

Abstract
DOCK2, a hematopoietic cell-specific, atypical guanine nucleotide exchange factor, controls lymphocyte migration through ras-related C3 botulinum toxin substrate (Rac) activation. Dedicator of cytokinesis 2–engulfment and cell motility protein 1 (DOCK2•ELMO1) complex formation is required for DOCK2-mediated Rac signaling. In this study, we identified the N-terminal 177-residue fragment and the C-terminal 196-residue fragment of human DOCK2 and ELMO1, respectively, as the mutual binding regions, and solved the crystal structure of their complex at 2.1-Å resolution. The C-terminal Pro-rich tail of ELMO1 winds around the Src-homology 3 domain of DOCK2, and an intermolecular five-helix bundle is formed. Overall, the entire regions of both DOCK2 and ELMO1 assemble to create a rigid structure, which is required for the DOCK2•ELMO1 binding, as revealed by mutagenesis. Intriguingly, the DOCK2•ELMO1 interface hydrophobically buries a residue which, when mutated, reportedly relieves DOCK180 from autoinhibition. We demonstrated that the ELMO-interacting region and the DOCK-homology region 2 guanine nucleotide exchange factor domain of DOCK2 associate with each other for the autoinhibition, and that the assembly with ELMO1 weakens the interaction, relieving DOCK2 from the autoinhibition. The interactions between the N- and C-terminal regions of ELMO1 reportedly cause its autoinhibition, and binding with a DOCK protein relieves the autoinhibition for ras homolog gene family, member G binding and membrane localization. In fact, the DOCK2•ELMO1 interface also buries the ELMO1 residues required for the autoinhibition within the hydrophobic core of the helix bundle. Therefore, the present complex structure reveals the structural basis by which DOCK2 and ELMO1 mutually relieve their autoinhibition for the activation of Rac1 for lymphocyte chemotaxis.
Keywords: X-ray crystallography, NMR, protein complex, CDM proteins, immunology
Dedicator of cytokinesis 2 (DOCK2), a large protein with a molecular mass of 213 kDa, is specifically expressed in hematopoietic cells (1). DOCK2 plays a critical role in lymphocyte migration and activation by regulating the actin cytoskeleton through ras-related C3 botulinum toxin substrate (Rac) activation (2, 3), and the deletion of DOCK2 enables long-term cardiac allograft survival (4). DOCK2 also controls various immunological functions, including helper T cell differentiation (5), neutrophil chemotaxis (6, 7), and type I interferon induction in plasmacytoid dendritic cells (8). DOCK2 belongs to the CDM family (Caenorhabditis elegans CED-5, mammalian DOCK180, and Drosophila melanogaster Myoblast city) of the evolutionally conserved, atypical guanine nucleotide exchange factors (GEFs) for the Rho-family GTPases. There are 11 mammalian members (DOCK180, DOCK2-11) of the CDM family. The CDM proteins share the DOCK-homology regions (DHR)-1 and DHR-2 (also known as Docker domains), and lack the Dbl homology (DH) and pleckstrin homology (PH) domains typically present in the mammalian Rho-family GEFs (6, 9, 10). DOCK2 mediates the Rac GEF reaction by means of the DHR-2 domain (6, 10, 11). DOCK2 also interacts with phosphatidylinositol 3,4,5-triphosphate [PtdIns(3,4,5)P3] through the DHR-1 domain (7, 12), to target the GEF activity to the plasma membrane. Recently, the crystal structure of the DHR-1 domain of DOCK180 was determined (13), as well as those of the DHR-2 domains of DOCK2 and DOCK9 bound with their substrates, Rac1 and cell division cycle 42 (Cdc42), respectively (14, 15).
DOCK2, like DOCK180 and DOCK3-5, has a Src-homology 3 (SH3) domain at its N terminus. The N-terminal 502-residue region, including the SH3 domain, of DOCK2 interacts with engulfment and cell motility protein 1 (ELMO1), a mammalian homologue of C. elegans CED-12 (16). ELMO1 contains the N-terminal ras homolog gene family, member G (RhoG)-binding region, the ELMO domain, the PH domain, and the C-terminal sequence with three PxxP motifs. The C-terminal region of ELMO1, including the Pro-rich sequence, binds the SH3-containing region of DOCK2, which is required for DOCK2 to activate Rac in vivo (16). Similarly, DOCK180 forms a ternary complex with ELMO1 and Rac (17, 18), which is essential for the in vivo catalytic activity (17, 19). ELMO1 binds to the N-terminal region (about 200 residues), including the SH3 domain, of DOCK180 (17, 19). However, an ELMO1 mutant lacking the Pro-rich sequence retains the ability to bind DOCK180 (20), suggesting that the region flanking the Pro-rich sequence of ELMO1 also participates in DOCK180 binding. Furthermore, the DOCK180•ELMO1 complex was proposed to function as a bipartite GEF for Rac, in which the DHR-2 domain of DOCK180 and the PH domain of ELMO1 cooperatively function (20). The crystal structure of the ELMO1 PH domain has recently been determined, and the unusual α-helical N-terminal extension of the PH domain in ELMO1 was found to be essential for DOCK180 binding and DOCK180-mediated Rac signaling (21).
