

Abstract
Conventional drug development for treatment of cocaine addiction is greatly hindered by the extreme difficulty in designing a selective cocaine antagonist. We employed anti-idiotypic (anti-Id) antibodies to generate cocaine antagonists. The purpose of this study was to investigate the feasibility of this alternative approach. Herein, we describe the molecular cloning, bacterial expression and functional properties of an anti-Id monoclonal antibody, designated K2-3f, which possesses an internal image of cocaine within its variable regions. The heavy and light chain variable domains of K2-3f were cloned by RT-PCR and a single chain antibody variable fragment (scFv) was assembled for expression in E. coli. The scFv bound to the human dopamine transporter with moderate affinity (Ka = 5.3 × 106 M−1) and excellent mimicry of the cocaine molecule, completely inhibited cocaine binding at a molar concentration closely resembling in vivo conditions while allowing approximately 90% of equimolar dopamine uptake. Our data suggest that the use of anti-Id antibody as a template for generation of a cocaine antagonist is a promising approach well worth pursuing. If this strategy is successful, it could be applied to potential ligand-receptor interactions in the treatment of other diseases.
Keywords: Anti-idiotypic antibody, Antigen mimicry, Cocaine, Cocaine analog, Dopamine transporter, Single chain antibody, scFv
INTRODUCTION
Cocaine addiction remains a major threat to public health worldwide. Until now, efforts to find effective therapies for cocaine addiction have been unsuccessful. Anti-cocaine drug development is greatly hindered by the extreme difficulty in designing a selective cocaine antagonist compound.
The addictive properties of cocaine are thought to result from inhibition of dopamine re-uptake by the dopamine transporter (DAT) in and around the synapses. By binding to DAT, cocaine inhibits dopamine re-uptake and thus prolongs signaling at key brain synapses. Despite intensive study, the precise molecular mechanism of dopamine transport and of its inhibition by cocaine remains unclear. Hydropathicity analysis of DAT reveals a topology of 12 putative transmembrane domains with intracellularly oriented amino and carboxyl termini [1 and 2]. Recent studies of DAT supporting the existence of differential sites for dopamine translocation and cocaine recognition [2, 3, 4 and 5] have given renewed incentive to the search for cocaine antagonists. The ideal antagonist should bind DAT at the site of cocaine binding, inhibiting cocaine binding, without interfering with dopamine uptake, thus avoiding the physiological and behavioral consequences of cocaine. However, none of the numerous structural classes of DAT ligands known was found to fulfill this role. Rothman and his colleagues [6] extended the search to analog peptides by searching a random peptide library. Up to now, the conventional search for the effective cocaine antagonist that would spare dopamine transport has not been successful.
An alternative approach for development of cocaine antagonists is based on anti-idiotypic (anti-Id) antibodies that possess an internal image of cocaine. We generated a panel of such anti-Id monoclonal antibodies (mAb) [7]. Several anti-Id mAbs bound to the human dopamine transporter (hDAT) and inhibited dopamine uptake at levels ranging from 40 to 90% of the inhibition given by cocaine itself [8]. We surmised that the different levels of inhibition produced by the various anti-Id mAbs may reflect the different ways in which each antibody mimics the cocaine molecule. It may also reflect the effect of steric hindrance: a physical interference with dopamine uptake by the large antibody molecule bound to the cocaine site.
The goal of this study was to investigate whether the anti-Id antibodies could be used as a template for generating cocaine antagonist peptides. In order to eliminate the possibility of steric hindrance by a large molecule of antibody, we chose to use the smallest fragments capable of retaining most binding properties of the original antibody molecules, the single chain antibody variable fragments (scFv). The anti-Id mAb (K2-3f) shown to least interfere with dopamine uptake was selected for producing scFv.
MATERIALS AND METHODS
Cell lines
A neuroblastoma N1E-115 cell line stably expressing the hDAT was generated and maintained in our laboratory as described previously [8].
Establishment of mAbs
Ab1 and anti-Id Ab2 mAbs were produced as described [7]. Ab2 “P1F5-1-A7” (K2-3f) hybridoma specific for Ab1 (K2-3) was cloned twice by limiting dilution. MAb isotypes were determined by Enzyme-linked Immunosorbent Assay (ELISA) with reagents from an isotyping kit (Southern Biotechnology, AL). MAbs were purified following the published protocol [7]. For in vitro binding assays, mAbs were dialyzed overnight at 4°C against PBS buffer.
Evaluation of cocaine from brain tissue by HPLC
The HPLC technique for extraction and evaluation of cocaine from mouse brain tissue followed the protocol described previously [7].
Confocal immunofluorescence microscopy
N1E-115 cells grown to confluence on a six-well Costar cell culture plate (Corning, NY) were rinsed with PBS and fixed with 1% paraformaldehyde at room temperature for 30 min. After washing with PBS-Tween buffer, cells were incubated in PBS-Tween 1% BSA buffer for 1 hour. Cells were then incubated with K2-3f (10 μg/ml) and/or goat polyclonal anti-hDAT IgG (sc-1433, Santa Cruz Biotechnology, CA) (20 μg/ml) for 1 hour, followed by three washes (5 min each) with PBS-Tween buffer. The anti-hDAT IgG recognizes an epitope mapping at the carboxyl terminus (amino acid number: 601 - 620) of hDAT. Cells were then incubated with FITC-conjugated anti-goat IgG (KPL, MD) and PE-conjugated anti-mouse IgG (BD PharMingen), both diluted 1:100, for 1 hour and washed again. All primary and secondary antibodies were diluted in PBS-Tween 1% BSA buffer. The bottom of each well was cut and mounted on a slide with cell side up. Confocal images were generated on an Olympus FluoView 300 confocal laser scanning system with an Olympus BX50 microscope at the Center for Microscopy and Imaging at College of Veterinary Medicine, University of Illinois at Urbana-Champaign.
