

Abstract
Mesoporous silica nanoparticles (MSNPs) have garnered a great deal of attention as potential carriers for therapeutic payloads. However, achieving triggered drug release from MSNPs in vivo has been challenging. Here, we describe the synthesis of stimulus-responsive polymer-coated MSNPs and the loading of therapeutics into both the core and shell domains. We characterize MSNP drug-eluting properties in vitro and demonstrate that the polymer-coated MSNPs release doxorubicin in response to proteases present at a tumor site in vivo, resulting in cellular apoptosis. These results demonstrate the utility of polymer-coated nanoparticles in specifically delivering an antitumor payload.
Nanotechnology has the potential to impact many long-standing challenges in medicine, such as selective drug delivery and sensitive detection of disease.1−4 In recent years, mesoporous silica nanoparticles (MSNPs) have attracted attention as a promising component of multimodal nanoparticle systems.5−9 MSNPs are excellent candidates for many biomedical applications owing to their straightforward synthesis, tunable pore morphologies, facile functionalization chemistries, low-toxicity degradation pathways in the biological milieu, and capacity to carry disparate payloads (molecular drugs, proteins, other nanoparticles) within the porous core.5−8,10−12
Despite their promise, however, recent reports highlight the potential toxicity of unmodified MSNPs due to interactions of surface silanols with cellular membranes.13−16 This toxicity can be reduced by coating the nanoparticle with a polymer shell.5,17−21 Polymer shells also provide colloidal stability, handles for chemoligation (targeting moieties) and improved blood circulation lifetimes, which are crucial for efficient in vivo drug delivery. Unfortunately, the polymer shell also limits both drug loading and release from MSNPs.
In order to address the drawbacks of coating, we developed an MSNP polymer that degrades in response to external stimuli. We explored both physical triggers, such as temperature, and biochemical triggers, such as proteases found in the tumor microenvironment. Loading and responsive drug release were explored using payloads incorporated into the MSNP core as well as the polymer shell.
In our attempt to coat MSNPs with polymers, we considered previously reported approaches, such as noncovalent assembly or surface-initiated polymerizations techniques.18,22−26 These methods, however, have limitations. Noncovalent strategies are prone to colloidal and biological instability, whereas covalent surface-initiated polymerization approaches typically result in larger particles and expose the MSNP to harsh reaction conditions. Furthermore, the existing methods do not provide the flexibility to allow drug loading in distinct compartments. We therefore developed a new strategy (Figure 1a) based on a core–shell architecture and an aqueous free radical polymerization technique. We first electrostatically adsorb an acrylamide to the MSNP surface and then utilize the acryl groups to synthesize a covalently cross-linked poly(ethylene glycol) (PEG)-based polymer shell. The covalent cross-linking can provide addition stability to the polymer shell.27−29 This synthetic strategy provides a covalent polymer shell without the use of catalysts and surfactants and requires mild conditions compatible with a variety of potential biomolecular payloads.
Figure 1.

Polymer-coated MSNPs: (a) Synthetic scheme for the polymer coating of MSNPs. (b) TEM micrographs of uncoated and PEG-coated MSNPs. Scale bar is 20 nm. (c) In vitro viability of HeLa cells in the presence of uncoated MSNPs and polymer-coated MSNPs (n = 3). (d) In vivo circulation lifetime of polymer-coated MSNPs after tail vein injections in Swiss Webster mice (n = 3). (e) Cellular uptake of PEG–MSNPs by HeLa cells. Red, Vivo Tag 680 conjugated to the polymer shell; Blue, DAPI. Scale bar is 10 μm.
MSNPs were synthesized via co-condensation of silicates, similar to previous reports.10,30 To coat the anionic surface of the MSNPs, we used bifunctional N-(3-aminopropyl) methacrylamide hydrochloride (APMA). The amino group was electrostatically bound to the nanoparticle surface, while the acrylamide group was available for radical polymerization. Subsequently, a covalently cross-linked polymer shell was synthesized at room temperature by radical polymerization of monomers, including N-isopropylacrylamide (NIPAm) or poly(ethylene glycol) diacrylate (PEGDA). The monomer concentrations during synthesis were kept low and in order to produce a dense polymer shell, it was necessary to perform a second polymerization step to yield a “double-coated” nanoparticle.