In the absence of ELMO1, the N-terminal region, including the SH3 domain, of DOCK180 interacts with the DHR-2 domain, which may facilitate the autoinhibition of the DOCK180 GEF activity (22). ELMO1 binding to the SH3 domain disrupts the SH3•DHR-2 interaction, and allows Rac binding to the DHR-2 domain for the GEF activity (22). This autoinhibition of DOCK180 is relieved by the mutation of Ile31 in the SH3 domain, even though the location of this residue is distant from the PxxP-binding pocket. ELMO1 by itself is also reportedly autoinhibited because of the interaction of the ELMO inhibitory domain (EID), lying between the C-terminal PxxP motif and the PH domain, with the ELMO autoregulatory domain (EAD) residing between the N-terminal RhoG-binding domain and the ELMO domain. Actually, EAD binding to DOCK180, as well as the mutations of Met692 and Glu693 in the EAD, disrupts this intramolecular inhibitory interaction, resulting in RhoG binding and membrane localization (23). Consequently, DOCK2 and ELMO1 mutually relieve their autoinhibitions by an unknown interaction mode involving the DOCK2 N-terminal and ELMO1 C-terminal regions, a scenario that is far more complicated than simple SH3•PxxP binding.
In the present study, we identified the regions required for the interaction between DOCK2 and ELMO1, and determined their complex structure. The SH3 domain of DOCK2 binds to the C-terminal sequence of ELMO1 much more extensively than other SH3 domains, which only bind the PxxP motif. Moreover, the α-helical region following the SH3 domain forms an intermolecular five-helix bundle with both the N- and C-terminal extensions of the PH domain of ELMO1. Thus, the entire region is assembled into a rigid structural unit. The residues required for the respective autoinhibitions (i.e., Ile31 of DOCK2 and Met692 of ELMO1) are buried in the hydrophobic core of the intermolecular assembly, thus revealing the structural basis of the mutual autoinhibition relief. Further mutagenesis demonstrated that this unique assembly mode is important for ELMO1 to relieve the autoinhibition of DOCK2, so it can participate in Rac activation in lymphocyte chemotaxis.
Results
NMR Analysis of the DOCK2 SH3 Domain Interaction with ELMO1 Peptides.
The C terminus of ELMO1 contains six Pro residues located close together (residues 707, 710, 711, 712, 714, and 717), constituting three potential PxxP motifs, which are the potential SH3 domain binding sites. To define the interaction between the DOCK2 SH3 domain and the ELMO1 Pro-rich sequence, we first generated various fusion constructs for cell-free protein synthesis (24–26) and obtained one sample suitable for structure determination by NMR spectroscopy (Fig. S1). The fusion construct includes a 26-residue ELMO1 peptide (residues 697–722) fused to the N terminus of the DOCK2 SH3 domain (residues 8–70) (Fig. 1A and Fig. S2A). For comparison, we also analyzed the DOCK2 SH3 domain alone and compared its chemical shifts for the backbone 15N and 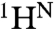 atoms with those in the fusion sample. The residues located in the arginine and threonine pair (RT) loop, the n-Src loop, β4, and the helix-like loop between β4 and β5 exhibited large chemical shift changes (Fig. S2B).
atoms with those in the fusion sample. The residues located in the arginine and threonine pair (RT) loop, the n-Src loop, β4, and the helix-like loop between β4 and β5 exhibited large chemical shift changes (Fig. S2B).
Fig. 1.

Mutually interactive regions of DOCK2 and ELMO1. (A) Domain organizations of DOCK2 and ELMO1. Known protein interaction and functional regions are indicated. Blue and red bars indicate the DOCK2 and ELMO1 regions included in the NMR construct. Green and yellow bars indicate the regions used for the crystal structure determination. ELMO1-interacting region (EIR) and DOCK2-interacting region (DIR) indicate the ELMO1-interacting region and the DOCK2-interacting region, respectively. (B) Overviews of the crystal structure of the DOCK2•ELMOl complex. Ribbon representations of the DOCK2•ELMOl complex. DOCK2 is colored green and ELMO1 is yellow. The six proline residues in the ELMO1 Pro-rich tail are colored red. The two views are related by a 90° rotation about the vertical axis.
We determined the solution structure of the DOCK2 SH3–ELMO1 peptide fusion complex (Table S1). The rmsd from the mean structure was 0.78 Å for the backbone atoms and 1.19 Å for all heavy (non-hydrogen) atoms in the well-ordered region (residues 704–722 for the ELMO1 peptide and residues 9–68 for the DOCK2 SH3 domain) (Fig. S2C). The topology of the DOCK2 SH3 domain resembles those of other SH3 domains, and consists of five β-strands packed in two β-sheets, which form a β-barrel-like structure (Fig. S2D). The ELMO1 peptide binds to the DOCK2 SH3 domain as a polyproline II helix in a class II orientation (27, 28). Tyr44 of DOCK2 is buried in the center of the interface and hydrophobically interacts with Pro714 and Pro717 of ELMO1. These analyses revealed that, among the three candidates, P714-x-x-P717 serves as the PxxP motif. On the other hand, the N-terminal seven residues of the ELMO1 peptide (residues 697–703) were poorly defined, probably because of high mobility, although they were necessary for the structure determination (Figs. S1 and S2C). This observation prompted us to examine whether any additional interactions, besides the SH3 domain–PxxP motif interaction, also occur between DOCK2 and ELMO1.
