

Abstract
PdxB catalyzes the second step in the biosynthesis of pyridoxal phosphate by oxidizing 4-phospho-D-erythronate (4PE) to 2-oxo-3-hydroxy-4-phospho-butanoate (OHPB) with concomitant reduction of NAD+ to NADH. PdxB is a nicotino-enzyme wherein the NAD(H) cofactor remains tightly bound to PdxB. It has been a mystery how PdxB performs multiple turnovers since addition of free NAD+ does not re-oxidize the enzyme-bound NADH following conversion of 4PE to OHPB. We have solved this mystery by demonstrating that a variety of physiologically available α-ketoacids serve as oxidants of PdxB to sustain multiple turnovers. In a coupled assay using the next two enzymes of the biosynthetic pathway for pyridoxal phosphate (SerC and PdxA), we have found that α-ketoglutarate, oxaloacetic acid, and pyruvate are equally good substrates for PdxB (kcat/Km values ~ 1 × 104 M-1s-1). The kinetic parameters for the substrate 4PE include a kcat of 1.4 s-1, a Km of 2.9 μM, and a kcat/Km of 6.7 × 106 M-1s-1. Additionally, we have characterized the stereochemistry of α-ketoglutarate reduction by showing that D-2-HGA, but not L-2-HGA, is a competitive inhibitor vs. 4PE and a noncompetitive inhibitor vs. α-ketoglutarate.
Vitamin B6 (pyridoxal, pyridoxine, pyridoxamine) is an essential metabolite in all known organisms. Pyridoxal phosphate (PLP) is required for >100 enzymatic reactions mostly related to amino acid metabolism (e.g. transamination, decarboxylation, β-elimination or –substitution, γ-elimination or –substitution). There exist three routes by which organisms acquire Vitamin B6 (1). In animals Vitamin B6 is acquired through the diet and is modified by salvage enzymes including transaminases, phosphatases, and kinases. Vitamin B6 deficiency in humans leads to a wide range of symptoms including increased excretion of xanthurenic acid, epileptic convulsions, dermatitis, and decreased lymphocyte counts, reflecting the many diverse functions of PLP-dependent enzymes (2). In most microorganisms and plants, PLP is synthesized from glutamine, ribose-5-phosphate, and glyceraldehyde-3-phosphate by the enzyme complex Pdx1/Pdx2 (3 - 5).
Surprisingly, PLP biosynthesis in E. coli is quite different from the more widely used Pdx1/Pdx2-dependent pathway (1). E. coli uses seven enzymes in a bifurcated pathway that converts pyruvate, glyceraldehyde-3-phosphate, and erythrose-4-phosphate to PLP via the intermediates 1-deoxy-D-xylulose 5-phosphate and 1-amino-propan-2-one-3-phosphate (Figure 1). This pathway is the only biosynthetic route to PLP in the γ-proteobacteria and a variant of this pathway is found in some α-proteobacteria such as Sinorhizobium meliloti. It seems that the γ-proteobacteria lost the ancestral and more universal Pdx1/Pdx2-dependent pathway at some point. The more recent acquisition of a PLP biosynthetic pathway in these bacteria appears to have occurred by recruitment of enzymes from other pathways. For example, the first enzyme in the pathway, Epd, is closely related to GapB (>40% identity), the glyceraldehyde-3-phosphate dehydrogenase found in glycolysis. The third enzyme in the pathway, SerC, functions as a glutamate-dependent transaminase in both the serine and PLP biosynthetic pathways.
Figure 1.
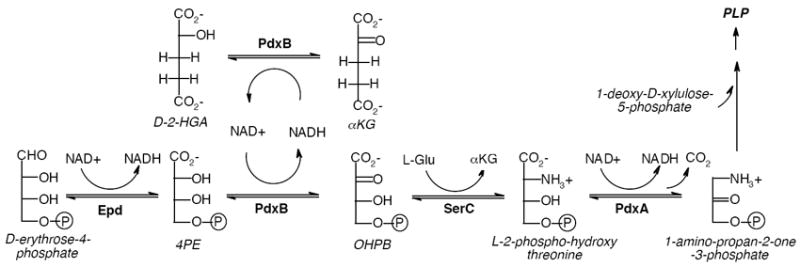
The initial stages of the biosynthetic pathway toward PLP in E. coli. OAA and pyruvate also serve to regenerate the NAD+-form of PdxB.
PdxB catalyzes the second step of the PLP biosynthetic pathway in E. coli, converting 4-phospho-D-erythronate (4PE) to 2-oxo-3-hydroxy-4-phospho-butanoate (OHPB). PdxB has been studied with some difficulty by Winkler and co-workers (6). PdxB contains tightly bound NAD+ and/or NADH that cannot be removed by extensive dialysis. Although single turnover reactions in the presence of 4PE can be detected by formation of NADH bound to PdxB, sustained multiple turnovers in the presence of exogenous NAD+ have not been observed. Addition of SerC to drive the thermodynamically unfavored reaction forward leads to a modest increase in formation of bound NADH, still without multiple turnovers. The lack of sustained catalytic activity was postulated to be due to strong product inhibition, a missing protein subunit, suboptimal reaction conditions, or interference by the His6-tag used to purify the protein (6).
We have further characterized PdxB and solved the conundrum of an enzyme that can apparently only perform a single turnover. Specifically, we have achieved multiple turnover activity by addition of various α-ketoacids. Using a coupled assay with the enzymes SerC and PdxA, we have characterized the kinetic properties of α-ketoglutarate (αKG), oxaloacetic acid (OAA), and pyruvate as co-substrates of PdxB. Surprisingly, all three are likely to be equally effective co-substrates in vivo. Additionally we have characterized the stereochemistry of the reaction with the co-substrate αKG.
Materials and Methods
Materials
Bugbuster, benzonase, and the expression plasmids pET21b and pET45b were obtained from EMD Biosciences. IPTG was obtained from Research Products International. Lysozyme, NAD+, NADH, guanidine HCl, urea, 2,6-dichloroindole phenol (DCIP), N-nitrosodimethylamine (NDMA), methylene blue, potassium ferricyanide, pyruvate, OAA, αKG, D-2-hydroxyglutarate, and L-2-hydroxyglutarate were obtained from Sigma-Aldrich. 4PE was synthesized by Yehor Novikov (procedure to be published elsewhere) and was characterized by 1H-NMR (500 MHz, D2O): d 4.08 (1H, d, J = 5.0 Hz), 3.97 (1H, m), 3.84 (1 H, m), 3.78 (1H, m). αKG was characterized by 1H-NMR (500 MHz, D2O): 2.96 (2H, t), 2.39 (2H, t). D-2-HGA was characterized by 1H-NMR (500 MHz, D2O): 1.82(1H, m), 1.95 (1H, m), 2.23 (2H, m), 3.99 (1H, dd).