Cloning of anti-Id mAb variable domains
Total mRNA was isolated from early passage K2-3f hybridoma cells (1 × 106) using the Quick Prep Micro mRNA Purification Kit (Amersham Pharmacia Biotech). Complementary DNA (cDNA) was produced from mRNA using First-Strand cDNA Synthesis Kit (Amersham Pharmacia Biotech) and random hexanucleotide primers. Genes coding for the variable domains were amplified from cDNA by PCR. Briefly, the heavy chain variable (VH) domain with its native signal sequence was amplified using the degenerate primers MVH3 (5′-gggaattcATGRAATGSASCTGGGTYWTYCTCTT-3′) and MVH4 (5′-cccaagcttCCAGGGRCCARKGGATARACIGRTGG-3′) with initial 10 min denaturation at 94°C followed by 30 cycles of 1 min denaturation at 94°C, 1 min annealing at 45°C, and 2 min extension at 72°C. EcoRI and HindIII restriction sites are underlined. Standard abbreviations have been used for mixed sites: K, G or T; M, A or C; N, A, C, G or T; S, C or G; R, A or G; V, A, C or G; W, A or T; Y, C or T. The light (κ) chain variable (VL) domain was amplified under the same conditions using the primer pair MVL1 (5′-gggaattcGAYATTGTGMTSACMCARWCTMCA-3′) and MVL2 (5′-ggtaagcttGGATACAGTTGGTGCAGCATC-3′). The VH or VL amplification product was digested with EcoR1 and HindIII and ligated into the EcoR1-HindIII sites of pETBlue-2 (Novagen) to yield plasmid pETBlue.K2-3f.VH or pETBlue.K2-3f.VL. The VH and VL inserts were sequenced using a 5′-primer (pETBlue2Up, 5′-GTCACGACGTTGTAAAACGACGGCC-3′) by the W. M. Keck Center for Comparative and Functional Genomics at the University of Illinois at Urbana-Champaign.
Sequence analysis
Sequence analyses were performed using the Biology Workbench 3.2 utility (http://workbench.sdsc.edu/). The International ImMunoGeneTics (IMGT) database (http://imgt.cines.fr:8104/) and the Kabat database (http://immuno.bme.nwu.edu/) were used to identify gene families and groups of VH or VL. The complementarity-determining regions (CDR) were assigned using the IMGT database. The A/G-G-C/T-A/T and A-G-C/T sequences were identified as hot spots for somatic hypermutation and affinity maturation.
Molecular modeling
The program WAM (http://antibody.bath.ac.uk/) [9] containing a combined knowledge-based and energy-based algorithm was used to generate the molecular model of K2-3f. Briefly, three methods were used to perform the final screen: the RMS deviation screen, the accessibility profile screen, or energy only (backbone + sidechains of the final model). For sidechain modeling, two methods, the dead-end elimination algorithm and the CONGEN iterative algorithm, were used. CONGEN was used for the accessibility profile screen and the VFF energy screen. Models made as PDB files were visualized and analyzed using the VMD (Visual Molecular Dynamics, Version 1.7) program (http://www.ks.uiuc.edu/Research/vmd/) developed by the Theoretical Biophysics Group at the Beckman Institute of the University of Illinois at Urbana-Champaign.
Assembly of scFv constructs
The scFv were constructed by a two-step overlap extension PCR (Fig. 3). Briefly, for the first step PCR, fragments representing VH-Linker-VL N terminus were amplified by PCR from K2-3f cDNA using primers MH105 (5′-gcggccatggatCAAGTGAAGCTGGAGCAGTCTGGAGCT-3′) and MH106 (5′-TGGAGATTGTGTCATCACAATATCGGAACCACCACCACCGGAACCACCACC ACCGGAACCACCACCACCATAGACAGATGGGGGTGTCGTTTT-3′) corresponding to the heavy chain framework region (FR) 1 (MH105) and γ1 constant region-linker-light chain FR1 (MH106). Accordingly, fragments representing VL-His6 were constructed by PCR amplification from K2-3f cDNA using primers MH107 (5′-gcgGATATTGTGATGACACAATCTCCA-3′) and MH104 (5′-gcgaagcttTCAGTGGTGGTGGTGGTGGTGGGATACAGTTGGTGCAGCATCAGC–3′) corresponding to the light chain FR1 (MH107) and κ constant region-His6 (MH104). NcoI and HindIII restriction sites are underlined. The cycling program started with 94°C for 10 min and continued with 30 cycles of the following thermocycling conditions: 94°C for 1 min, 68°C for 1 min and 72°C for 2 min per cycle using Taq DNA polymerase (Life Technologies). The amplified gene fragments were purified (Qiagen) on 1% (w/v) agarose. For the second step overlap extension PCR, both 1st PCR products, VH-Linker-VL N terminus and VL-His6, were used as templates. Fragments representing the full-length scFv were constructed by PCR amplification from the mixture of the first PCR products using primers MH105 and MH104. The cycling program started with 94°C for 10 min and continued with 30 cycles of the following thermocycling conditions: 94°C for 1 min, 68°C for 1 min and 72°C for 2 min per cycle. After purification, PCR products of scFv fragments were digested and inserted into the NcoI-HindIII sites of pETBLue-2 (pETBlue.K2-3f.scFv) for sequence verification.
FIG. 3.

(A) Schematic presentation of the genetic construction of scFv K2-3f. (B) Deduced amino acid sequence of the K2-3f scFv (GenBank accession number AF479583). Arrow represents the cleavage site of signal peptide pelB. Hot spots within CDR regions are indicated in italic type.