Transmission electron microscopy (TEM) images (Figure 1b and Figure S1c, Supporting Information) indicated that the synthesis yielded individually encapsulated MSNPs displaying a thin polymeric shell, which is absent on uncoated nanoparticles. TEM measurements indicated that the diameters of the uncoated and polymer-coated MSNPs were 70 ± 8 and 94 ± 12 nm, respectively. Dynamic light scattering measurements showed that the hydrodynamic diameter of the MSNPs increased upon addition of single or double pNIPAm-co-PEG (9:1 molar ratio) shells by ∼20–40% (Figure S1, Supporting Information). The DLS and TEM data indicate that this polymer coating procedure avoids agglomeration of the nanoparticles into the larger (micrometer-scale) aggregates that have been previously observed with other coating techniques.22
We next investigated the drug-loading capacity of the polymer-coated MSNPs by comparing the total amount of drug loaded before and after polymer coating. We chose doxorubicin (Dox) as a model payload due to its well-characterized spectral characteristics, its use in chemotherapy, and its affinity for the negatively charged surface of the silica nanoparticles, which enhances loading into the MSNP pores.31 The loading of Dox in polymer-coated MSNPs was only slightly lower (∼50% of total Dox added) compared with the uncoated MSNPs (∼60% of total Dox added) (Figure S3a, Supporting Information). This suggests that the polymer shell does not reduce the drug loading capacity of MSNPs as drastically as other reported polymer shell–MSNP systems, which is an additional advantage of our technique over previously reported coating methods.17,32 We also observed that the polymer shell provided colloidal stability at low pH and prevented aggregation of the MSNPs, which are prone to interparticle hydrogen bonding (Figure S4a, Supporting Information). The synthesis allows facile incorporation of comonomers that can add additional functionality to the shell (Figure S4b, Supporting Information).
To assess the in vitro safety and biocompatibility of the polymer-coated MSNPs, we analyzed mitochondrial activity of HeLa cells following incubation with differing concentrations of polymer-coated MSNPs. No significant in vitro cytotoxicity was observed for nanoparticle concentrations of 0.01–1 mg/mL and with a range of PEG content in the polymer shell (Figure 1c). By contrast, the uncoated MSNPs exhibited signs of cytotoxicity at concentrations of 1 mg/mL.13,33
To study the biological trafficking of the polymer-coated MSNPs, the outer polymer shell was tagged following polymerization with the near-infrared (near-IR) dye Vivo Tag 680. For this formulation, the inner shell consisted of PEGDA and the outer shell was a copolymer of 10 mol % APMA with PEGDA. The dye was conjugated to the free amine side chains on the APMA comonomer. Labeled MSNPs were used to study blood circulation properties in vivo and cellular uptake of the nanoparticles in vitro. Compared with uncoated MSNPs, PEGylated MSNPs have been shown to possess a longer blood-circulation lifetime and lower excretion of degradation products in the urine.14 In the present work, increasing the mole percent of PEG from 2 to 90 in the polymer shell increased the circulation lifetime of MSNPs, measured by quantifying Vivo Tag 680 fluorescence in capillary blood draws (Figure 1d). Cellular uptake studies (Figure 1e) confirmed that the 90 mol % PEG-coated MSNP formulation (PEG–MSNPs) was internalized by HeLa cells after 4 h of incubation.
In addition to providing colloidal stability, functional groups for chemical ligation, and improved biocompatibility and blood circulation lifetimes, the polymer shell can be used to impart stimuli responsive characteristics to MSNPs. Stimulus-responsive nanoparticles have been shown to be extremely useful for controlled drug release.17,34,35 These systems overcome several current delivery challenges in therapy because they can be utilized for sustained drug delivery and co-delivery of multiple drugs with distinct release profiles. In this work, we engineered these long-circulating, biocompatible polymer-coated MSNPs to respond to temperature and the biological microenvironment (protease) for controlled drug delivery.