Purification of enzymes
The genes encoding PdxB and PdxA from E. coli strain JW2317 were cloned into pET45b using the restriction enzymes KpnI and BamHI to generate expression clones with a His6-tag at the N-terminus. The gene encoding SerC was cloned into pET21b using the restriction enzymes NdeI and XhoI to generate an expression clone with a His6-tag at the C-terminus. All clones were verified by DNA sequencing.
PdxB, SerC, and PdxA were expressed in E. coli BL21(DE3). A single colony from a fresh transformation was used to inoculate a 10 mL culture that was grown overnight at 37 °C in Luria Broth supplemented with 50 μg/mL ampicillin. The starter culture was then transferred to 1 L of Luria Broth and grown to mid-log phase (0.5 – 0.7 at OD600) at room temperature. Protein expression was induced by addition of IPTG to a final concentration of 0.2 mM and the culture was shaken at 200 rpm at room temperature for an additional 4 h. Cells were harvested by centrifugation at 3,500 × g at 4 °C for 20 min and cell pellets were stored at -80 °C.
For purification of the enzymes, cell pellets were resuspended and incubated for 20 min at room temperature in Bugbuster (5 mL/g of cell paste) with added benzonase (20 U/mL) and lysozyme (1 mg/mL). The extract was then centrifuged for 30 min at 4 °C at 20,000 × g. The supernatant was loaded onto a 5 mL Ni-agarose column (Amersham Biosciences) that had been equilibrated with 50 mM potassium phosphate (pH 6.8) containing 0.5 M KCl and 20 mM imidazole. The column was washed with 200 mL of the same buffer at 1 mL/min using an AKTA FPLC. Proteins were eluted with a linear gradient from 0 to 500 mM imidazole in the same buffer. Fractions were collected and analyzed by SDS-PAGE. The purest fractions were pooled and concentrated using a centrifugal ultra-filtration unit (Millipore, 30 kDa cut-off). Enzymes were exchanged into 50 mM Tris-HCl (pH 8.0) containing 50 mM NaCl (TN buffer) by extensive dilution and re-concentration. Purified enzymes were stored at -80 °C following addition of glycerol to 20% (v/v). Protein concentrations were determined by BioRad protein assay using bovine serum albumin as a standard. No contaminating bands were observed for PdxB. SerC and PdxA were judged to be >85% pure by SDS-PAGE and shown to be free of 4PE dehydrogenase activity.
Extraction and identification of the NAD+/NADH cofactor from PdxB
The tightly bound NAD+/NADH cofactor was extracted from PdxB using three different methods. First, heat treatment (120 °C, 20 s) of PdxB (5 mg/mL in TN buffer) was followed by centrifugation to remove the precipitated protein (20,000 × g for 3 min). Second, PdxB (39 mg/mL) was brought to 5.5 M guanidine HCl by addition of solid guanidine HCl under constant stirring for 20 min at room temperature. The samples were then diluted 10 – 30-fold into TN buffer and centrifuged to remove the precipitated protein as above. Third, PdxB (39 mg/mL) was brought to 7.8 M urea by addition of solid urea under constant stirring for 20 min at room temperature. The samples were then diluted 10-fold into ethanol and centrifuged to remove the precipitated protein as above. All three methods yielded a protein-free preparation of the cofactor as monitored by UV-Vis spectroscopy (absorbance maxima at 260 and/or 340, but not 280 nm).
To confirm the identity of heat-extracted NAD+ and NADH, co-migration with authentic samples was demonstrated using HPLC. Injection of samples onto a Microsorb- MV 300-5 C18 (Varian) column (40 × 4.6 mm) with isocratic elution using 100 mM potassium phosphate buffer (pH 6.5) yielded retention times of 5.2 – 5.4 min and 9.0 – 9.3 min for NAD+ and NADH, respectively. Authentic NAD+ and NADH samples remained intact through the brief heat treatment used to extract the cofactor from PdxB.
Attempts to exchange NAD+ for the enzyme-bound NADH were attempted by partially denaturing PdxB. PdxB (~5 mg/mL) was incubated with varying concentrations of guanidine HCl (0.5 – 4 M), urea (4.5 – 7.5 M), or 3.5 M MgCl2 in the presence of 1 mM NAD+ at 4°C for 1 h in TND buffer (TN buffer containing 1 mM DTT). The samples were then twice diluted 2-fold in TND buffer containing 1 mM NAD+, with 30 min incubations between dilutions. The denaturant and free NAD+ were then removed by successive dilution (>10-fold) and re-concentration using a centrifugal ultra-filtration unit until the denaturant and NAD+ concentrations were below 10 mM and 1 μM, respectively. Samples were evaluated by monitoring the absorbance of protein (ε280 of 28,420 M-1cm-1 for PdxB; ProtParam tool at ExPASy) and NADH (ε340 of 6,220 M-1cm-1).
Assay of PdxB with artificial oxidants
Assays of PdxB (20 nM – 100 μM) with artificial oxidants were performed in TN buffer at room temperature in the presence or absence of 1 mM 4PE. Concentrations and extinction coefficients of the oxidants were as follows: DCIP (0.1 mM, ε600= 16,100 M-1cm-1), NDMA (35 μM, ε440= 35,400 M-1cm-1), methylene blue (36 μM, ε660= 71,547 M-1cm-1), and potassium ferricyanide (1.2 mM, ε420= 1,020 M-1cm-1). Reactions were followed by monitoring the time-dependent change in absorbance of the oxidant at its λmax using a HP8452 spectrophotometer.