Bacterial expression and protein purification
The K2-3f scFv sequence was isolated from pETBlue.K2-3f.scFv as a Nco1-HindIII fragment and inserted in frame to Nco1-HindIII site of bacterial expression vector pET22b(+) (Novagen). The resulting pET22b.scFv.K2-3f construct encodes a fusion protein consisting of the E. coli pelB signal peptide, the K2-3f scFv sequence and a hexahistidine tag. The scFv constructs were sequenced and confirmed using a pair of 5′- and 3′-primers (pET22bUp, 5′-TGCTGCTCCTCGCTGCCCAG–3′; pET22bDown, 5′-GCCAACTCAGCTTCCTTTCG-3′). Plasmid pET22b.scFv.K2-3f was transformed into the E. coli strain BL21(DE3)pLysS. Bacterial clones weregrown in 1 liter LB medium containing 100 μg/ml ampicillin. When induction was performed, bacterial cells transformed with pET22b.scFv.K2-3f werefirst grown to an A600 of 0.7 at 37 °C, then 1 mM isopropyl β-D-thiogalactoside (IPTG) was added. After 3 h of growth at 37 °C, cellswere pelleted by centrifugation and resuspended in 10 ml of BugBuster buffer (Novagen) containing 10 μl Benzonase nuclease. The cell suspension was incubated on a shaking platform at a slow setting for 20 min at room temperature. Insoluble cell debris were removed by centrifugation at 16,000 × g for 20 min at 4°C. The soluble extract was applied to a nickel-chelated agarose affinitycolumn that had been equilibrated with a nickel-binding buffer [20mM Tris/HCl (pH7.9), 0.5M NaCl and 5mM imidazole]. The column was extensively washed with a wash buffer [20mM Tris/HCl (pH7.9), 0.5M NaCl and 60mM imidazole], the protein was eluted with elution buffer [20mM Tris/HCl (pH7.9), 0.5M NaCl, 1M imidazole] at a flow rate of 0.5ml/min, and 1ml fractions were collected. A sample (25μl) was removed from each fraction and analyzed by ELISA for the ability to bind Ab1 (K2-3). Fractions containing binding activity were pooled, transferred to a dialysiscassette (molecular weight 10,000, Pierce) and dialyzed against PBS buffer (pH 7.0) for 12h at 4°C. Protein concentration was determined using a Bio-Rad protein assay kit with BSA as a standard. Purified proteins were further analyzed by SDS-polyacrylamide gel electrophoresis, Western blot, and mass spectrometry.
SDS-polyacrylamide gel electrophoresis and Western blot
Samples were electrophoresed through a 10% polyacrylamide gel under reducing conditions and electroblotted onto a nitrocellulose transfer membrane (NitroPure, MSI, MA) with a Bio-Rad Mini Transblot apparatus by standard techniques. After blocking the membrane with 5% non-fat dry milk in PBS containing 0.1% Tween 20, protein was detected with horseradish peroxidase (HRP)-labeled anti-His tag (specific for the His-His-His-His-His-His at C-terminus, Invitrogen) diluted 1:2500 followed by ABTS substrate (KPL, MD). Blots werethen washed three times, and processed for chemiluminescence detection using ECL reagent following the manufacturer’s (Amersham Pharmacia Biotech) instructions. Photographic exposures of 10s to 2 min were made.
Mass spectrometry
Mass spectra were obtained on a Voyager mass spectrometer by using matrix-assisted laser desorption/ionization (MALDI). Samples were dialyzed against 1mM potassium phosphate buffer, pH 8.0, and concentrated to 30 pmol/μl. Analysis was performed by the Mass Spectrometry Laboratory, School of Chemical Sciences, University of Illinois at Urbana-Champaign.
ELISA
Flat bottom microtiter plates (Immulon 2, Dynatech, VA) were coated with 10 μg/ml Ab1 K2-3 in carbonate buffer (15 mM Na2CO3, 35 mM NaHCO3, 0.02% NaN3, pH 9.5) and incubated overnight at 4°C. Non-specific binding sites were blocked by 1 h incubation at room temperature in SuperBlock solution (Pierce, IL). Fifty μl of the appropriate concentration scFv derived from the anti-Id Ab2 mAb K2-3f were added to the wells and incubated for 1 hour at room temperature. The presence of scFv binding to the Ab1 K2-3 was detected with an HRP-labeled mAb to the His tag diluted 1:3000, followed by ABTS substrate (KPL, MD). After 20 min, plates were read at 410 nm with a Dynatech MR5000 ELISA reader (Dynatech, VA). For the inhibition assay, soluble cocaine or dopamine at the appropriate concentration was added to Ab1 K2-3-coated wells and incubated for 1 hour at room temperature. The scFv was then added and incubated for 1 hour at room temperature. The assay was completed with the detection system described above.
Binding of the scFv K2-3f on hDAT and competitive inhibition assays with cocaine were performed essentially as described above but on cell-coated plates instead of mAb-coated plates. hDAT-transfected neuroblastoma cells (6C6) grown at confluence in flasks were collected and seeded in flat-bottom 96-well tissue culture plates. Forty-eight hrs later, the medium was discarded and the wells washed. The cells were then fixed with 1% paraformaldehyde (100 μl/well) for 30 minutes at room temperature.
Determination of affinity constant (Ka)
The affinity constant of anti-Id mAb or scFv for the DAT was determined by Scatchard plot [10] following the protocol described by Bator and Reading [11] using Fab or scFv instead of the whole antibody molecule. Scatchard plots were performed with Prism (Version 3.0.2, GraphPad Software, CA).
Dopamine uptake assays
Measurement of dopamine uptake was performed essentially as previously described (8). hDAT-transfected cells were grown in 75-cm2 flasks for 3 days. After reaching confluence, the cells were collected by trypsinization, counted, distributed in 24 well plates (1 × 105 per well) and allowed to re-attach for 24 hours. Following removal of media from the wells, cells were washed at room temperature with 1 ml Krebs Phosphate (KP) buffer containing 5 mM Tris, 7.5 mM HEPES, 120 mM NaCl, 5.4 mM KCl, 1.2 mM CaCl2, 1.2 mM MgSO4, 1 mM ascorbic acid, 5 mM glucose, at pH 7.1. Tritiated dopamine (ICN) in 0.5 ml of KP buffer at a final concentration of 50 nM was added to wells, and incubated for 5 min at 37°C, after which, the assay solution was promptly removed. The cells were washed twice with 1 ml ice-cold KP buffer and dissolved in 0.4 ml of 1% SDS. The entire liquid content of each well was transferred to a scintillation vial with 5 ml Bio-SafeII biodegradable cytoscint cocktail (RPI, IL) and assayed for radioactivity in a liquid scintillation counter (Liquid Scintillation Analyzer, Packard Instruments, Downers Grove, IL). For the inhibition assay, soluble cocaine, anti-Id scFv K2-3f or an irrelevant antibody Fab (anti-OVA) at the appropriate concentration was added to cell-coated wells and incubated for 10 min at room temperature. The [3H] dopamine was then added and incubated for 5 min at 37°C. The assay was completed as described above. Computer analysis of dopamine uptake data and IC50 of competitors were performed using Prism 3.0a program for Macintosh (GraphPad Software, San Diego, CA).