The thermally responsive system was synthesized from NIPAm–PEG (9:1)-coated MSNPs, and doxorubicin was used as the test drug. It has been shown that pNIPAm, which possesses a lower critical solution temperature (LCST) of ∼31 °C, can provide temperature-triggered release of drugs from various nanoparticles.35 Consistent with these prior results, at temperatures greater than the LCST (37 °C), about 50% more Dox was released within the first 2 h of incubation, compared with the same formulation maintained at room temperature (Figure 2a). By comparison, uncoated MSNPs released the same quantity of Dox at either temperature.
Figure 2.

Controlling drug release from polymer-coated MSNPs: (a) Temperature-triggered release of doxorubicin from pNIPAM-co-PEG coated MSNPs. Inset shows release after 2 h. (b) Doxorubicin release profile in PBS at 37 °C for uncoated MSNPs and core-loaded and shell-loaded PEG–MSNPs. (c) Dox-induced chemotoxicity on J2-3T3 fibroblasts from MMP-degradable PEG–MSNPs (HD, highly degradable; LD, low degradability), PEG–MSNPs, and Dox-loaded MSNPs (MSNP-Dox) (n = 3).
We further investigated the influence of the polymer shell on the drug release profiles from MSNPs in which the drug was loaded in either the inorganic core or the polymer shell of the nanoparticle. To compare these two loading strategies, we measured the drug loading efficiencies and characterized the drug release profiles in core-loaded and shell-loaded PEG–MSNPs. For the core-loaded PEG–MSNPs, doxorubicin was loaded in the MSNPs with a single PEGDA shell and a second covalent polymer shell (PEG-co-APMA) was synthesized after drug loading to provide a diffusion barrier. For the shell-loaded PEG–MSNPs, Dox was loaded after the synthesis of the second PEG-co-APMA shell. Excess unloaded Dox was removed by centrifugation of the loaded nanoparticles. Compared with uncoated MSNPs, the loading efficiency of the polymer-coated MSNPs was reduced somewhat but was not dramatically different (p = 0.043 by ANOVA tukey analysis, Figure S3b, Supporting Information). Interestingly, the polymer-coated MSNPs held comparable amounts of Dox in either core- or shell-loaded formulations (Figure S3b, Supporting Information). Core-loaded PEG–MSNPs displayed the slowest rate of Dox release; only ∼20% of the drug was released after 24 h (Figure 2b). The shell-loaded PEG–MSNPs released ∼40% of the drug in the first 2 h and ∼60% after 24 h. Initially, the uncoated MSNPs released Dox at a slightly slower rate compared with the shell-loaded PEG–MSNPs but a larger total quantity of drug was released after 24 h (∼85% vs 63% of the loaded drug, respectively). The initial rapid rate of drug release from the shell-loaded PEG–MSNPs compared with the uncoated MSNPs suggested that Dox was loaded both in the core and the polymer shell in this PEG–MSNP formulation. The initial relatively rapid rate of release observed is attributed to diffusion of Dox loaded in the PEG shell. The release of Dox from a formulation in which the drug was loaded exclusively in the MSNP core and then coated with a polymer shell (core-loaded PEG–MSNPs) was much slower. Thus, the polymer shell provides a facile means to tune the drug release profile. The core-loaded PEG–MSNPs display a very slow drug release profile with no premature (“burst”) release, both desirable attributes for a “triggered” formulation designed to respond to specific tumoral extracellular signatures, such as proteases. For instance, it is known that matrix metalloproteinases (MMPs), which have the ability to degrade the extracellular matrix, are up-regulated in tumor environments because of secretion by rapidly dividing cancer cells and stromal cells.34
We therefore investigated whether MMP proteases could trigger drug release from the polymer-coated MSNPs. For the protease-sensitive polymer shell, we used PEGDA–peptide macromer possessing MMP substrate polypeptides with a highly degradable (HD-MMP) and a low-degradability (LD-MMP) sequence.36 We investigated protease-triggered drug release from core-loaded or shell-loaded PEG–peptide (HD-MMP and LD-MMP)-coated MSNPs by analyzing the chemotoxicity of Dox released during 48 h of incubation with 3T3-J2 fibroblasts.