Coupled assays of PdxB activity
PdxB was assayed using the coupling enzymes SerC and PdxA. Standard reaction conditions contained in 500 μL: 4PE (100 μM), αKG (2 mM), L-Glu (2 mM), NAD+ (1 mM), SerC (2.6 μM), PdxA (2.0 μM), and PdxB (20 nM) in TND buffer. All components were incubated for 20 min at room temperature prior to initiation of the reaction by addition of PdxB. Background rates in the absence of 4PE, αKG, or PdxB were <10% of rates for reactions containing all components. For determinations of Kms, assays were performed in triplicate using at least seven different substrate concentrations: 4PE (0.8 – 50 μM), αKG (5 – 2000 μM), pyruvate (10 – 1000 μM), OAA (5 – 400 μM). The concentration of 4PE was held constant at 29 μM for the determinations of the Kms for various α-keto acids and the concentration of αKG was held constant at 2 mM for the determination of the Km for 4PE. The concentration of 4PE was quantitated by an end-point assay. Inhibition experiments with D-2-HGA were performed at varying concentrations of 4PE (4 – 75 μM) at a fixed concentration of αKG (250 μM) or varying concentrations of αKG (15 – 300 μM) at a fixed concentration of 4PE (6 μM). All reactions were monitored using an HP8452 spectrophotometer by following formation of NADH (ε340= 6,220 M-1cm-1) over 1 – 3 min. Observed reaction rates were linear with respect to time and not limited by the coupling enzymes SerC or PdxA. Observed reaction rates depended linearly on the concentration of PdxB. Data fitting to the Michaelis-Menten equation was performed using weighted least-squares in Excel using the Solver module.
Results
PdxB contains a tightly bound NADH
PdxB was overexpressed with a His6-tag in E. coli and purified to >95% purity by nickel affinity chromatography. The protein migrated as an apparent dimer by gel filtration chromatography, in agreement with the dimeric form observed in the crystal structure of the PdxB homolog from Pseudomonas aeruginosa (7). As purified, the enzyme exhibits a prominent spectral signal at 322 nm (Figure 2), suggestive of a nicotino-protein in the reduced state wherein the typical 340 nm absorbance of NADH is significantly blue-shifted in the apolar environment of the protein (8). This blue-shift is comparable to that of other nicotino-proteins for which λmax varies from 320 – 330 nm (9 - 11). The bound cofactor could be extracted from PdxB by treatment with heat, guanidine HCl, or urea to yield a protein-free preparation with absorbance maxima at 260 and 340 nm but not 280 nm. After heat treatment to extract the cofactor, the ratio of A259/A340 of 2.5 – 2.7 suggests that the sample contained primarily NADH (ε259 = 16,900 M-1cm-1; ε340 = 6,220 M-1cm-1; ratio A259/A340 = 2.7 (12)). The released cofactor was shown to be 10% NAD+ and 90% NADH by HPLC analysis, consistent with the absorbance spectrum (Table 1). Assuming an ε280 of 28,420 M-1cm-1 for PdxB and using an ε259 of 16,900 M-1cm-1 for NAD(H) (12), there exist 0.75 – 0.9 equivalents of cofactor per subunit of PdxB. Thus PdxB, as isolated, exists primarily with bound cofactor in the reduced state.
Figure 2.
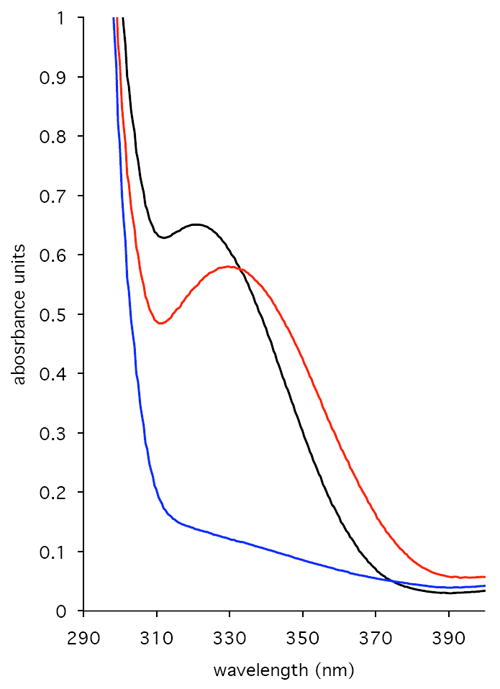
UV-Vis absorbance spectra of PdxB (130 uM). Black line: as isolated containing tightly bound NADH. Red line: after addition of 4PE (0.32 mM). Blue line: after addition of αKG (0.58 mM).
Table 1.
Spectroscopic properties of PdxB and its tightly bound cofactor.
| PdxB treatment | λmax (nm) on protein | λmax (nm) after heat extraction and protein precipitation | Ratio NADH:NAD+ (by HPLC) |
|---|---|---|---|
| As isolated | 280, 322 | 260, 340 | 90:10 |
| + 4PE | 280, 333 | 260, 340 | 90:10 |
| + αKG | 280 | 260 | 2:98 |
Addition of the substrate 4PE to PdxB caused a significant change in the absorbance properties of PdxB, with a 10 nm red-shift and an apparent increase in absorbance at 340 nm (Figure 2). However, heat extraction and HPLC analysis of the cofactor following incubation with 4PE revealed no additional NADH formation; the sample still contained 90% NADH (Table 1). This suggests the formation of a non-productive PdxB:4PE:NADH complex that has a less shielded protein environment for the bound NADH.
We attempted to exchange the bound NADH on PdxB for NAD+ in order to study the forward enzymatic reaction with the substrate 4PE. Partial denaturation in the presence of NAD+ has been used previously to replace varying levels of reduced NADH on nicotino-proteins such as UDP-galactose 4-epimerase (13). However, in the presence of high concentrations of denaturant (guanidine HCl, urea, or MgCl2) and after extended incubation with NAD+, PdxB was always recovered with a significant fraction of bound NADH (0.4 – 0.9 equivalents; see Experimental section). We conclude that NADH is very tightly bound to PdxB.
The NADH bound to PdxB is highly unreactive with a variety of external oxidants
Because some nicotino-proteins have been shown to use external NAD+ to re-oxidize tightly bound NADH (14 - 15), we attempted to assay PdxB in the presence of NAD+. Incubation of 4PE and NAD+ (each at 1 mM) with PdxB (20 nM – 100 uM) did not lead to accumulation of NADH, as measured spectroscopically at 340 nm. This lack of catalytic turnover has been previously described (6). Similarly, NADP+ was not reduced in the presence of PdxB and 4PE. Further, in the presence or absence of 4PE, the tightly bound NADH could not be re-oxidized in a single-turnover experiment by addition of NAD+ or NADP+.