Crystal Structure of the Complex of the DOCK2 and ELMO1 Interacting Domains.
First, we determined the ELMO1-interacting region of DOCK2 and the DOCK2-interacting region of ELMO1 that are suitable for crystallographic studies (see SI Text). We generated longer fragments of DOCK2 (residues 1–177; the SH3 domain and the flanking region) and ELMO1 (residues 532–727; including the PH domain and the Pro-rich region) by cell-free protein synthesis (Fig. S3). The DOCK2 fragment precipitated during the synthesis, but it was prepared as a soluble protein complex by cell-free coexpression with the ELMO1 fragment. The purified complex between the DOCK2 and ELMOl fragments (hereafter referred to as the DOCK2•ELMOl complex) was stable and monomeric with 1∶1 stoichiometry, as determined by analytical ultracentrifugation (Fig. S4A).
We determined the crystal structure of the DOCK2•ELMOl complex (Table S2). The asymmetric unit contained two virtually identical DOCK2•ELMOl complexes. Overall, DOCK2 and ELMO1 form a large, rigid complex involving the entire regions of the two protein fragments. The DOCK2 structure is composed of the N-terminal SH3 domain (residues 1–69) and a three-helix bundle domain (residues 81–167; Dα1–3). The electron density of the 11-residue region connecting the two domains was not observed. The ELMO1 structure is composed of the PH domain (residues 560–674), its N- and C-terminal extended helices Eα1 and Eα3 (residues 532–558 and 681–697, respectively), and the C-terminal Pro-rich tail (residues 700–727) (Fig. 1B and Fig. S3). The DOCK2 SH3 domain and the ELMO1 Pro-rich tail interact extensively with each other. Furthermore, Dα1–3 of DOCK2 and Eα1 and Eα3 of ELMO1 form an intermolecular five-helix bundle (Fig. 1B). A surface area of approximately 2,370 Å2 is buried in the interface between DOCK2 and ELMO1.
The SH3 Domain of DOCK2 and the Pro-Rich Tail of ELMO1.
The overall fold of the DOCK2 SH3 domain and the binding manner of the PxxP-motif (Pro714–Pro717) are consistent with those defined by our NMR analysis. However, the SH3 domain reinforces the interaction with the regions flanking the PxxP motif of ELMO1 (Fig. 2A). The overall intermolecular binding mode of the DOCK2 SH3 domain with ELMOl, burying approximately 1,450 Å2, is by far the most extensive among the SH3 domain complexes characterized to date (SI Text and Fig. S5). The interactions occur in three regions. First, the 28-residue Pro-rich tail (Asp700–Asn727) winds around the SH3 domain, burying approximately 1,310 Å2. The DOCK2 SH3 domain forms extensive hydrophobic interactions and several hydrophilic interactions with the ELMO1 tail. At the center of the interface, the side-chain imino group of Trp44 of DOCK2 hydrogen bonds with the main-chain carbonyl group of Lys715, within the PxxP motif of ELMO1. The DOCK2 SH3 domain forms six hydrogen bonds that interact with the C-terminal half (Asn719–Asn727), following the PxxP motif, of the ELMO1 tail. In contrast, the DOCK2 SH3 domain binds the N-terminal half (Asp700–Ile713), mostly through hydrophobic interactions.
Fig. 2.

DOCK2•ELMO1 interactions around the SH3 domain. (A) Interface between the DOCK2 SH3 domain and the ELMO1 Pro-rich tail. The six proline residues (Pro707, Pro710, Pro711, Pro712, Pro714, and Pro717) in the ELMO1 Pro-rich tail are colored red, and the labels of the two Pro residues forming the PxxP motif are underlined. Hydrogen bonds are indicated by dashed lines. The DOCK2 and ELMO1 residues are labeled in black and red, respectively. (B) Interface between the DOCK2•ELMO1 five-helix bundle and the DOCK2 SH3 domain. The DOCK2 and ELMO1 residues are labeled in black and red, respectively.
Second, the DOCK2 SH3 domain also interacts with Eα1 and Eα3 of ELMO1, through its RT loop (Fig. 2B). Among the RT-loop residues, Tyr17 stacks its side chain between those of Pro532 of Eα1 and Pro710 in the ELMO1 tail, whereas Ile31 and Gly32 hydrophobically interact with Ile533 of Eα1, Leu698 of Eα3, and Leu701 and Ile706 of the tail. In addition, Asn18 hydrogen bonds with Pro532 and Elu535 of Eα1, whereas Gln30 hydrogen bonds with Arg697 of Eα3. Within the SH3 domain, Ile31, corresponding to Ile32, which is reportedly required for the autoinhibition of DOCK180 (21), is located at the center of the interactions (Fig. 2B).
Third, DOCK2 interacts with the boundary between Eα3 and the ELMO1 tail. Lys67, at the C terminus of the SH3 domain, hydrogen bonds with Leu699 of Eα3 and Asp700 in the tail. Arg128 and Ser129 of Dα1 form hydrogen bonds with Leu699 of Eα3 and Glu702 in the tail. The residues involved in these interactions in the three regions are well conserved in the CDM family (Fig. S3).