Cocaine binding inhibition assays
Cells were grown and allowed to attach to 24-well plates as described above. Following removal of media from the wells, cells were washed at room temperature with 1 ml KP buffer. Tritiated cocaine (provided by NIDA, NIH through Research Triangle Institute) in 0.5 ml of KP buffer at a final concentration of 50 nM was added to wells, and incubated for 5 min at 37°C, after which, the assay solution was promptly removed. Cells were washed twice with 1 ml ice-cold KP buffer and dissolved in 0.4 ml of 1% SDS. The entire liquid content of each well was transferred to a scintillation vial and assayed for radioactivity. For the inhibition assay, soluble cocaine, anti-Id scFv or an irrelevant Fab (anti-OVA) at the appropriate concentration was added to wells and incubated for 10 min at room temperature. The [3H] cocaine was then added and incubated for 5 min at 37°C. The assay was completed as described above. Computer analysis of cocaine binding data and IC50 of competitors were performed using Prism 3.0a program for Macintosh (GraphPad Software, San Diego, CA).
RESULTS
Production and characterization of anti-Id mAb K2-3f
The murine anti-Id mAb K2-3f (Ab2, hybridoma P1F5-1-A7, IgGκγ1) was elicited by mAb K2-3 (Ab1, hybridoma 2B:B4) (Fig. 1A). Ab1 K2-3 was induced against the cocaine hapten K2 coupled to keyhole limpet haemocyanin (KLH) [7]. Conjugation to KLH occurred at position 1N of the cocaine molecule; therefore, K2-KLH presented to the immune system the methyl ester in the 2β-position and the benzoyl in the 3β-position. It contains an intact tropane ring (Fig. 1B).
FIG. 1.

(A) Schematic presentation of Ab1 mAb K2-3, anti-Id mAb (Ab2) K2-3f, and scFv K2-3f generation. Hybridomas and scFv were produced as described in the Materials and Methods section. (B) Structures of cocaine and the K2 hapten.
The anti-Id nature of K2-3f was confirmed by both in vitro and in vivo analyses: K2-3f bound to the K2-3 that elicited it and its binding was inhibited by 1 mg/ml soluble cocaine. When injected into Balb/c mice, K2-3f (Ab2) induced high titers K2-3 (Ab1)-like Ab3 antibody specific for cocaine. This response was sufficient to decrease (up to 60%) the amount of cocaine targeting the brains of immunized mice after cocaine challenge compared to the amount of cocaine reaching the brain of mice injected with an irrelevant (anti-OVA) mAb.
Binding of K2-3f anti-Id mAb to hDAT: specificity and affinity
To ascertain whether the K2-3f anti-Id mAb could specifically bind to hDAT, an ELISA assay was performed using a mouse neuroblastoma cell line (N1E-115) transfected with the hDAT.
K2-3f showed the highest avidity to hDAT among a panel of several Ab2 tested. The specificity and relative avidity of K2-3f for hDAT can be surmised from the inhibition patterns of its binding. Partial inhibition (20–41%) was obtained with a high concentration of monomeric cocaine (5 mg/ml); a more substantial level of inhibition (60–66%) was obtained in the presence of a much lower concentration of polymeric cocaine in the form of cocaine-BSA conjugate (100 μg/ml). Ten mg/ml of BSA or a high concentration of dopamine (5 mg/ml) did not produce any inhibition. These findings suggest that K2-3f binds hDAT at the site of cocaine binding and that their avidity for hDAT is stronger than that of soluble cocaine. The K2-3f binding is also inhibited 87–93% by the K2-3 Ab1 mAb that elicited it, confirming that K2-3f binds hDAT with its antigen combining-sites. The binding affinity constants of K2-3f (Ka = 2.7 × 107 M−1) for hDAT were determined by Scatchard plot analysis using Fab fragments.
The binding of K2-3f to hDAT was then confirmed by confocal immunofluorescence microscopy. Figure 2 distinctly visualizes not only the binding, but also the position of hDAT on the cell body. In B, the localization of the K2-3f molecules incubated with the hDAT-transfected N1E-115 neuronal cells are visualized by the addition of a PE-conjugated anti-mouse IgG. In C, the same hDAT-transfected N1E neuronal cells are stained with an anti-hDAT polyclonal antibody and FITC-conjugated anti-goat IgG. In F, double staining with the two systems unmistakably confirms the co-localization of the hDAT and the K2-3f molecules. Most fluorescence appears to be situated on the cell membrane of the cell body, indicating that the hDAT molecules, at high density, were mostly located on the cell surface of N1E-115 cell bodies. Almost no fluorescence was observed on the axon or dendrites.
FIG. 2.

Confocal Immunofluorescence Microscopy demonstrating the binding of K2-3f to the mouse neuroblastoma N1E-115 cell line transfected with and stably expressing hDAT. Cells were fixed with 1% paraformaldehyde (A) and incubated with: 10 μg/ml K2-3f followed by PE -conjugated anti-mouse IgG (B), 20 μg/ml of polyclonal goat anti-hDAT IgG followed by FITC-conjugated anti-goat IgG (C), both (F), anti-OVA as a control (E). Untransfected N1E-115 cells as a background control were incubated with anti-hDAT antibody and then FITC-conjugated anti-goat IgG antibody (D). Confocal images were generated on an Olympus FluoView 300 confocal laser scanning system with an Olympus BX50 microscope. Bar, 50 μm.