Because some nicotino-enzymes have been shown to use artificial oxidants to re-oxidize tightly bound NADH and allow multiple turnovers (10, 16 - 18), we attempted to achieve multiple turnovers of PdxB using 2,6-dichloroindole phenol (DCIP), N-nitrosodimethylamine (NDMA), methylene blue, and potassium ferricyanide. DCIP, NDMA and methylene blue could not be reduced in the presence of PdxB and 4PE. Neither could any of these reagents oxidize the tightly bound NADH in the absence of 4PE in a single-turnover experiment. NADH extracted from PdxB using urea denaturation did react with DCIP, and quantitation indicated 0.9 equivalents of NADH/subunit, consistent with the HPLC results described above. Potassium ferricyanide could be used to oxidize PdxB in a single turnover, albeit slowly. Over the course of ~30 min, 1.0 – 1.2 enzyme equivalents of ferricyanide could be reduced to ferrocyanide as detected by the loss of absorbance at 420 nm (data not shown). Addition of 4PE to the reaction had no effect, indicating that multiple turnovers could not be sustained by addition of this oxidant. Thus, the tightly bound NADH is highly protected and unreactive within its binding pocket on PdxB.
Tightly-bound NADH on PdxB can be oxidized by αKG, but by itself αKG does not promote multiple turnovers
Based on the reported ability of SerA, the closest characterized homolog of PdxB, to reduce αKG (19), we tested the ability of PdxB to reduce αKG to 2-hydroxyglutarate (2- HGA). Addition of αKG to PdxB leads to a rapid (<5 s) loss in absorbance at 322 nm (Figure 2), suggesting oxidation of the bound NADH to NAD+. Given the lack of reactivity of reduced PdxB to numerous other external oxidants (see above), this suggests a specific interaction between PdxB and αKG. Heat extraction of the cofactor confirmed the loss of absorbance at 340 nm compared to that of cofactor derived from untreated enzyme. Additionally, HPLC analysis of the extracted cofactor showed the presence of 95 – 98% NAD+ (Table 1). The reactivity of PdxB with αKG is perhaps not surprising, as 4PE closely resembles the reduced product 2-HGA, particularly in the spacing of the negative charges at the extremities of the molecule. It is also interesting to note that αKG is generated as a product of SerC, the next enzyme in the biosynthetic pathway of PLP (Figure 1).
We next used NMR to test whether incubation of PdxB with 4PE and αKG could yield successive cycles of oxidation of 4PE to form OHPB and reduction of αKG to form 2-HGA. Despite using high concentrations of enzyme (4 - 40 uM) and substrates (0.5 - 2 mM) and extended incubation (1 min - overnight), only 4PE and αKG could be detected by NMR. Specifically, the doublet representing the proton on C-2 of 4PE at 4.08 ppm remained at equal intensity compared to the other proton signals of 4PE. Additionally, no new signals at 3.99 ppm due to the C-2 proton of 2-HGA or below 2.0 ppm due to the C-3 or C-4 protons could be detected. These results suggest that the reaction catalyzed by PdxB, like the very similar reaction catalyzed by SerA (oxidation of 3-phosphoglycerate to 3-phosphohydroxypyruvate) is thermodynamically unfavorable (20 - 21). We were unable to probe the reaction in the reverse direction because OHPB is not readily available.
PdxB can be made to perform multiple turnovers by coupling with SerC and PdxA
We next coupled the PdxB reaction to the next two enzymes in the PLP biosynthetic pathway, SerC and PdxA (Figure 1). This allowed convenient monitoring of the progress of the reaction by following at 340 nm the reduction of NADH catalyzed by PdxA. (The cycling of NAD+/NADH on PdxB is expected to be spectroscopically silent.) NADH formation was observed only in the presence of all the following components: 4PE and αKG as substrates of PdxB, L-Glu as a substrate for SerC, and NAD+ as a substrate for PdxA. Using this coupled assay we determined the kinetic parameters for 4PE and αKG (Figure 3, Table 2). The Km of 2.9 μM and kcat/Km of 6.7 × 106 M-1s-1 for 4PE suggest a well-evolved enzyme – substrate interaction. The Km for αKG of 93 μM is below expected intracellular concentrations (22), suggesting that αKG is a physiologically relevant substrate. The kcat/Km of 1.1 × 104 M-1s-1 for αKG is within the range expected for typical metabolic enzymes.
Figure 3.
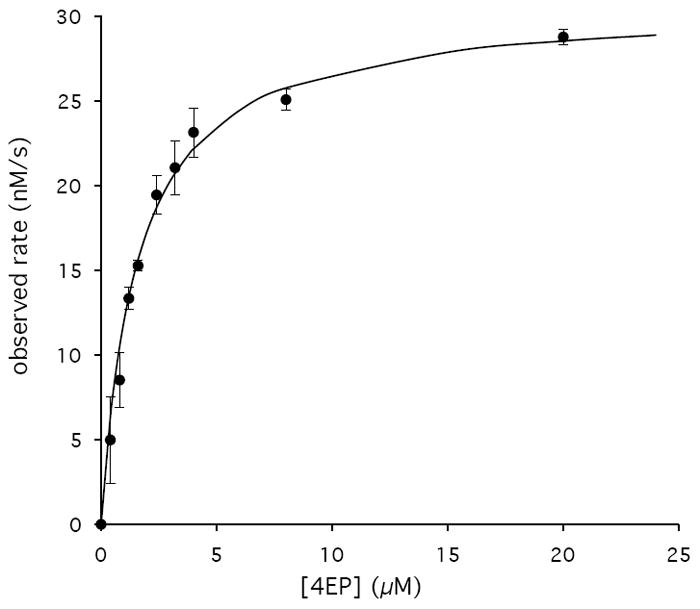
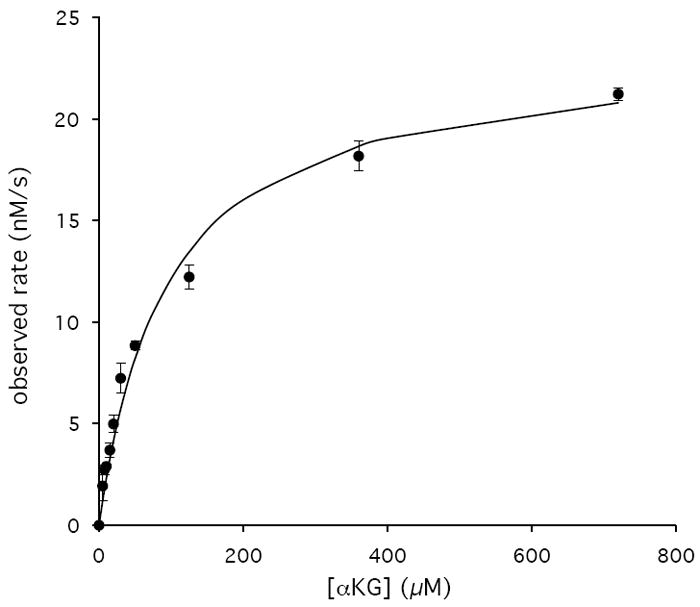
Representative Km and kcat determinations for PdxB of 4PE (A) and αKG (B) performed in triplicate with error bars indicated for each data point and the lines representing best fits to the Michaelis-Menten equation.