Five-Helix Bundle.
The intermolecular five-helix bundle consists of Dα1–3 of DOCK2 and Eα1 and Eα3 of ELMO1 (Fig. 3 and Fig. S4 B and C). In the DOCK2 three-helix bundle domain, Dα2 is arranged in an antiparallel manner relative to Dα1 and Dα3. Almost all of the interactions between the three helices are hydrophobic, involving a large number of aliphatic side chains of Ile, Val, and Leu residues and those of Trp98 and Met120 (Fig. S4B). Similarly, the ELMO1 Eα1 and Eα3 helices are arranged in an antiparallel manner and form multiple hydrophobic interactions involving the side chains of Ile and Leu residues (Fig. S4C). On the other hand, Dα1 and Dα2 of DOCK2 and Eα1 and Eα3 of ELMO1 form the interface between the DOCK2 and ELMO1 helix bundles, burying approximately 930 Å2 (Fig. 3). The interface involves not only a number of hydrophobic interactions, but also several hydrophilic interactions. The residues Leu95, Trp96, Trp102, Val107, Phe114, Met121, and Met125 of DOCK2 hydrophobically interact with Ile544, Leu547, Ile548, Leu681, Leu689, Met692, Leu696, and Leu699 of ELMO1. Met692 of ELMO1, which is required for ELMO autoinhibition (23), is buried at the center of the five-helix bundle. In addition, the Glu693 side chain of ELMO1 hydrogen bonds with the Lys103 side chain of DOCK2. Similarly, the Tyr106 side chain of DOCK2 hydrogen bonds with the Asp685 side chain of ELMO1, and the Arg128 side chain of DOCK2 hydrogen bonds with the Leu699 main-chain carbonyl group of ELMO1.
Fig. 3.

DOCK2•ELMO1 five-helix bundle formation. Interface within the DOCK2•ELMO1 five-helix bundle, showing the interactions between the DOCK2 and ELMO1 helices. Hydrogen bonds are indicated by dashed lines.
The hydrophobic residues within the three helices of DOCK2 are highly conserved among the CDM family members (Fig. S3A). Actually, the mutations of such hydrophobic residues, L96D/W99E and I132D/L133D in DOCK180 (L95/W98 and L131/L132, respectively, in DOCK2), reportedly disrupt the ability to bind ELMO1 (21). Therefore, DOCK180 forms a three-helix bundle similar to that of DOCK2, and uses it to interact with ELMO1. On the other hand, nearly all of the hydrophobic interactions between the two helices are conserved in the ELMO family (Fig. S3B). The DOCK2 residues on the interface with ELMO1 are well conserved with those of DOCK180, whereas three residues, Trp96, Met121, Met125, are not conserved (Fig. S3A). As for ELMO1, the present interaction mode with DOCK2 (Fig. 3) agrees well with the results of a previous mutational study, which showed that Leu547 and Ile548 of ELMO1 are required for DOCK180 binding (21). Therefore, DOCK180 is also likely to form a five-helix bundle, as observed in the present DOCK2•ELMO1 complex.
In addition, the C terminus of Eα1 of ELMO1 forms hydrophobic interactions with the Eα2 helix within the PH domain (Fig. S4D). These hydrophobic residues are strictly conserved in the ELMO family (Fig. S3B). The Eα1•Eα2 interactions fix the arrangement of the PH domain relative to the five-helix bundle (Fig. 1B). The core structure of the PH domain of ELMO1 in the DOCK2•ELMOl complex is essentially the same as that of the ELMO1 fragment consisting of the PH domain and the Eα1 helix (21). However, Eα1 significantly bends, at the central Pro542, and interacts with Eα3 to form the five-helix bundle with DOCK2, whereas Eα1 was straight in the previous structure, due to crystal packing (21) (Fig. S4E).
Characterization of DOCK2 and DOCK180 Mutants.
On the basis of the DOCK2•ELMO1 interactions described above, we mutated several ELMO1-interacting residues in the full-length DOCK2 protein. First, six putative hydrogen-bonding residues in the SH3 domain (Fig. 2 A and B) were changed to Ala (Fig. 4A). Although the Q30A and S62A mutations had no effect, the W4A, E39A, W44A, and R46A mutations significantly reduced the ELMO1 binding. In particular, the Ala mutation of Trp44, which is located in the center of the SH3•PxxP interface (Fig. 2A), had the most profound effect on ELMO1 binding (Fig. 4A). Therefore, the hydrogen bonds of Trp44 with the PxxP motif, and those of Trp4, Glu39, and Arg46 with the C-terminal region downstream of the PxxP motif, are important for ELMO1 binding. On the other hand, the point mutations of Trp102 and Tyr106 of DOCK2 (Fig. 3) to Glu (Y106E and W102E) substantially reduced ELMO1 binding (Fig. 4B). Similarly, the R128A mutation nearly completely abolished the ELMO1 binding, whereas the S129A mutation did not affect it (Fig. 4A). Arg128 is located at the C terminus of the Dα2 helix, in the vicinity of the SH3 domain, and interacts with the main chain at the border between the Eα3 helix and the Pro-rich tail of ELMO1 (Figs. 2B and 3). These results indicate that both binding interfaces, the SH3 domain and the helix bundle, are important for the DOCK2•ELMO1 complex formation.
