

Abstract
Resistance to endocrine therapy agents has presented a clinical obstacle in the treatment of hormone-dependent breast cancer. Our laboratory has initiated a study of microRNA regulation of signaling pathways that may result in breast cancer progression on aromatase inhibitors (AI). Microarray analysis of hormone refractory cell lines identified 115 differentially regulated microRNAs, of which 49 microRNAs were believed to be hormone-responsive. A group of microRNAs were inversely expressed in the AI-resistant lines versus LTEDaro and tamoxifen-resistant. We focused our work on hsa-miR-128a which was hormone-responsive and selectively up-regulated in the letrozole-resistant cell lines. Human miR-128a was predicted to target the TGFβ signaling pathway and indeed sensitivity to TGFβ was compromised in the letrozole-resistant cells, as compared to parental MCF-7aro. Human miR-128a was shown to negatively target TGFβRI protein expression by binding to the 3’UTR region of the gene. Inhibition of endogenous miR-128a resulted in re-sensitization of the letrozole-resistant lines to TGFβ growth inhibitory effects. This data suggests that the hormone-responsive miR-128a can modulate TGFβ signaling and survival of the letrozole-resistant cell lines. To our knowledge, this is the first study to address the role of microRNA regulation as well as TGFβ signaling in AI-resistant breast cancer cell lines. We believe that in addition to estrogen-modulation of gene expression, hormone-regulated microRNAs may provide an additional level of post-transcriptional regulation of signaling pathways critically involved in breast cancer progression and AI-resistance.
Keywords: Aromatase Inhibitor Resistance, microRNA, miR-128a, TGFβ, Estrogen
INTRODUCTION
Approximately 70% of breast cancers are hormone-dependent diseases. ER is critical in mediating estrogen-dependent transcription of genes involved in cell survival and proliferation of breast cancer cells (1, 2). In addition, aromatase is a cytochrome P450 enzyme that is responsible for the production of estrogen from an androgen substrate, which further drives breast cancer progression (3, 4). Endocrine therapy agents have been developed to target hormone-dependent signaling pathways, either by antagonizing ER through the use of tamoxifen, or by inhibiting estrogen synthesis via aromatase inhibitors (AI) letrozole, anastrozole and exemestane. Endocrine therapy agents have shown excellent clinical efficacy (5-8), though the main hindrance with these agents is the development of acquired resistance and consequent loss of regulated growth. Previous studies addressing resistance to tamoxifen and AIs have implicated heightened crosstalk between growth factor and ER signaling pathways (9-12), resulting in activated ER signaling without the need for hormone (13, 14). This study utilizes a microarray approach to further investigate novel signaling pathways regulated by microRNAs, and the resulting effects on growth of AI-resistant breast cancer cell lines.
MicroRNAs (miRNAs) are non-coding RNAs whose mature products are small single-stranded species that range between 21-22 nucleotides in length (15). MicroRNAs play critical roles in gene silencing. Binding of microRNAs to their target genes occurs either by perfect match or mismatch base pairing to complementary sequences within the 3’ untranslated region (UTR), resulting in mRNA degradation (16, 17) or translational repression (18-20). In terms of cancer progression, microRNAs can function as onco-miRs or tumor suppressors by regulating apoptotic factors (21), Ras (22), E2F1 (23), and factors involved in metastasis (24, 25).
The transforming growth factor beta (TGFβ) family encompasses a large network of polypeptide growth factors. The effects of TGFβ ligands are propagated by transmembrane serine/threonine receptor TGFβRII which recruits and activates TGFβRI (26). TGFβRI is responsible for initiating downstream signaling cascades that are mediated by SMAD transcription factors (SMADs 2, 3 and 4), which form a complex and bind DNA to activate transcription of TGFβ-responsive genes (27, 28). TGFβ has been suggested to enhance tumor cell migration, as overexpression of TGFβI or TGFβRI in murine mammary glands can promote metastatic potential of breast tumors (29, 30). In contrast, TGFβ signaling has also been reported to inhibit growth of epithelial cells (31), and a hormone-independent variant of MCF-7 cells was reported to lose sensitivity to TGFβ (32). This data suggests that TGFβ can inhibit and promote breast cancer progression. To date, the TGFβ signaling pathway has not been reported to play a role in AI-resistance, though studies have suggested differing roles for TGFβ in tamoxifen-resistance in vivo and in vitro (33, 34).
We previously carried out affymetrix microarray analysis to elucidate gene expression profiles associated with endocrine therapy resistant breast cancer cell lines (13). To further address changes beyond gene expression, our laboratory investigated post-transcriptional microRNA regulation of signaling pathways which could exacerbate AI-resistance. Microarray analysis was performed to determine differential expression patterns of microRNAs in the parental MCF-7aro breast cancer cells versus derivative resistant cell lines. This analysis identified a role for human miR-128a in the negative regulation of TGFβRI expression, resulting in loss of sensitivity to the growth inhibitory effects of TGFβ in letrozole-resistant breast cancer cells.
MATERIALS AND METHODS
Cell culture and resistant cell line generation
The human breast cancer epithelial cell line MCF-7 was stably transfected to overexpress the aromatase gene (MCF-7aro), and previously reported by our laboratory (35). MCF-7aro cells were maintained as previously described (13). In addition to the parental cell line, the testosterone-only (T-only) cells were generated as a hormone-only control, where T was converted to 17β-estradiol (E2) by the expressed aromatase. Cells resistant to all three AIs, letrozole, anastrozole and exemestane were referred to as T+LET R, T+ANA R and T+EXE R, respectively. The tamoxifen-resistant (T+TAM R) cells and long-term estrogen deprived (LTEDaro) lines were also generated for comparison to the AI-resistant cell lines. MCF-7aro and derivative resistant cells were previously characterized by our laboratory (13).