Table 2.
Kinetic parameters for PdxB using the coupled assay
| substrate | kcat (s-1) | Km (μM) | kcat/Km (M-1s-1) | Intracellular concentration | Relative flux through PdxB |
|---|---|---|---|---|---|
| 4PE | 1.4 ± 0.32 | 2.9 ± 1.3 | (6.7 ± 2.8) × 106 | n.a. | n.a. |
| αKG | 1.0 ± 0.24 | 93 ± 19 | (1.1 ± 0.24) × 104 | 0.1 – 0.9 mM* | 1.1 – 9.9 s-1 |
| OAA | 1.1 ± 0.31 | 86 ± 19 | (1.3 ± 0.21) × 104 | 0.15 – 0.25 mM‡ | 2.0 – 3.3 s-1 |
| pyruvate | 1.3 ± 0.14 | 128 ± 20 | (9.9 ± 0.16) × 103 | 0.4 mMˆ | 4.0 s-1 |
To establish the stereochemistry of αKG reduction, we used D-2-HGA and L-2-HGA as inhibitors of PdxB in the coupled assay. D-2-HGA yielded an IC50 of 102 μM, whereas for L-2HGA the IC50 was greater than 2 mM (Figure 4). These results suggest that αKG is reduced to D-2-HGA, which is consistent with the stereochemistry of the 4PE substrate for the forward reaction. Further kinetic investigations showed that D-2-HGA was competitive with respect to 4PE (Ki = 30 μM) and noncompetitive with respect to αKG (Kic = 17 μM, Kiu = 208 μM) (Figure 5).
Figure 4.
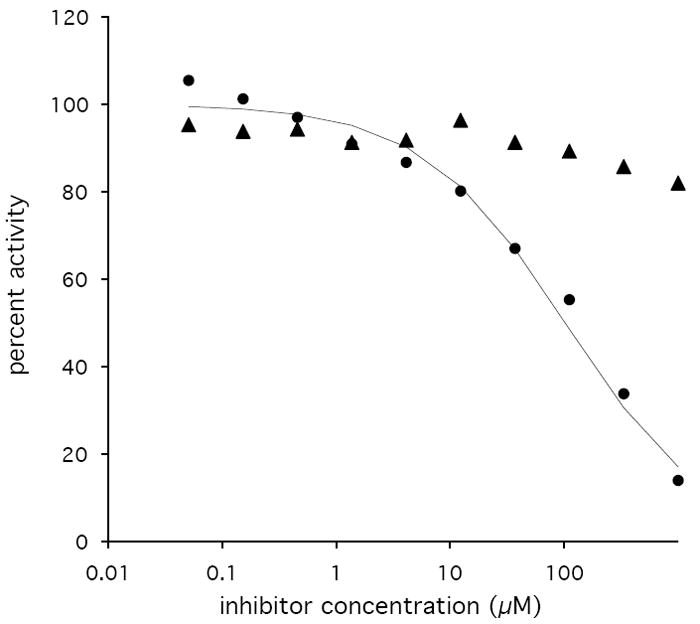
Inhibition of PdxB by L-2-HGA (triangles) and D-2-HGA (circles) using 4PE (6 μM) and αKG (250 μM) as substrates. The curve for inhibition by D-2-HGA is the best fit using the equation: percent activity = 100/(1 + [I]/IC50).
Figure 5.
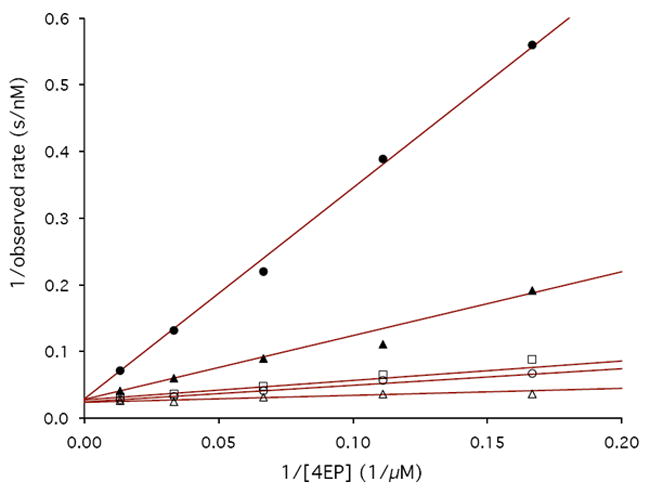
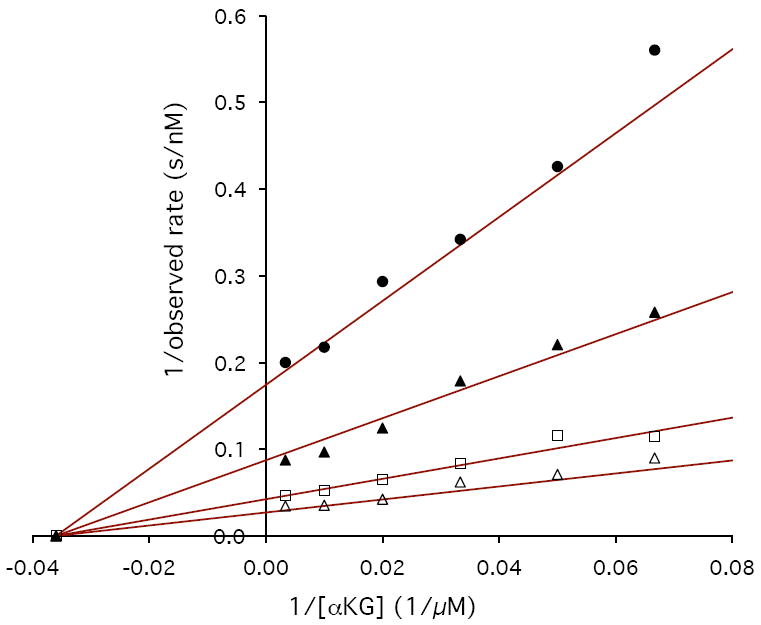
Lineweaver-Burke plots demonstrating the inhibition of PdxB by D-2-HGA. A) 4PE vs. D-2-HGA at concentrations of 0 μM (open triangles), 50 μM (open circles), 70 μM (open squares), 300 μM (solid triangles), and 1 mM (solid circles). B) αKG vs. D-2-HGA at concentrations of 0 μM (open triangles), 70 μM (open squares), 300 μM (solid triangles), and 1 mM (solid circles).