Fig. 4.

The SH3–PxxP interaction is essential for ELMO1-binding in DOCK2. (A–C) Interaction of the ELMO1 binding residues with DOCK2. HEK293T cell lysates expressing HA-tagged DOCK2 (WT and mutants) were incubated with GST-tagged ELMO1. The DOCK2-HA in the pull-downs and total cell lysates was detected by immunoblotting with an anti-HA antibody. The results were quantified by densitometry and are expressed as the ratio of ELMO1 bound to total DOCK2 after normalization to the WT value, with an arbitrary value of 1. The results indicate mean ± SD of three independent experiments. (D) Chemotactic response of BW5147α-β- cells expressing DOCK2 (WT or mutant). BW5147α-β- cells (−) stably expressing HA-tagged DOCK2 (clones 4B3 and 4C2) or HA-tagged DOCK2 W44A (clones 2B9 and 3G10) were allowed to migrate on stromal cells prepared from mouse lymph nodes. The migration speed was calculated by analyzing at least 148 cells per clone. **P < 0.01. (E) The effect of the W44A mutation on Rac activation. BW5147α-β- cells (−) expressing wild-type DOCK2 (clone 4C2) or the W44A mutant (clone 3G10) were stimulated with CXCL12 (400 ng/mL) before the assay. Data are representative of three independent experiments. (F) The effect of the W44A mutation on ELMO1-mediated relief of DOCK2 autoinhibition. The GFP-tagged DOCK2 N-terminal fragment (Met1–Glu200), with or without the W44A mutation, and the FLAG-tagged DOCK2 C-terminal fragment (Ser1195–Met1828) were expressed in HEK293T cells in the presence or absence of the HA-tagged ELMO1. TCL, total cell lysate. The association between the N- and C-terminal fragments was analyzed by reciprocal immunoprecipitations with the relevant antibodies. Data are representative of two independent experiments.
In contrast to the marked reduction in ELMO1 binding by the DOCK2 W44A mutation (Fig. 4A), the point mutations of the corresponding Trp residue of DOCK180 (W45A and W45K) reportedly had minimal effects on ELMO1 binding (21, 22). Consequently, we confirmed the observed difference by mutating the corresponding Trp residues to Lys in DOCK2 and DOCK180. The W44K mutation almost completely abolished the ability of DOCK2 to bind to ELMO1, whereas the DOCK180 W45K mutation retained it (Fig. 4C). Therefore, these mutagenesis results indicated that DOCK2 and DOCK180 differ in their requirements for the SH3•PxxP interaction in ELMO1 binding. This finding led us to examine the functional significance of the SH3•PxxP interaction in DOCK2-mediated chemotaxis. For this purpose, we stably expressed wild-type DOCK2 and the W44A mutant in BW5147α-β- cells, which lack the endogenous expression of DOCK2 (2). As compared with the nontransfected cells, the expression of wild-type DOCK2 in BW5147α-β- cells significantly increased the migration speed (Fig. 4D). However, the expression of the W44A mutant failed to restore the motility in two independent clones, although its expression level was comparable to that of the wild-type DOCK2 (Fig. 4D). Consistent with this finding, chemokine-induced Rac activation was readily detected in BW5147α-β- cells expressing the wild-type DOCK2, but not the W44A mutant (Fig. 4E). Collectively, these results indicate that the SH3•PxxP interaction is required for DOCK2-mediated Rac activation.
The SH3 domain of DOCK180 has been implicated in its autoinhibition by binding to the catalytic DHR-2 domain and thus blocking Rac access to it (22). To explore the mechanism by which the SH3•PxxP interaction regulates the DOCK2-mediated Rac activation, we transiently expressed the DOCK2 N-terminal fragment (Met1–Glu200), with or without the W44A mutation, and the C-terminal fragment (Ser1195–Met1828), including the DHR-2 domain, in HEK293T cells. Regardless of the presence of the W44A mutation, the N-terminal fragment of DOCK2 associated with the C-terminal fragment, suggesting that a similar autoinhibitory mechanism occurs in DOCK2 (Fig. 4F). However, upon the coexpression of ELMO1, this association was disrupted in the case of the wild-type DOCK2, but not the W44A mutant (Fig. 4F). These results suggest that the SH3•PxxP interaction is required to relieve the autoinhibition of DOCK2 and to allow Rac access to the catalytic DHR-2 domain.
Discussion
DOCK2 is a key regulator of the immune system. In this study, we determined the crystal structure of the complex of the DOCK2 and ELMO1 interacting regions, which revealed a large, rigid assembly. Three α-helices of DOCK2 and two α-helices of ELMO1 form a helix bundle. The SH3 domain of DOCK2 interacts with the long Pro-rich tail and the two α-helices of ELMO1 in an unusually extensive manner. Together, these structural elements, as well as the PH domain of ELMO1, accrete to form a rigid assembly. A mutational analysis demonstrated that the entire assembly of these two regions is required for the full-length DOCK2 and ELMO1 proteins to form a complex and activate Rac in vivo.