In a typical experiment, 3T3-J2 fibroblasts were incubated with nanoparticles and assayed for cell viability using alamarBlue 48 h later. We observed high levels of Dox-induced chemotoxicity (∼15–20% cell viability) in all shell-loaded nanoparticles, regardless of their polymeric shell (PEGDA, LD-MMP, or HD-MMP; Figure 2c). This level of chemotoxicity was comparable to Dox-loaded uncoated MSNPs and free drug (for the same quantity of Dox administered) and is in accordance with the fast drug release profile of shell-loaded and uncoated MSNPs. In contrast, the Dox-induced chemotoxicity of core-loaded PEG–MSNPs was dependent on polymer shell composition. Low level of chemotoxicity (∼60–70% cell viability) was observed for core-loaded PEG–MSNP and LD-PEG–MSNPs, suggesting that in 48 h, the quantity of drug released from low- and nondegradable PEG nanoparticles was limited. Residual levels of drug release from LD-PEG–MSNPs is attributed to leaching of the drug and not cleavage of the PEG–peptide shell. In contrast, the chemotoxicity of core-loaded HD-PEG–MSNPs was high (30% cell viability), signifying that rapid Dox release resulted from cleavage of the PEG–peptide shell by endogenous MMPs in the cellular medium. Indeed, blocking the endogenous MMPs secreted by the fibroblasts with the inhibitor batimastat lowered the chemotoxicity of the HD-MMP–MSNPs (Figure S5b, Supporting Information). These results demonstrate that protease-triggered release can be achieved with polymer-coated MSNPs. The various polymer coatings on MSNPs thus allowed both spatial control over loading and temporal control over release of Dox in vitro.
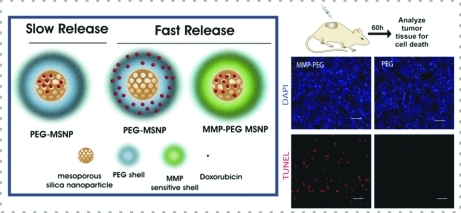
Finally, we conducted in vivo studies in subcutaneous xenograft mouse models to test the protease-triggered release of Dox from the polymer-coated MSNPs. We injected a human sarcoma cell line (HT-1080), known to have elevated levels of MMPs,37 subcutaneously in flanks of immune-compromised mice (Figure 3a). Two weeks later, core-loaded HD-PEG–MSNPs, core-loaded PEG–MSNPs, uncoated MSNPs and Dox-loaded uncoated MSNPs were normalized to a drug concentration of 2 mg/kg and injected into well-defined tumors. The tumors were removed after 60 h and analyzed for Dox-induced apoptosis by measurement of TUNEL staining and caspase levels (Figure 3b,c). Tumor cell lysates were analyzed for apoptosis markers procaspase-9 and cleaved caspase-9 by immunoblotting (Figure 3b). While the GAPDH levels were similar for each sample, exposure to the core-loaded HD-PEG–MSNP formulation and Dox-loaded uncoated MSNPs generated higher levels of the caspases. In contrast, the core-loaded PEG–MSNPs showed lower caspase levels, similar to those of saline-treated samples. Interestingly, core-loaded PEG–MSNPs generated lower levels of caspases compared with unloaded uncoated MSNPs. This finding is in accordance with in vitro studies suggesting that the polymer shell reduces inherent nanoparticle toxicity. TUNEL staining of the tumor sections (Figure 3c) indicated that the core-loaded HD-PEG–MSNPs mediated significantly higher Dox-induced cell death compared with PEG–MSNPs. Taken together, these results show that the core-loaded MSNPs with an MMP-sensitive PEG shell exhibit higher Dox-induced chemotoxicity than those with a non-MMP-sensitive PEG shell. We conclude that this higher chemotoxicity is due to efficient release of Dox triggered by the MMPs in vivo. This system takes advantage of the ability of the PEG polymer shell to reduce the inherent toxicity of MSNPs, and incorporation of a protease-cleavable moiety achieves triggered delivery that accelerates tumor-localized drug release.
Figure 3.