PdxB utilizes multiple small molecule oxidants
We tested other α-keto acids for their ability to re-oxidize PdxB and support multiple turnovers in the coupled assay. Despite their shorter length, OAA and pyruvate were also able to support multiple turnovers of PdxB in the coupled assay with SerC and PdxA. Kinetic characterization yielded kcat/Km values for OAA and pyruvate that were essentially identical to that for αKG (Table 2). Given intracellular concentrations for αKG of 100 – 900 μM (22), OAA of 150 – 250 μM (23), and pyruvate of 400 μM (24), it appears that all of these compounds can serve as physiological re-oxidants of PdxB.
Discussion
Nicotino-enzymes contain tightly bound NAD(P)(H) that does not dissociate to exchange with the pool of free NAD(P)(H) in solution. Nicotino-enzymes are not only involved in oxidation of alcohols (18), but can also perform redox-silent reactions such as epimerizations (e.g. UDP-galactose epimerase) and more complex reactions (e.g. dehydroquinate synthase). Thus, the NAD(P)(H) bound to a nicotino-protein can be thought of as a cofactor (analogous to flavin or PLP) instead of as a coenzyme (analogous to acetyl-CoA). The tightly bound NAD(P)(H) cofactor in nicotino-enzymes often exists in a highly buried protein environment (25). The crystal structure of PdxB from Pseudomonas aeruginosa (7) (45% identity with PdxB from E. coli) yields some insight into the nature of the tight binding interaction between PdxB and NAD. NAD binds in an extended conformation in the nucleotide binding domain using numerous specific interactions with PdxB and is completely shielded from the surface. The nicotinamide ring is in a pocket containing both hydrophobic and hydrophilic residues, but no water. The two different subunits in the dimer have slightly different conformations in the active site cleft, with subunit A being more closed than subunit B. The more buried NAD in subunit A has an inorganic phosphate bound nearby, but there is no room for a carbohydrate substrate. This closed conformation in subunit A is likely responsible for the blue-shifted NADH-bound form of PdxB (Figure 2). In subunit B of the crystal structure the active site is opened to allow the substrate analog L-(+)-tartrate to bind next to the nicotinamide ring. The more open conformation is consistent with the red-shift in absorbance seen upon addition of 4PE to the isolated enzyme (Figure 2).
Nicotino-proteins that perform a net oxidation reaction generally fall into four categories in terms of the mechanism by which the bound NADH is re-oxidized. Most simply, enzymes such as mammalian acetaldehyde dehydrogenase (15) catalyze their reactions in the presence of external NAD+ that is capable of re-oxidizing the tightly bound NADH. SerA, the closest homolog to PdxB, may fall into this first category, although it is not clear whether SerA is a nicotino-enzyme (19, 20, 27). In a variation of this first case, hydride transfer from external NADH to the enzyme-bound NAD+ in methanol dehydogenase from Bacillus methanolicus is facilitated by another protein (27). Enzymes such as carveol dehydrogenase from Rhodococcus erythropolis (10) and alcohol dehydrogenase from Amycolatopsis methanolica (16) instead use artificial external oxidants such as DCIP and NDMA. In a physiological setting it is thought these enzymes transfer their reducing equivalents directly to the electron transport chain, as no other small-molecule oxidants have been discovered. Enzymes such as formaldehyde dismutase (11) generate a mixture of methanol and formic acid from formaldehyde in the absence of any other external oxidant or reductant. That is, enzyme-bound NADH formed by oxidation of formaldehyde can be oxidized by reaction with a second formaldehyde to yield methanol and NAD+. Finally, enzymes such as malate-lactate transhydrogenase (28, 29) and glucose-fructose oxidoreductase (30) exchange reducing equivalents between a variety of small molecules via the tightly bound NAD(P)H cofactor. Our observation that PdxB can utilize a number of physiologically available α-keto acids suggests that PdxB belongs to this fourth type of nicotino-enzyme.
By knowing the intramolecular concentrations of αKG, OAA, and pyruvate, it is possible to compare the relative fluxes of these three α-keto acids through PdxB (Table 2). In vivo, all three are likely to contribute to regeneration of the NAD+ form of PdxB that is required to oxidize 4PE. By comparison, SerA has been reported to have low activity with OAA (5% compared to αKG) and no activity with pyruvate (19). Although low levels of promiscuous activity are quite common for many enzymes, most metabolic enzymes show much greater specificity for their physiological substrates (31). That is, broad specificity for a variety of substrates, while common for house-keeping enzymes such as cytochrome P450s and transaminases, is rare among enzymes in biosynthetic or metabolic pathways. It is unclear whether this broad specificity towards α-keto acids in PdxB evolved to fulfill some physiological need for versatility or instead is an unrefined practical solution. Also, it is unclear what benefit the cell derives by having PdxB depend on α-keto acids instead of NAD+ for catalytic activity. Perhaps low ratios of NAD+ vs. NADH during conditions that favor growth and biosynthetic processes means that α-keto acids are more readily available than NAD+ to provide oxidizing equivalents.