Cloning and construction of K2-3f scFv
PCR products of the expected size (350–400 base pairs) were obtained from amplification of K2-3f hybridoma cDNA with VH (MVH3/4) and VL (MVL1/2) primers (see Materials and Methods). The nucleotide sequences have been submitted to the GenBank with accession numbers AF420003 (K2-3f VL), AF420004 (K2-3f VH). The VH gene was cloned behind the pelB signal sequence, and the VL gene was cloned in front of a sequence that encodes a 6-histidine (His6) carboxyl-terminal tag (Fig. 3A). The His6 tag provides a convenient means of detecting K2-3f scFv protein with the anti-His mAb. The scFv is flanked by 5′ NcoI and 3′ HindIII sites in order to facilitate in-frame cloning of scFv genes into pET22b(+). The predicted amino acid sequences of K2-3f scFv are shown in Fig. 3B.
Sequence analysis and molecular modeling
Sequence analysis using the IMGT database and the Kabat database revealed that the amino acid sequences of the CDR1 and CDR2 of the K2-3f VH are very similar to those of the IgHV1 germline gene (GenBank accession number AAG39174); however, its CDR3 region (CDR-H3) carries an insertion of two Arg, two Tyr and two Phe (Fig. 3B). It was reported that somatic insertions are usually clustered at hot-spots in the CDR regions [12 and 13] and the inserted sequence is often a direct duplication of an adjacement sequence (in most cases, only one or two amino acids). In our case, a hot spot corresponding to amino acids Gly-Asn was identified between two Tyr and two Phe which were likely generated by somatic insertion during the affinity maturation in vivo, clearly indicating the importance of these inserted residues in antigen binding. The amino acid sequence of the K2-3f VL, except for one amino acid in FR1, is identical to that of the IgκV12 germline gene (GenBank accession number AJ235956). The VH and VL of K2-3f belong to the VH VII gene family (subgroup IIA) and VL XVI (V), respectively. The primary sequence analysis seems to indicate that the internal image of cocaine is located within the CDR-H3 region of K2-3f. In future work, focused on therapeutic applications, hot spots identified in the CDR regions, especially the one in CDR-H3, may be a good candidate for in vitro affinity maturation of K2-3f [14].
In addition to the primary sequence analysis, we generated the three-dimensional molecular model (Fig. 4) for K2-3f VH and VL regions using the WAM program [9]. The VFF energy screen was chosen for K2-3f. For sidechain modeling, the CONGEN iterative algorithm was used. The long CDR-H3 loop of K2-3f clearly has a major structural implication for its combining site. Docking experiments using the DockVision program, a combined Monte Carlo and simulated annealing algorithm, revealed K2-3f fitted snugly onto Ab1 mAb K2-3 with Tyr211 and Tyr212 at the tip of the CDR H3 loop interacting with the Ile207 and Tyr214 cleft of K2-3 (data not shown).
FIG. 4.
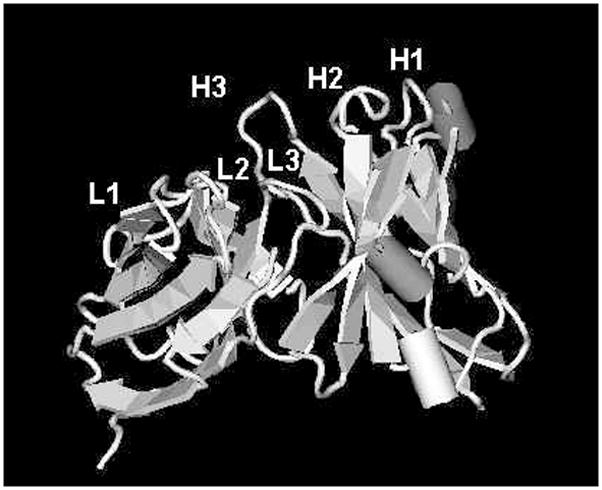
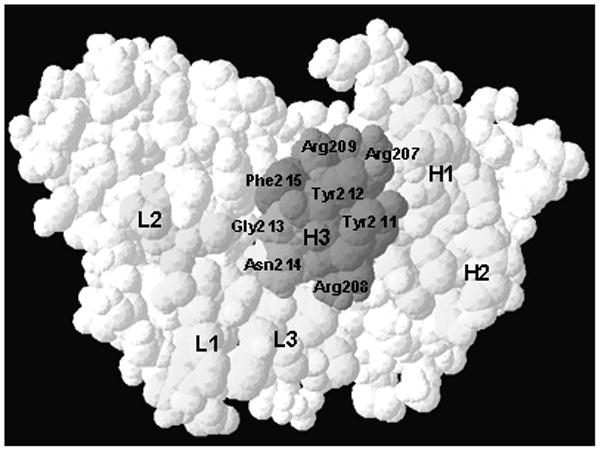
(A) Structural model of K2-3f V region. H1, 2, 3: heavy chain CDR1, 2, 3; L1, 2, 3: light chain CDR 1, 2, 3. (B) Surface view of K2-3f V region. The H3 region, especially Tyr211–Tyr212, may be the critical component of the internal image of cocaine
Expression and purification of K2-3f scFv
The pelB signal sequence was intended to facilitate folding and purification by targeting the protein to the E. coli periplasmic space. Prolonged (> 3h) IPTG induction caused the release of scFv into the media. Media from small scale cultures were tested by ELISA on a K2-3 (Ab1)-coated plate to establish that the presence of scFv could be detected by the peroxidase-conjugated anti-His antibody. A single positive clone (3F1) was selected for large scale fermentation and characterization.
K2-3f scFv was purified by nickel affinity chromatography (Novagen) (see Materials and Methods). One ml fractions were collected. A pool of fractions #1 and #2 containing the highest concentration of elute was further characterized by reduced SDS-PAGE gels and Western blot analysis with the anti-His antibody (Fig. 5A). A major overexpressed protein was present in the preparations. This protein stained positive with the anti-His antibody by Western blotting.
FIG. 5.

(A) SDS-PAGE analysis, and Western blot of K2-3f scFv expressed in E. coli. The purified scFv (lane 1) and molecular weight standards (lane M, BioRad) were electrophoresed under reducing condition. In Western blot, the protein was recognized by a peroxidase-labeled anti-His mAb (lane 2) as described in the Materials and Methods section. (B) The precise molecular mass was determined by MALDI mass spectrometry.