Protease-triggered release of Dox in vivo: (a) Schematic for evaluating protease-triggered release from core-loaded MSNPs. (b) Immunoblots for protein levels of caspases (apoptotic markers) and GAPDH from tumor lysates of animals 60 h after treatment. (c) TUNEL staining for apoptotic cells in tumor sections. Red, TUNEL; blue, DAPI. Scale is 50 μm.
In conclusion, we reported a facile and versatile method for coating MSNPs with responsive, biocompatible polymers. The polymer shell not only enables functionalization of the MSNPs with various ligands but also provides colloidal stability, temperature sensitivity, imaging capability, longer blood circulation, high payload capacity, and the opportunity to tune the loading and release of small molecules. Furthermore, we demonstrated that the polymer shell can be used to achieve predetermined, temporal control over drug release; the appropriately modified polymer can be responsive to endogenous proteases allowing triggered, localized drug release in vitro and in vivo. The polymer coatings also allow spatial control of payload loading within the nanostructure of the MSNP. This capacity is important for applications requiring multiple payloads with specifically timed release profiles from a single nanoparticle system.
Acknowledgments
We acknowledge financial support from the NIH through Grants R01-CA124427, U54-CA119349, and U54-CA119335. This material is based upon work supported by the NSF Grant No. DMR-0806859. We thank Swanson Biotechnology Center at the KI-MIT. We thank Justin Lo for discussions about the manuscript and figures. S.N.B. is an HHMI Investigator. A.K. acknowledges support from Amgen-UROP Scholars Program at MIT.
Supporting Information Available
Detailed experimental procedures, figures and complete refs (5) and (37). This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Farokhzad O. C.; Langer R. ACS Nano 2009, 3, 16. [DOI] [PubMed] [Google Scholar]
- Shi J.; Votruba A. R.; Farokhzad O. C.; Langer R. Nano Lett. 2010, 10, 3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y. Nat. Mater. 2008, 7, 758. [DOI] [PubMed] [Google Scholar]
- Byrne J. D.; Betancourt T.; Brannon-Peppas L. Adv. Drug Delivery Rev. 2008, 60, 1615. [DOI] [PubMed] [Google Scholar]
- Ashley C. E.; et al. Nat. Mater. 2011, 10, 389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slowing I. I.; Trewyn B. G.; Giri S.; Lin V. S. Y. Adv. Funct. Mater. 2007, 17, 1225. [Google Scholar]
- Slowing I. I.; Vivero-Escoto J. L.; Trewyn B. G.; Lin V. S. Y. J .Mater. Chem. 2010, 20, 7924. [Google Scholar]
- Wu S.-H.; Lin Y.-S.; Hung Y.; Chou Y.-H.; Hsu Y.-H.; Chang C.; Mou C.-Y. ChemBioChem 2008, 9, 53. [DOI] [PubMed] [Google Scholar]
- Liong M.; Angelos S.; Choi E.; Patel K.; Stoddart J. F.; Zink J. I. J. Mater. Chem. 2009, 19, 6251. [Google Scholar]
- Trewyn B. G.; Slowing I. I.; Giri S.; Chen H.-T.; Lin V. S. Y. Acc. Chem. Res. 2007, 40, 846. [DOI] [PubMed] [Google Scholar]
- Vivero-Escoto J. L.; Slowing I. I.; Trewyn B. G.; Lin V. S. Y. Small 2009, 6, 1952. [DOI] [PubMed] [Google Scholar]