The SH3 domain of DOCK180 reportedly interacts with the catalytic DHR-2 domain, which may sterically block Rac access to the DHR-2 domain and thereby cause the autoinhibition of DOCK180 (22). Actually, in the present study, we observed the binding of the N-terminal ELMO-interacting region with the C-terminal DHR-2 containing region of DOCK2 and its reduction by the assembly with ELMO (Fig. 4F). Therefore, a similar mechanism underlies the autoinhibition of the DOCK2 GEF activity. In the case of DOCK180, the mutation of the conserved Ile residue within the SH3 domain (I32K) disrupts the SH3•DHR-2 interaction, and relieves DOCK180 of the autoinhibition (22). This Ile residue corresponds to Ile31 in DOCK2, within the RT loop of the SH3 domain. Ile31 is not close to the PxxP-binding site, but is buried at the center of the hydrophobic interface with the Eα1 and Eα3 helices and the following peptide segment of ELMO1 (Fig. 2B).
On the other hand, the ELMO1 protein also has an autoinhibitory mechanism (23). The EID binds to the EAD in the autoinhibited form, and EAD binding with DOCK180 relieves ELMO1 of its autoinhibition. Therefore, DOCK2/DOCK180 and ELMO1 mutually relieve their autoinhibited forms (Fig. 5A). The double mutation of Met692 and Glu693 to Ala within the EAD reportedly disrupts the interaction between the EID and the EAD. Correspondingly, Met692 is buried at the center of the hydrophobic core of the intermolecular five-helix bundle, whereas the Glu693 side chain interacts with the Lys103 side chain of DOCK2, as revealed by the present structure. As described above, the hydrophobic core of the five-helix bundle also buries Ile31 of DOCK2, which is the putative key residue for the DOCK2 autoinhibition. Therefore, the tight assembly between the ELMO1-interacting region of DOCK2 and the DOCK2-interacting region of ELMO1 deprives these regions of their respective intramolecular autoinhibitory interactions. Thus, the structural basis by which DOCK2 and ELMO1 mutually relieve their autoinhibited forms has now been revealed (Fig. 5A).
Fig. 5.

Hypothetical model of the DOCK2•ELMO1•Rac1 ternary complex. (A) The schematic model of the mutual relief of DOCK2 and ELMO1 from their autoinhibited forms for Rac activation. EIR, ELMO1-interacting region; DIR, DOCK2-interacting region. (B) Side view, showing the membrane attachment of the DOCK2•ELMO1•Rac1 ternary complex. The membrane is shown as a light yellow bar. The membrane attachment site of Rac1 at the C terminus is depicted by a red dashed line. (C) Top view from the membrane side. The present structure of the DOCK2•ELMO1 complex (green and yellow) is overlaid onto the structures of the DOCK180 DHR-1 domain (orange, PDB code 3L4C) (13), the dimeric DOCK2 DHR-2 domain•Rac1 complex (blue and red, PDB code 3B13), and importin subunit β-1 (ARM, armadillo-like repeat) (gray, PDB code 2QNA) (30). The two views are related by a 90° rotation about the horizontal axis.
The PH domain of ELMO1 is firmly fixed, relative to the five-helix bundle, in the rigid DOCK2•ELMO1 complex. The ELMO1 PH domain reportedly cooperates with DOCK180 to activate Rac (20). The mutations of the PH domain residues, such as G559A and W665A, in ELMO1 abrogate the formation of the functional ternary complex with DOCK180 and nucleotide-free Rac, although they do not affect the DOCK180 binding (20). In the present DOCK2•ELMO1 structure, Gly559 and Trp665 of ELMO1 are involved in intramolecular interactions between the Eα1 and Eα2 helices (Fig. S4D). Therefore, their mutations are expected to impair the proper positioning of the ELMO1 PH domain relative to the other portions of the DOCK•ELMO complex. Thus, the fixed arrangement of the ELMO1 PH domain may be important for functional ternary complex formation with DOCK and Rac.
In the typical mammalian RhoGEFs, such as Trio and Dbs, the DH and PH domains act as a “cassette” and coordinately bind their cognate Rho GTPases (Rac1 and Cdc42) for efficient activation (29). By analogy to the DH–PH domains, the PH domain of ELMO1 may facilitate Rac1 binding by the DHR-2 domain. A structural model of the DOCK180•Rac complex has been proposed (13), and thus we integrated the present DOCK2•ELMO1 structure into a similar model of the DOCK2•Rac1 complex (Fig. 5 B and C, and Table S3). In this preliminary model, the two DOCK2•ELMO1 complexes might interact with two Rac1 molecules on the flat surface of the plasma membrane. The PH domain of ELMO1 may be arranged similarly to that in the Rac1•Trio DH–PH cassette (Protein Data Bank code 2NZ8).
The ELMO1-interacting residues of DOCK2 are also highly conserved in DOCK180. In fact, the present structure indicates that their overall interaction modes may be similar to each other. Nevertheless, we found an interesting difference between DOCK2 and DOCK180. The mutation of Trp44, within the SH3 domain of DOCK2, abolished the ELMO1 binding ability, but the corresponding mutation of Trp45 of DOCK180 did not affect it (Fig. 4C). Phe63, within the SH3 domain of DOCK2, forms hydrophobic interactions with the ELMO1 PxxP-tail, but it is replaced with Tyr in DOCK180. Similarly, in the binding interface in the five-helix bundle, Trp96, Met121, and Met125 of DOCK2 are replaced with Arg, Ile, and Ile, respectively, in DOCK180 (Fig. S3A). These amino acid replacements may alter the ELMO-binding affinity between DOCK2 and DOCK180. Alternatively, an additional region of DOCK180, including Gly171, whose mutation reduces ELMO1 binding (22), may contribute to the tighter binding of ELMOl to DOCK180, as compared to DOCK2. In the current DOCK2•ELMOl complex structure, the corresponding Gly residue of DOCK2 (Gly172) is located within a disordered C-terminal region (residues 168–177), and thus is not involved in ELMO1 binding.