Mass spectrometry analysis indicated that the major protein had a mass of 28,727.28 Da (Fig. 5B). The molecular mass is consistent with the theoretical molecular mass of the K2-3f scFv monomer (28.7 KDa) predicted by the SAPS program (Biology WorkBench), assuming that the pelB signal sequence at the N-terminus has been cleaved. The observed reactivity on a Western blot with the anti-His antibody (Fig. 5A) may suggest that the C-terminus is intact. The estimated yield of nickel-purified K2-3f scFv from a 1-liter equivalent of fermentation is approximately 800 μg.
Binding properties of K2-3f scFv
The binding of purified scFv K2-3f was tested by ELISA on a K2-3 (Ab1)-coated plate and its binding affinity was determined (Ka = 1.4 × 107 M−1). This suggests full translation of the N-terminus of the native K2-3f’s antigen-combining site. The scFv was recognized by anti-His antibody, also suggesting full translation of the C-terminus of the recombinant protein. ScFv K2-3f (10 μg/ml, 0.3 μM) binding can be inhibited by cocaine (1 mg/ml, 3 mM) but not by dopamine (1mg/ml, 5 mM) (Fig. 6A). This clearly indicates that the recombinant protein has retained the internal image of cocaine, which characterizes the native anti-Id mAb (K2-3f). We also found that both the unpurified preparation of scFv (data not shown) and the purified scFv bound specifically to the hDAT expressed on the surface of a transfected neuroblastoma cell line in a manner similar to the native K2-3f. Purified scFv K2-3f bound hDAT with a binding affinity (Ka = ~ 5.3 × 106 M−1), approximately 5 fold lower than its native K2-3f Fab fragments. Nevertheless, the binding of K2-3f scFv to DAT (10 μg/ml, 0.3μM) was inhibited by a high concentration of cocaine (3 mg/ml, 9 mM), suggesting that scFv bound DAT at the site of cocaine binding and that its affinity for the DAT was stronger than that of cocaine. Interestingly, we observed that the binding of K2-3f could also be inhibited by a very high concentration of dopamine (3 mg/ml, 15 mM) at some degree (Fig. 6B). The unexpected dopamine inhibition was likely caused by oxidation and precipitation of dopamine on the N1E-115 cells during the prolonged incubation in ELISA assays (Dr. Jianyong Li, personal communication). In fact, we observed light or dark brown colors on neuroblastoma cells in the wells of dopamine at high concentrations. However, dopamine at a concentration lower than 0.5 mg/ml (2.5 mM) did not inhibit K2-3f binding to hDAT (data not shown).
FIG. 6.

(A) Inhibition of scFv K2-3f binding to Ab1 mAb K2-3 by cocaine. (B) Cocaine inhibition of scFv K2-3f binding to the hDAT expressed on the surface of a transfected neuroblastoma N1E-115 cell line.
Inhibition of cocaine binding DAT by scFv K2-3f
To ascertain whether the K2-3f scFv could block cocaine binding to DAT without interfering with dopamine uptake, two assays, dopamine uptake and cocaine binding inhibition, were performed (see Materials and Methods). Although there is no widely accepted and validated assay for detecting a cocaine antagonist, several strategies have been used to obtain “uptake-to-binding ratios”. One major strategy to determine such ratio is to compare the IC50 value of a test agent for inhibition of [3H]dopamine uptake and its IC50 value for inhibition of the binding of a transporter ligand such as cocaine or a cocaine analog. The goal of such a comparison is to identify a selective agent which has a high “uptake-to-binding” ratio. In our system, we observed that K2-3f IC50 for [3H]dopamine uptake was 550 ± 102 nM and 7.0 ± 1.4 nM for [3H]cocaine binding while cocaine IC50 for [3H]dopamine uptake was 32 ± 1.6 nM and 85 ± 14 nM for [3H]cocaine binding. Therefore, the “uptake-to-binding ratio” of K2-3f was approximately 80. It is noteworthy that in the present study, following an approach originally proposed by Rothman et al. [15], we conducted the dopamine uptake and cocaine binding assays under identical conditions. We found that our cocaine IC50 values for uptake and binding were lower than those (95nM for uptake and 104 nM for binding) obtained in a similar system but under different conditions [16]. Nevertheless, such IC50-based ratios must be evaluated with caution because experimental conditions used in these assays are often not close to in vivo situation. Therefore, a better approach must be to conduct the assays in a condition similar to in vivo setting [15]. Previous studies using cocaine or cocaine analog compounds have indicated that the concentration and occupancy of the dopamine transporter in humans: 13–47 nM [17] or 60 nM [18]. To resemble as much as possible in vivo conditions, we chose to use 50 nM for both dopamine uptake and cocaine binding assays. As shown in Figure 7A, 50 nM K2-3f scFv completely inhibited 50 nM cocaine binding to DAT while allowing about 90% equimolar (50 nM) dopamine uptake (Fig. 7B). In contrast, 50 nM cocaine allowed only about 30% equimolar dopamine uptake. More interestingly, while 100 μM cocaine alone completely inhibited 50 nM dopamine uptake, the presence of 50 nM K2-3f scFv significantly neutralized the effect of cocaine by allowing about 60% dopamine uptake (Fig. 7C).
FIG. 7.

(A) Inhibition of cocaine (50 nM) binding to hDAT by 50 nM K2-3f scFv. A hundred percent inhibition was defined with 100 μM unlabeled cocaine (147–487 cpm). The hundred percent cocaine binding was defined with KP buffer only (1203–1921 cpm). (B) Effects of 50 nM K2-3f scFv or cocaine on 50 nM dopamine uptake. Zero percent dopamine uptake was defined with 100 μM unlabeled cocaine (881–1640 cpm). The hundred percent dopamine uptake was defined with KP buffer only (6111 – 10314 cpm). (C) Effect of 50 nM K2-3f scFv on 50 nM dopamine uptake in the presence of 100 μM cocaine. Error bars represent the standard deviation of 3 determinations. Within each experiment, comparison of raw data of the scFv K2-3f group to its control (A, C: anti-OVA mAb Fab; B: cocaine) gave P values of *P<0.005 **P<0.001 (Student’s t-test, SigmaPlot, Version 8.0).
