1997. E2F1-induced apoptosis requires DNA binding but not transactivation and is inhibited by the retinoblastoma protein through direct interaction. Genes Dev. 11:1840–1852 [DOI] [PubMed] [Google Scholar]
- 18. Ianari A, et al. 2009. Proapoptotic function of the retinoblastoma tumor suppressor protein. Cancer Cell 15:184–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Inoue Y, Kitagawa M, Taya Y. 2007. Phosphorylation of pRB at Ser612 by Chk1/2 leads to a complex between pRB and E2F-1 after DNA damage. EMBO J. 26:2083–2093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Irwin M, et al. 2000. Role for the p53 homologue p73 in E2F-1-induced apoptosis. Nature 407:645–648 [DOI] [PubMed] [Google Scholar]
- 21. Irwin MS, et al. 2003. Chemosensitivity linked to p73 function. Cancer Cell 3:403–410 [DOI] [PubMed] [Google Scholar]
- 22. Isaac CE, et al. 2006. The retinoblastoma protein regulates pericentric heterochromatin. Mol. Cell. Biol. 26:3659–3671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Julian LM, Palander O, Seifried LA, Foster JE, Dick FA. 2008. Characterization of an E2F1-specific binding domain in pRB and its implications for apoptotic regulation. Oncogene 27:1572–1579 [DOI] [PubMed] [Google Scholar]
- 24. Knudsen ES, Buckmaster C, Chen T-T, Feramisco JR, Wang JYJ. 1998. Inhibition of DNA synthesis by RB: effects on G1/S transition and S-phase progression. Genes Dev. 12:2278–2292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Knudsen KE, et al. 2000. RB-dependent S-phase response to DNA damage. Mol. Cell. Biol. 20:7751–7763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Komori H, Enomoto M, Nakamura M, Iwanaga R, Ohtani K. 2005. Distinct E2F-mediated transcriptional program regulates p14ARF gene expression. EMBO J. 24:3724–3736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kontaki H, Talianidis I. 2010. Lysine methylation regulates E2F1-induced cell death. Mol. Cell 39:152–160 [DOI] [PubMed] [Google Scholar]
- 28. Lazzerini Denchi E, Helin K. 2005. E2F1 is crucial for E2F-dependent apoptosis. EMBO Rep. 6:661–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lin WC, Lin FT, Nevins JR. 2001. Selective induction of E2F1 in response to DNA damage, mediated by ATM-dependent phosphorylation. Genes Dev. 15:1833–1844 [PMC free article] [PubMed] [Google Scholar]
- 30. Liu K, Lin FT, Ruppert JM, Lin WC. 2003. Regulation of E2F1 by BRCT domain-containing protein TopBP1. Mol. Cell. Biol. 23:3287–3304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu K, Luo Y, Lin FT, Lin WC. 2004. TopBP1 recruits Brg1/Brm to repress E2F1-induced apoptosis, a novel pRb-independent and E2F1-specific control for cell survival. Genes Dev. 18:673–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lomazzi M, Moroni MC, Jensen MR, Frittoli E, Helin K. 2002. Suppression of the p53- or pRB-mediated G1 checkpoint is required for E2F-induced S-phase entry. Nat. Genet. 31:190–194 [DOI] [PubMed] [Google Scholar]
- 33. Ma Y, Cress WD, Haura EB. 2003. Flavopiridol-induced apoptosis is mediated through up-regulation of E2F1 and repression of Mcl-1. Mol. Cancer Ther. 2:73–81 [PubMed] [Google Scholar]
- 34. Mann R, Mulligan RC, Baltimore D. 1983. Construction of a retrovirus packaging mutant and its use to produce helper-free defective retrovirus. Cell 33:153–159 [DOI] [PubMed] [Google Scholar]
- 35. Markham D, Munro S, Soloway J, O'Connor DP, La Thangue NB. 2006. DNA-damage-responsive acetylation of pRb regulates binding to E2F-1. EMBO Rep. 7:192–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Martinez-Balbas MA, Bauer UM, Nielsen SJ, Brehm A, Kouzarides T. 2000. Regulation of E2F1 activity by acetylation. EMBO J. 19:662–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Marzio G, et al. 2000. E2F family members are differentially regulated by reversible acetylation. J. Biol. Chem. 275:10887–10892 [DOI] [PubMed] [Google Scholar]
- 38. Munro S, Khaire N, Inche A, Carr S, La Thangue NB. 2010. Lysine methylation regulates the pRb tumour suppressor protein. Oncogene 29:2357–2367 [DOI] [PubMed] [Google Scholar]
- 39. Pediconi N, et al. 2009. hSirT1-dependent regulation of the PCAF-E2F1-p73 apoptotic pathway in response to DNA damage. Mol. Cell. Biol. 29:1989–1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pediconi N, et al. 2003. Differential regulation of E2F1 apoptotic target genes in response to DNA damage. Nat. Cell Biol. 5:552–558 [DOI] [PubMed] [Google Scholar]
- 41. Ramadan S, et al. 2005. p73 induces apoptosis by different mechanisms. Biochem. Biophys. Res. Commun. 331:713–717 [DOI] [PubMed] [Google Scholar]
- 42. Saddic LA, et al. 2010. Methylation of the retinoblastoma tumor suppressor by SMYD2. J. Biol. Chem. 285:37733–37740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stevens C, Smith L, La Thangue NB. 2003. Chk2 activates E2F-1 in response to DNA damage. Nat. Cell Biol. 5:401–409 [DOI] [PubMed] [Google Scholar]
- 44. Talluri S, et al. 2010. A G1 checkpoint mediated by the retinoblastoma protein that is dispensable in terminal differentiation but essential for senescence. Mol. Cell. Biol. 30:948–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Thillainadesan G, et al. 2008. Genome analysis identifies the p15ink4b tumor suppressor as a direct target of the ZNF217/CoREST complex. Mol. Cell. Biol. 28:6066–6077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Todaro GJ, Green H. 1963. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J. Cell Biol. 17:299–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Urist M, Tanaka T, Poyurovsky MV, Prives C. 2004. p73 induction after DNA damage is regulated by checkpoint kinases Chk1 and Chk2. Genes Dev. 18:3041–3054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang B, Liu K, Lin FT, Lin WC. 2004. A role for 14-3-3 tau in E2F1 stabilization and DNA damage-induced apoptosis. J. Biol. Chem. 279:54140–54152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yang SZ, Lin FT, Lin WC. 2008. MCPH1/BRIT1 cooperates with E2F1 in the activation of checkpoint, DNA repair and apoptosis. EMBO Rep. 9:907–915 [DOI] [PMC free article] [PubMed] [Google Scholar]