Our observation that DOCK2•ELMO1 complex formation is essential for the DOCK2 function makes it an attractive therapeutic target for immunologic disorders, such as autoimmune diseases and graft rejection, caused by tissue infiltration by lymphocytes. Although the mechanism by which DOCK180 maintains the SH3 domain-independent, tight binding of ELMO1 remains elusive, the observed difference raises the possibility that ELMO1 binding might be specifically disrupted by small compounds that bind to the interface in DOCK2, but not DOCK180. In this context, our structure will provide important information for the development of an inhibitor that selectively disrupts DOCK2•ELMO1 complex formation.
Materials and Methods
The protein samples for structure determination were produced by the cell-free synthesis method. The solution structure of the DOCK2 SH3–ELMO1 fusion protein [Protein Data Bank (PDB) code 2RQR] was determined by NMR. The crystal structures of the DOCK2•ELMOl (PDB code 3A98) and DOCK2-DHR2•Rac1 (PDB code 3B13) complexes were determined by the single-wavelength anomalous dispersion and molecular replacement methods, respectively. Detailed experimental procedures are provided in the SI Text.
Supplementary Material
Acknowledgments.
We thank Y. Tomo, M. Aoki, E. Seki, M. Ikari, K. Hanada, T. Matsuda, N. Matsuda, Y. Motoda, Y. Fujikura, T. Harada, S. Watanabe, and T. Nagira for preparing proteins for NMR experiments. We also thank H. Niwa, K. Katsura, S. Tojo, M. Ueno, K. Honda, M. Goto, M. Inoue, M. Aoki, A. Sakamoto, M. Toyama, Y. Terazawa, T. Fujimoto, M. Wakiyama, A. Shimada, and Y. Fujii for technical assistance. We are grateful to Dr. Osamu Ohara (Kazusa DNA Research Institute, Japan) for the DOCK2 clone (KIAA0209). This work was supported by the RIKEN Structural Genomics/Proteomics Initiative, the National Project on Protein Structural and Functional Analyses, and by the Targeted Proteins Research Program, the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 2RQR, 3A98, 3B13).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1113512109/-/DCSupplemental.
References
- 1.Nishihara H, et al. Non-adherent cell-specific expression of DOCK2, a member of the human CDM-family proteins. Biochim Biophys Acta. 1999;1452:179–187. doi: 10.1016/s0167-4889(99)00133-0. [DOI] [PubMed] [Google Scholar]
- 2.Fukui Y, et al. Haematopoietic cell-specific CDM family protein DOCK2 is essential for lymphocyte migration. Nature. 2001;412:826–831. doi: 10.1038/35090591. [DOI] [PubMed] [Google Scholar]
- 3.Sanui T, et al. DOCK2 is essential for antigen-induced translocation of TCR and lipid rafts, but not PKC-theta and LFA-1, in T cells. Immunity. 2003;19:119–129. doi: 10.1016/s1074-7613(03)00169-9. [DOI] [PubMed] [Google Scholar]
- 4.Jiang H, et al. Deletion of DOCK2, a regulator of the actin cytoskeleton in lymphocytes, suppresses cardiac allograft rejection. J Exp Med. 2005;202:1121–1130. doi: 10.1084/jem.20050911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tanaka Y, et al. T helper type 2 differentiation and intracellular trafficking of the interleukin 4 receptor-alpha subunit controlled by the Rac activator Dock2. Nat Immunol. 2007;8:1067–1075. doi: 10.1038/ni1506. [DOI] [PubMed] [Google Scholar]
- 6.Kunisaki Y, et al. DOCK2 is a Rac activator that regulates motility and polarity during neutrophil chemotaxis. J Cell Biol. 2006;174:647–652. doi: 10.1083/jcb.200602142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nishikimi A, et al. Sequential regulation of DOCK2 dynamics by two phospholipids during neutrophil chemotaxis. Science. 2009;324:384–387. doi: 10.1126/science.1170179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gotoh K, et al. Selective control of type I IFN induction by the Rac activator DOCK2 during TLR-mediated plasmacytoid dendritic cell activation. J Exp Med. 2010;207:721–730. doi: 10.1084/jem.20091776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Côté JF, Vuori K. GEF what? Dock180 and related proteins help Rac to polarize cells in new ways. Trends Cell Biol. 2007;17:383–393. doi: 10.1016/j.tcb.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meller N, Irani-Tehrani M, Kiosses WB, Del Pozo MA, Schwartz MA. Zizimin1, a novel Cdc42 activator, reveals a new GEF domain for Rho proteins. Nat Cell Biol. 2002;4:639–647. doi: 10.1038/ncb835. [DOI] [PubMed] [Google Scholar]