Based on the inhibition data with D-2-HGA (Figures 3 and 4), the products of the PdxB-dependent reduction of αKG, OAA, and pyruvate are most likely the less common D-enantiomers that are not part of normal metabolic processes such as the citric acid cycle. So how does the cell recycle these unusual metabolites? E. coli has D-lactate and decarboxylating D-malate specific dehydrogenases that both generate pyruvate (32, 33). In the case of 2-HGA, only an L-specific 2-HGA oxidase has been reported in E. coli (34). Thus, for E. coli to recycle D-2-HGA, it must rely on some as yet unknown D-2-HGA dehydrogenase or use the activity of a different dehydrogenase such as SerA (kcat/Km for D-2-HGA is 1.9 × 103 M-1s-1 (19). Alternatively, D-2-HGA could be a waste by-product given the relatively low requirement for PLP (5.5 μM/hr; Kim et al., submitted for publication).
It seems likely that the highly homologous PdxBs from other γ-proteobacteria (>40% identity) also use α-keto acids to ensure multiple turnovers. How catalysis is effected in PdxR, the functional equivalent of PdxB in Sinorhizobium meliloti, is less certain (35). PdxR contains a tightly bound flavin instead of NAD(H). Although PdxR is not homologous to PdxB, belonging to a family of flavin-dependent oxidoreductases, it does complement the pdxB knock-out in E. coli. PdxR has been shown to perform multiple turnovers of the substrate 4PE in the presence of artificial electron acceptors such as DCIP, ferricyanide or cytochrome c from equine heart, but not NAD(P)+. Thus, no physiological oxidant has been discovered for PdxR. It is interesting to speculate that PdxR and other flavo- or nicotino-proteins depend on as yet undiscovered co-substrates to support the multiple turnovers required for true enzymatic catalysis.
Acknowledgments
Supported by NIH RO1 GM083285 to S.D.C.
Abbreviations
- PLP
pyridoxal phosphate
- 4PE
4-phospho-D-erythronate
- OHPB
2-oxo-3-hydroxy-4-phospho-butanoate
- αKG
α-ketoglutarate
- OAA
oxaloacetic acid
- 2-HGA
2-hydroxyglutaric acid
- TN buffer
50 mM Tris-HCl (pH 8.0) with 50 mM NaCl
- TND buffer
TN buffer with 1 mM DTT
- DCIP
2,6-dichloroindole phenol
- NDMA
N-nitrosodimethylamine
References
- 1.Fitzpatrick TB, Amrhein N, Kappes B, Macheroux P, Tews I, Raschle T. Two independent routes of de novo vitamin B6 biosynthesis: not that different afer all. Biochem J. 2007;407:1–13. doi: 10.1042/BJ20070765. [DOI] [PubMed] [Google Scholar]
- 2.Wilson JA. Disorders of vitamins: deficiencies, excess, and errors of metabolism. In: Petersdorf RG, editor. Harrison’s principles of internal medicine. New York: McGraw-Hill; 1982. pp. 461–470. [Google Scholar]
- 3.Burns KE, Xiang Y, Kinsland CL, McLafferty FW, Begley TP. Reconstitution and biochemical characterization of a new pyridoxal-5’-phosphate biosynthetic pathway. J Am Chem Soc. 2005;127:3682–3683. doi: 10.1021/ja042792t. [DOI] [PubMed] [Google Scholar]
- 4.Ehrenshaft M, Bilski P, Li MY, Chignell CF, Daub ME. A highly conserved sequence is a novel gene involved in de novo vitamin B6 biosynthesis. Proc Natl Acad Sci USA. 1999;96:9374–9378. doi: 10.1073/pnas.96.16.9374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Strohmeier M, Raschle T, Mazurkiewicz J, Rippe K, Sinning I, Fitzpatrick TB, Tews I. Structure of a bacterial pyridoxal 5’-phosphate synthase complex. Proc Natl Acad Sci USA. 2006;103:19284–19289. doi: 10.1073/pnas.0604950103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao G, Pease AJ, Bharani N, Winkler ME. Biochemical characterization of gapB-encoded erythrose 4-phosphate dehydrogenase of Escherichia coli K-12 and its possible role in pyridoxal 5’-phosphate biosynthesis. J Bacteriol. 1995;177:2804–2812. doi: 10.1128/jb.177.10.2804-2812.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ha JY, Lee JH, Kim KH, Kim DJ, Lee HH, Kim H-K, Yoon H-J, Suh SW. Crystal structure of D-erythronate-4-phosphate dehydrogenase complexed with NAD. J Mol Biol. 2007;366:1294–1304. doi: 10.1016/j.jmb.2006.12.038. [DOI] [PubMed] [Google Scholar]
- 8.Fischer P, Fleckenstein J, Hoenes J. Spectroscopic investigation of dihydronicotinamides-I: Conformation, absorption, and fluorescence. Photochem Photobiol. 1988;47:193. doi: 10.1111/j.1751-1097.1988.tb02714.x. [DOI] [PubMed] [Google Scholar]
- 9.Piersma SR, Visser AJWG, de Vries S, Duine JA. Optical spectroscopy of nicotinoprotein alchohol dehydrogenase from Amycopatopsis methanolica: A comparison with horse liver alchohol dehydrogenase and UDP-galactose epimerase. Biochemistry. 1998;37:3068–3077. doi: 10.1021/bi972115u. [DOI] [PubMed] [Google Scholar]
- 10.van der Werf MJ, van der Ven C, Barbirato F, Eppink MHM, de Bon JAM, van Berkel WJH. Stereselective carveol dehydrogenase from Rhodococcus erythropolis DCL14. J Biol Chem. 1999;274:26296–26304. doi: 10.1074/jbc.274.37.26296. [DOI] [PubMed] [Google Scholar]
- 11.Kato N, Yamagami T, Shimao M, Sakazawa C. Formaldehyde dismutase, a novel NAD-binding oxidoreductase from Pseudomonas putida F61. Eur J Biochem. 1986;156:59–64. doi: 10.1111/j.1432-1033.1986.tb09548.x. [DOI] [PubMed] [Google Scholar]
- 12.Dawson RMC, Elliott DC, Elliott WH, Jones KM. Data for biochemical research. Clarendon Press; Oxford: 1986. [Google Scholar]