DISCUSSION
We report here the production and characterization of an anti-Id scFv (K2-3f). The K2-3f scFv mimics the configuration of the cocaine molecule and binds hDAT at the site of cocaine binding with higher affinity than cocaine. More importantly, the scFv, at a low molar concentration similar to in vivo conditions, by binding hDAT completely inhibits cocaine binding while allowing equimolar dopamine uptake. This makes it a possible novel cocaine antagonist.
The idiotypic network theory implies that the “internal image” of anti-Id antibodies can mimic “any” antigen in the “outside world” [19 and 20]. During the past two decades, numerous anti-Id antibodies mimicking protein antigens have been obtained [reviewed in 21]. In contrast, only a few anti-Id antibodies with the internal images of non-protein organic compounds have been reported [22, 23, 24 and 25]. In the present study, we confirm that the scFv engineered from anti-Id mAb K2-3f mimics the configuration of cocaine (339.8 Da), maintains the characteristics of the native anti-Id mAb and binds to DAT, the major target of cocaine in the brain. Obviously amino acid sequence homology between such a small non-protein antigen and an antibody’s internal image cannot be expected. Mimicry must therefore be based on conformation or electrochemical effects rather than sequence homology [26]. The finding that a polypeptide can mimic cocaine may possibly suggest that cocaine may act as a peptidomimetic compound that interferes with the function of an unknown endogenous polypeptide in the brain.
Sequence analysis and structural modeling indicated that CDR3 of K2-3f (ARRRTYYGNFFD), especially Tyr211–Tyr212, might be the critical component of the internal image of cocaine. Further studies using appropriate peptides [27] derived from this putative internal image of cocaine will be needed to confirm this prediction.
Initially, two anti-Id mAbs (K1-4c and K2-3f) chosen for construction of the scFv molecules, because of their low interference with dopamine uptake [8], were successfully constructed and expressed in the same bacterial system. However, scFv K1-4c, while still weakly bound to the Ab1 mAb (K1-4) used to generate it, lost most of the binding affinity shown by its native mAb for hDAT (data not shown). These results may indicate that not all scFv constructs keep the internal images of their native anti-Id mAbs faithfully. The non-linear complexity of the internal image structure may be critical for the success of this approach.
The use of the K2-3f scFv as a therapeutic cocaine inhibitor may be a debatable issue. However, cutting edge research suggests that the delivery of scFv to the brain through the Bood-Brain Barrier (BBB) may be a realistic goal. Although peptides with the appropriate physicochemical characteristics can cross the BBB [28], generally, most antibodies (150 kDa) or their F(ab′)2 (100 kDa) or Fab (50 kDa) fragments cannot be delivered without modification. Current research efforts in the field of drug delivery to the brain have developed some strategies to facilitate the passage of peptides such as Fab fragments through the BBB. Among them, cationization, which increases a peptide’s positive electrical charge, facilitates peptide binding to the negative charges on the surface of the endothelial cells and has been widely described as a promising technique [29]. It is also possible that in the clinical situation, the passage of anti-Id scFv through the BBB will not present any problem, since cocaine abuse and the consequent opening of the BBB have been implicated in the observed increase in neuroinvasion of the HIV-1 virus and monocyte migration in the brain of chronic cocaine users [30].
On the other hand, the use of anti-Id antibody as a template for generation of effective cocaine analogs is a promising approach well worth pursuing. If this strategy is successful, it could be applied to potential ligand-receptor interactions in the treatment of other drug addictions (e.g., nicotine, heroin) and diseases. In fact, the use of internal image for drug design has been largely overlooked. This study addresses the intriguing possibility of using the internal images of our anti-Id antibody molecules for the design of cocaine antagonists.
We also think that the internal image of cocaine may serve as a novel and unique molecular probe towards the elucidation of the interaction of cocaine with hDAT. Functional properties of K2-3f presented in this report suggest the existence of a distinct cocaine binding site (possibly not involved in dopamine uptake) recognized by this anti-Id mAb. Binding domain mapping of K2-3f on hDAT is currently in progress in our laboratory. Since the precise binding site of cocaine on DAT has not yet been determined, mapping the binding site of K2-3f on hDAT would possibly greatly enhance our understanding of the complicated interaction of cocaine with the transporter.
Acknowledgments
M. Ho was supported by National Research Service Award Individual Fellowship F31-DA14484 from the National Institute on Drug Abuse at the National Institutes of Health and University of Illinois Fellowship (1-1-10742) and presented this work as partial fulfillment of the PhD degree. This work was supported by the University of Illinois Research Board (1-2-68205) to M. S. and the University of Illinois On-Campus Dissertation Research Grant (1-2-16449) to M. H. We gratefully acknowledge Dr. Naoya Tsurushita (Protein Design Labs, Inc., CA) for providing expert advice in antibody expression, Dr. Nick Whitelegg (University of Bath, UK) and John Sanders at the University of Illinois School of Chemical Sciences in molecular modeling, Lou Ann Miller for assistance in confocal laser scanning microscopy, and Dr. Elizabeth Greeley for critical reading of the manuscript.