- Tasciotti E.; Liu X.; Bhavane R.; Plant K.; Leonard A. D.; Price B. K.; Cheng M. M.-C.; Decuzzi P.; Tour J. M.; Robertson F.; Ferrari M. Nat. Nanotechnol. 2008, 3, 151. [DOI] [PubMed] [Google Scholar]
- Chang J.-S.; Chang K. L. B.; Hwang D.-F.; Kong Z.-L. Environ. Sci. Technol. 2007, 41, 2064. [DOI] [PubMed] [Google Scholar]
- He Q.; Zhang Z.; Gao F.; Li Y.; Shi J. Small 2010, 7, 271. [DOI] [PubMed] [Google Scholar]
- He Q.; Zhang Z.; Gao Y.; Shi J.; Li Y. Small 2009, 5, 2722. [DOI] [PubMed] [Google Scholar]
- Lin Y.-S.; Haynes C. L. J. Am. Chem. Soc. 2010, 132, 4834. [DOI] [PubMed] [Google Scholar]
- Gao Q.; Xu Y.; Wu D.; Sun Y.; Li X. J. Phys. Chem. C 2009, 113, 12753. [Google Scholar]
- Muhammad F.; Guo M.; Qi W.; Sun F.; Wang A.; Guo Y.; Zhu G. J. Am. Chem. Soc. 2011, 133, 8778. [DOI] [PubMed] [Google Scholar]
- Song S. W.; Hidajat K.; Kawi S. Chem. Commun. 2007, 4396. [DOI] [PubMed] [Google Scholar]
- Thornton P. D.; Heise A. J. Am. Chem. Soc. 2010, 132, 2024. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Trewyn B. G.; Slowing I. I.; Lin V. S. Y. J. Am. Chem. Soc. 2009, 131, 8398. [DOI] [PubMed] [Google Scholar]
- Huang S.; Fan Y.; Cheng Z.; Kong D.; Yang P.; Quan Z.; Zhang C.; Lin J. J. Phys. Chem. C 2009, 113, 1775. [Google Scholar]
- Lu J.; Liong M.; Zink J. I.; Tamanoi F. Small 2007, 3, 1341. [DOI] [PubMed] [Google Scholar]
- Popat A.; Hartono S. B.; Stahr F.; Liu J.; Qiao S. Z.; Qing Lu G. Nanoscale 2011, 3, 2801. [DOI] [PubMed] [Google Scholar]
- Sun J.-T.; Hong C.-Y.; Pan C.-Y. J. Phys. Chem. C 2010, 114, 12481. [Google Scholar]
- Yang Y.; Yan X.; Cui Y.; He Q.; Li D.; Wang A.; Fei J.; Li J. J. Mater. Chem. 2008, 18, 5731. [Google Scholar]
- Joralemon M. J.; O’Reilly R. K.; Hawker C. J.; Wooley K. L. J. Am. Chem. Soc. 2005, 127, 16892. [DOI] [PubMed] [Google Scholar]
- Choi W. I.; Yoon K. C.; Im S. K.; Kim Y. H.; Yuk S. H.; Tae G. Acta Biomater. 2010, 6, 2666. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Bansal V.; Zelikin A. N.; Caruso F. Nano Lett. 2008, 8, 1741. [DOI] [PubMed] [Google Scholar]
- Lai C.-Y.; Trewyn B. G.; Jeftinija D. M.; Jeftinija K.; Xu S.; Jeftinija S.; Lin V. S. Y. J. Am. Chem. Soc. 2003, 125, 4451. [DOI] [PubMed] [Google Scholar]
- Prokopowicz M.; Przyjazny A. J. Microencapsulation 2007, 24, 694. [DOI] [PubMed] [Google Scholar]
- Shi X.; Wang Y.; Varshney R. R.; Ren L.; Zhang F.; Wang D.-A. Biomaterials 2009, 30, 3996. [DOI] [PubMed] [Google Scholar]
- He Q.; Zhang J.; Shi J.; Zhu Z.; Zhang L.; Bu W.; Guo L.; Chen Y. Biomaterials 2010, 31, 1085. [DOI] [PubMed] [Google Scholar]
- Danhier F.; Feron O.; Préat V. J. Controlled Release 2010, 148, 135. [DOI] [PubMed] [Google Scholar]
- Yavuz M. S.; Cheng Y.; Chen J.; Cobley C. M.; Zhang Q.; Rycenga M.; Xie J.; Kim C.; Song K. H.; Schwartz A. G.; Wang L. V.; Xia Y. Nat. Mater. 2009, 8, 935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J. S.; Shen C. J.; Legant W. R.; Baranski J. D.; Blakely B. L.; Chen C. S. Biomaterials 2010, 31, 3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albright C. F.; et al. Mol. Cancer Ther. 2005, 4, 751. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.