- 13.Liu Y, Vanhooke JL, Frey PA. UDP-galactose 4-epimerase: NAD+ content and charge-transfer band associated with the substrate-induced conformational transition. Biochemistry. 1996;35:7615–7620. doi: 10.1021/bi960102v. [DOI] [PubMed] [Google Scholar]
- 14.Klepp J, Oberfrank M, Retey J, Tritsch D, Biellmann J-F, Hull WE. Nature of coenzyme binding by glyceraldehyde-3-phosphate dehydrogenase: carbon-13 NMR studies with oxidized [4-13C]nicotinamide adenine dinucleotide. J Am Chem Soc. 1989;111:4440–4447. [Google Scholar]
- 15.Ramsey AJ, Hill JP, Dickinson FM. Some comparisons of pig and sheep liver cytosolic aldehyde dehydrogenases. Comp Biochem Physiol. 1999;93B:77–83. doi: 10.1016/0305-0491(89)90219-8. [DOI] [PubMed] [Google Scholar]
- 16.Opheim PW, van Beeumen J, Duine JA. Nicotinoprotein [NAD(P)-containing] alcohol/aldehyde oxidoreductases. Eur J Biochem. 1993;212:819–826. doi: 10.1111/j.1432-1033.1993.tb17723.x. [DOI] [PubMed] [Google Scholar]
- 17.Blandino A, Caro I, Cantero D. Effect of culture conditions on the aldehyde dehydrogenase activity of Acetobacter uceti cytoplasmic extracts. Biotechnol Lett. 1996;18:63–68. [Google Scholar]
- 18.Hektor H, Kloosterman, Dijkhuizen L. Nicotinoprotein methanol dehydrogenase enzymes in Gram-positive methylotrophic bacteria. J Molec Catal B: Enzymatic. 2000;8:103–109. [Google Scholar]
- 19.Zhao G, Winkler ME. A novel α-ketoglutarate reductase activity of the serA-encoded 3-phosphoglycerate dehydrogenase of Escherichia coli K-12 and its possible implications for human 2-hydroxyglutaric aciduria. J Bacteriol. 1996;178:232–239. doi: 10.1128/jb.178.1.232-239.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pizer L. The pathway and control of serine biosynthesis in Escherichia coli. J Biol Chem. 1963;238:3934–3944. [PubMed] [Google Scholar]
- 21.Sugimoto E, Pizer LI. The mechanism of end product inhibition of serine biosynthesis. J Biol Chem. 1968;243:2081–2089. [PubMed] [Google Scholar]
- 22.Bennett BD, Kimball EH, Gao M, Osterhout R, Van Dien SJ, Rabinowitz JD. Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat Chem Biol. 2009;5:593–599. doi: 10.1038/nchembio.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoque MA, Ushiyama H, Tomita M, Shimizu K. Dynamic responses of the intracellular metabolite concentrations of the wild type and pykA mutant Escherichia coli against pulse addition of glucose or NH3 under those limiting continuous cultures. Biochem Engineer J. 2005;26:38–49. [Google Scholar]
- 24.Lowry OH, Carter J, Ward JB, Glaser L. The effect of carbon and nitrogen sources on the level of metabolic intermediates in Escherichia coli. J Biol Chem. 1971;246:6511–6521. [PubMed] [Google Scholar]
- 25.Tanaka N, Kusakabe Y, Ito K, Yoshimoto T, Nakamura KT. Crystal structure of formaldehyde dehydrogenase from Pseudomonas putida: the structural origin of the tightly bound cofactor in nicotinoprotein dehydrogenases. J Mol Biol. 2002;324:519–533. doi: 10.1016/s0022-2836(02)01066-5. [DOI] [PubMed] [Google Scholar]
- 26.Dubrow R, Pizer LI. Transient kinetic and deuterium isotope effect studies on the catalytic mechanism of phosphoglycerate dehydrogenase. J Biol Chem. 1977;252:1539–1551. [PubMed] [Google Scholar]
- 27.Arfman HJ, van Beeumen JJ, de Vries GE, Harder W, Dijkhuizen L. Purification and characterization of an activator protein for methanol dehydrogenase from thermotolerant Bacillus spp. J Biol Chem. 1991;266:3955–3960. [PubMed] [Google Scholar]
- 28.Dolin MI, Phares EF, Long MV. Bound pyridine nucleotide of malic-lactic transhydrogenase. Biochem Biophys Res Commun. 1965;21:303–310. doi: 10.1016/0006-291x(65)90193-2. [DOI] [PubMed] [Google Scholar]
- 29.Allen SHG. The isolation and characterization of malate-lactate transhydrogenase from Micrococcus lactilyticus. J Biol Chem. 1966;241:5266–5275. [PubMed] [Google Scholar]
- 30.Zachariou M, Scopes RK. Glucose-fructose oxidoreductase, a new enzyme isolated from Zymomonas mobilis that is responsible for sorbitol production. J Bacteriol. 1986;167:863–869. doi: 10.1128/jb.167.3.863-869.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khersonsky O, Roodveldt C, Tawfik DS. Enzyme promiscuity: evolutionary and mechanistic aspects. Curr Opin Chem Biol. 2006;10:498–508. doi: 10.1016/j.cbpa.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 32.Lukas H, Reimann J, Kim OB, Grimpo J, Unden G. Regulation of aerobic and anaerobic D-malate metabolism of Escherichia coli by the LysR-type regulator DmlR (YeaT) J Bacteriol. 2010;192:2503–2511. doi: 10.1128/JB.01665-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tarmy EM, Kaplan NO. Kinetics of Escherichia coli D-lactate dehydrogenase and evidence for pyruvate-controlled change in conformation. J Biol Chem. 1968;243:2587–2596. [PubMed] [Google Scholar]
- 34.Kalliri E, Mulrooney SB, Hausinger RP. Identification of Escherichia coli YgaF as an L-2-hydroxyglutarate oxidase. J Bacteriol. 2008;190:3793–3798. doi: 10.1128/JB.01977-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tazoe M, Ichikawa K, Hoshino T. Flavin adenine dinucleotide-dependent 4-phospho-D-erythronate dehydrogenase is responsible for the 4-phosphohydroxy-L-threonine pathway in vitamin B6 biosynthesis in Sinorhizobium meliloti. J Bacteriol. 2006;188:4635–4645. doi: 10.1128/JB.01999-05. [DOI] [PMC free article] [PubMed] [Google Scholar]