- 11.Miyamoto Y, Yamauchi J, Sanbe A, Tanoue A. Dock6, a Dock-C subfamily guanine nucleotide exchanger, has the dual specificity for Rac1 and Cdc42 and regulates neurite outgrowth. Exp Cell Res. 2007;313:791–804. doi: 10.1016/j.yexcr.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 12.Côté JF, Motoyama AB, Bush JA, Vuori K. A novel and evolutionarily conserved PtdIns(3,4,5)P3-binding domain is necessary for DOCK180 signalling. Nat Cell Biol. 2005;7:797–807. doi: 10.1038/ncb1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Premkumar L, et al. Structural basis of membrane targeting by the Dock180 family of Rho family guanine exchange factors (Rho-GEFs) J Biol Chem. 2010;285:13211–13222. doi: 10.1074/jbc.M110.102517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kulkarni K, Yang J, Zhang Z, Barford D. Multiple factors confer specific Cdc42 and Rac activation by Dock exchange factors. J Biol Chem. 2011;286:25341–25351. doi: 10.1074/jbc.M111.236455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang J, Zhang Z, Roe SM, Marshall CJ, Barford D. Activation of Rho GTPases by DOCK exchange factors is mediated by a nucleotide sensor. Science. 2009;325:1398–1402. doi: 10.1126/science.1174468. [DOI] [PubMed] [Google Scholar]
- 16.Sanui T, et al. DOCK2 regulates Rac activation and cytoskeletal reorganization through interaction with ELMO1. Blood. 2003;102:2948–2950. doi: 10.1182/blood-2003-01-0173. [DOI] [PubMed] [Google Scholar]
- 17.Brugnera E, et al. Unconventional Rac-GEF activity is mediated through the Dock180-ELMO complex. Nat Cell Biol. 2002;4:574–582. doi: 10.1038/ncb824. [DOI] [PubMed] [Google Scholar]
- 18.Gumienny TL, et al. CED-12/ELMO, a novel member of the CrkII/Dock180/Rac pathway, is required for phagocytosis and cell migration. Cell. 2001;107:27–41. doi: 10.1016/s0092-8674(01)00520-7. [DOI] [PubMed] [Google Scholar]
- 19.Grimsley CM, et al. Dock180 and ELMO1 proteins cooperate to promote evolutionarily conserved Rac-dependent cell migration. J Biol Chem. 2004;279:6087–6097. doi: 10.1074/jbc.M307087200. [DOI] [PubMed] [Google Scholar]
- 20.Lu M, et al. PH domain of ELMO functions in trans to regulate Rac activation via Dock180. Nat Struct Mol Biol. 2004;11:756–762. doi: 10.1038/nsmb800. [DOI] [PubMed] [Google Scholar]
- 21.Komander D, et al. An alpha-helical extension of the ELMO1 pleckstrin homology domain mediates direct interaction to DOCK180 and is critical in Rac signaling. Mol Biol Cell. 2008;19:4837–4851. doi: 10.1091/mbc.E08-04-0345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu M, et al. A Steric-inhibition model for regulation of nucleotide exchange via the Dock180 family of GEFs. Curr Biol. 2005;15:371–377. doi: 10.1016/j.cub.2005.01.050. [DOI] [PubMed] [Google Scholar]
- 23.Patel M, et al. An evolutionarily conserved autoinhibitory molecular switch in ELMO proteins regulates Rac signaling. Curr Biol. 2010;20:2021–2027. doi: 10.1016/j.cub.2010.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kigawa T, Matsuda T, Yabuki T, Yokoyama S. In: Cell-free Protein Synthesis. Spirin Alexander S, Swartz James R., editors. New York: Wiley; 2007. pp. 83–97. [Google Scholar]
- 25.Kigawa T, et al. Preparation of Escherichia coli cell extract for highly productive cell-free protein expression. J Struct Funct Genomics. 2004;5:63–68. doi: 10.1023/B:JSFG.0000029204.57846.7d. [DOI] [PubMed] [Google Scholar]
- 26.Matsuda T, et al. Improving cell-free protein synthesis for stable-isotope labeling. J Biomol NMR. 2007;37:225–229. doi: 10.1007/s10858-006-9127-5. [DOI] [PubMed] [Google Scholar]
- 27.Cesareni G, Panni S, Nardelli G, Castagnoli L. Can we infer peptide recognition specificity mediated by SH3 domains? FEBS Lett. 2002;513:38–44. doi: 10.1016/s0014-5793(01)03307-5. [DOI] [PubMed] [Google Scholar]
- 28.Mayer BJ. SH3 domains: Complexity in moderation. J Cell Sci. 2001;114:1253–1263. doi: 10.1242/jcs.114.7.1253. [DOI] [PubMed] [Google Scholar]
- 29.Chhatriwala MK, Betts L, Worthylake DK, Sondek J. The DH and PH domains of Trio coordinately engage Rho GTPases for their efficient activation. J Mol Biol. 2007;368:1307–1320. doi: 10.1016/j.jmb.2007.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wohlwend D, Strasser A, Dickmanns A, Ficner R. Structural basis for RanGTP independent entry of spliceosomal U snRNPs into the nucleus. J Mol Biol. 2007;374:1129–1138. doi: 10.1016/j.jmb.2007.09.065. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.