Abbreviations
- CDR
complementarity-determining region(s)
- DAT
dopamine transporter
- hDAT
human dopamine transporter
- ELISA
Enzyme-linked Immunosorbent Assay
- Fab
fragment of antibody containing the antigen-binding site, generated by cleavage of the antibody with papain
- FR
framework region
- H
heavy chain
- HRP
horseradish peroxidase
- Id
idiotype, idiotypic
- IPTG
isopropyl-β-D-thiogalactopyranoside
- KLH
keyhole limpet haemocyanin
- L
light chain
- mAb
monoclonal antibody
- MALDI
matrix-assisted laser desorption/ionization
- PCR
polymerase chain reaction
- RT
reverse transcription
- scFv
single chain antibody variable fragment(s)
- V
variable region
References
- 1.Giros B, el Mestikawy S, Godinot N, Zheng K, Han H, Yang-Feng T, Caron MG. Mol Pharmacol. 1992;42:383–390. [PubMed] [Google Scholar]
- 2.Itokawa M, Lin Z, Cai NS, Wu C, Kitayama S, Wang JB, Uhl GR. Mol Pharmacol. 2000;57:1093–1103. [PubMed] [Google Scholar]
- 3.Kitayama S, Shimada S, Xu H, Markham L, Donovan DM, Uhl GR. Proc Natl Acad Sci USA. 1992;89:7782–7785. doi: 10.1073/pnas.89.16.7782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Giros B, Wang YM, Suter S, Mcleskey SB, Pifl C, Caron MG. J Biol Chem. 1994;269:15985–15988. [PubMed] [Google Scholar]
- 5.Lee FJ, Pristupa ZB, Ciliax BJ, Levey AI, Niznik HB. J Biol Chem. 1996;271:20885–20894. doi: 10.1074/jbc.271.34.20885. [DOI] [PubMed] [Google Scholar]
- 6.Rothman RB, Baumann MH, Dersch CM, Appel J, Houghten RA. Synapse. 1999;33:239–246. doi: 10.1002/(SICI)1098-2396(19990901)33:3<239::AID-SYN8>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 7.Schabacker DS, Kirschbaum KS, Segre M. Immunology. 2000;100:48–56. doi: 10.1046/j.1365-2567.2000.00004.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ho M, Segre M. Brain Res. 2000;872:231–235. doi: 10.1016/s0006-8993(00)02491-4. [DOI] [PubMed] [Google Scholar]
- 9.Whitelegg NR, Rees AR. Protein Eng. 2000;13:819–824. doi: 10.1093/protein/13.12.819. [DOI] [PubMed] [Google Scholar]
- 10.Scatchard G. Ann NY Acad Sci. 1949;51:660. [Google Scholar]
- 11.Bator JM, Reading CL. J Immunol Methods. 1989;125:167–176. doi: 10.1016/0022-1759(89)90090-2. [DOI] [PubMed] [Google Scholar]
- 12.Wilson PC, de Bouteiller O, Liu YJ, Potter K, Banchereau J, Capra JD, Pascual V. J Exp Med. 1998;187:59–70. doi: 10.1084/jem.187.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Wildt RM, van Venrooij WJ, Winter G, Hoet RM, Tomlinson IM. J Mol Biol. 1999;294:701–710. doi: 10.1006/jmbi.1999.3289. [DOI] [PubMed] [Google Scholar]
- 14.Chowdhury PS, Pastan I. Nat Biotechnol. 1999;17:568–572. doi: 10.1038/9872. [DOI] [PubMed] [Google Scholar]
- 15.Rothman RB, Becketts KM, Radesca LR, de Costa BR, Rice KC, Carroll FI, Dersch CM. Life Sci. 1993;53:PL267–272. doi: 10.1016/0024-3205(93)90602-y. [DOI] [PubMed] [Google Scholar]
- 16.Zhang L, Elmer LW, Little KY. Brain Res Mol Brain Res. 1998;59:66–73. doi: 10.1016/s0169-328x(98)00138-7. [DOI] [PubMed] [Google Scholar]
- 17.Little KY, Kirkman JA, Carroll FI, Breese GR, Duncan GE. J Neurochem. 1993;61:1996–2006. doi: 10.1111/j.1471-4159.1993.tb07435.x. [DOI] [PubMed] [Google Scholar]
- 18.De Keyser J, De Backer JP, Ebinger G, Vauquelin G. J Neurochem. 1989;53:1400–1404. doi: 10.1111/j.1471-4159.1989.tb08530.x. [DOI] [PubMed] [Google Scholar]
- 19.Jerne NK. Ann Immunol (Paris) 1974;125C:373–389. [PubMed] [Google Scholar]
- 20.Jerne NK. Immunol Rev. 1984;79:5–24. doi: 10.1111/j.1600-065x.1984.tb00484.x. [DOI] [PubMed] [Google Scholar]
- 21.Bona CA. Proc Soc Exp Biol Med. 1996;213:32–42. doi: 10.3181/00379727-213-44033. [DOI] [PubMed] [Google Scholar]
- 22.Schreiber AB, Couraud PO, Andre C, Vray B, Strosberg AD. Proc Natl Acad Sci USA. 1980;77:7385–7389. doi: 10.1073/pnas.77.12.7385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Glasel JA, Myers WE. Life Sci. 1985;36:2523–2529. doi: 10.1016/0024-3205(85)90149-3. [DOI] [PubMed] [Google Scholar]
- 24.Cacalano NA, Cleveland WL, Erlanger BF. J Immunol. 1991;147:3012–3017. [PubMed] [Google Scholar]
- 25.Leu JG, Chen BX, Diamanduros AW, Erlanger BF. Proc Natl Acad Sci USA. 1994;91:10690–10694. doi: 10.1073/pnas.91.22.10690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fields BA, Goldbaum FA, Ysern X, Poljak RJ, Mariuzza RA. Nature. 1995;374:739–742. doi: 10.1038/374739a0. [DOI] [PubMed] [Google Scholar]
- 27.Saphire EO, Parren PW, Pantophlet R, Zwick MB, Morris GM, Rudd PM, Dwek RA, Stanfield RL, Burton DR, Wilson IA. Science. 2001;293:1155–1159. doi: 10.1126/science.1061692. [DOI] [PubMed] [Google Scholar]
- 28.Kastin AJ, Pan W, Maness LM, Banks WA. Brain Res. 1999;848:96–100. doi: 10.1016/s0006-8993(99)01961-7. [DOI] [PubMed] [Google Scholar]
- 29.Girod J, Fenart L, Regina A, Dehouck M, Hong G, Scherrmann J, Cecchelli R, Roux F. J Neurochem. 1999;73:2002–2008. [PubMed] [Google Scholar]
- 30.Fiala M, Gan XH, Zhang L, House SD, Newton T, Graves MC, Shapshak P, Stins M, Kim KS, Witte M, Chang SL. Adv Exp Med Biol. 1998;437:199–205. doi: 10.1007/978-1-4615-5347-2_22. [DOI] [PubMed] [Google Scholar]