MicroRNA microarray analysis
Microarray analysis was performed using the Agilent human miRNA microarray chips, 8×15K format. For microarray analysis, 100ng total RNA was used for Cy3 labeling and hybridization to Agilent miRNA array chips. The Agilent scanner and Feature Extraction (FE) software were used for data collection subsequent to sample hybridization. Data analysis was performed using Partek Genomics Suite, version 6.4. Background correction, quantile normalization and data summary were generated using Robust Multichip Average (RMA) normalization. For all data analysis, parental MCF-7aro cells were considered as baseline and fold-change of microRNAs from all other cell lines were compared relative to the control cells. One-way ANOVA analysis, using treatment as a parameter, was performed to select 115 significant microRNAs based on a false discovery rate (FDR) of less than 1% (p-value equivalent to 0.003). For hierarchical clustering analysis, average linkage with Pearson’s dissimilarity was applied for data visualization. Prediction software to identify microRNA target genes included TargetScan (version 5.0) and miRBase Targets (version 5.0) available through the Sanger database. Gene targets identified by TargetScan and miRBase were subsequently loaded into GeneSpring 10 to identify which signaling pathways are predominantly regulated by microRNAs.
cDNA synthesis and quantitative real-time PCR analysis
For detection of microRNAs, the miScript reverse transcription kit was used from Qiagen (Valencia, CA), according to manufacturer’s protocol. SYBR green reagent for microRNA detection was obtained from Qiagen, and real-time PCR analysis was carried out according to the exact manufacturer’s recommendations. For detection of mRNA levels (and not small RNA species), 5 μg of total RNA was used for reverse transcription, as previously described (13). The following primers were used for transcript detection: U6 forward primer (5’-CTCGCTTCGGCAGCACA-3’), U6 reverse primer (5’-AACGCTTCACGAATTTGCGT-3’), TGFβR1 forward primer (5’-GAGCATGGATCCCTTTTTGA-3’), TGFβR1 reverse primer (5’-TATGAGCAATGGCTGGCTTT-3’), β-actin forward primer (5’-AGAAGGAGATCACTGCCCTGGCACC-3’) and β-actin reverse primer (5’-CCTGCTTGCTGATCCACATCTGCTG-3’).
Cell proliferation assays
MCF-7aro parental cells and resistant cell lines were seeded in triplicate into 96-well plates overnight in steroid-depleted medium (CD FBS MEM). For dose-response studies with TGFβ1, concentrations ranged between 0.1ng/ml and 2ng/ml TGFβ1. To assess cell growth and viability, 0.5mg/ml MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) powder was mixed with MEM media for 1hour at 37°C. MTT solution was then removed and replaced with DMSO. The absorbance values at 570nm were read on a SpectraMax M5 spectrophotometer plate reader from Molecular Devices (Sunnyvale, CA). For the microRNA cell proliferation assays, cells were seeded simultaneously with reverse transfection reagent siPORT Neofx (Ambion, Foster City, CA) and synthetic microRNA (Ambion, Foster City, CA) for 24 hours, according to manufacturer’s protocol. After 24 hour reverse transfection, media was replaced with MEM plus 1ng/ml TGFβ1 and allowed to grow up to 5 days. For all proliferation assays, time points were taken at 1, 3, and 5 days and experiments were performed three independent times. TGFβ1 was purchased from R&D systems (Minneapolis, MN). MTT powder was purchased from Sigma-Aldrich (St. Louis, MO).
Lucife rase reporter assays
To assess TGFβ1 response, parental and T+LET R lines were transfected with a SMAD luciferase reporter in 12 well plates, at a cell density of 1.2×105 cells/well. Cells were seeded overnight and subsequently transfected with 0.3μg pGL3-CAGA12 lux SMAD luciferase reporter plasmid, using Lipofectamine 2000 transfection reagent (Invitrogen, Carlsbad, CA), according to manufacturer’s protocol. For transfection control purposes, the pRL-CMV renilla reporter was cotransfected with the SMAD luciferase reporter. After 5 hours, OptiMEM (Invitrogen, Carlsbad, CA) transfection medium was replaced with CD FBS MEM and supplemented with 1ng/ml TGFβ1 for 8 hours, in the absence of hormone. For reverse transfection with the microRNA mimic, siPORT Neofx (Ambion, Foster City, CA) transfection agent was mixed with the microRNA (100nM) and 0.1μg WT-128a-βR1/psiCheck or MT-128a-βR1/psiCheck luciferase reporter vectors and overlaid with 1.2 ×105 cells/12-well. Reverse transfection was allowed to proceed for 24 hours and media was changed to CD FBS MEM for 3 days post transfection. The Dual Luciferase Assay System (Promega, Madison, WI) was used to read both firefly and renilla luciferase activities, with three independent experiments performed.
Western blotting analysis
For TGFβ1 responsive western analysis, cells were serum starved overnight and treated with 1-2ng/ml TGFβ1 in serum-free MEM for 15-30 minutes. For the microRNA mimic effect by western, microRNAs (100nM) were reverse transfected with siPORT Neofx, according to the manufacturer’s protocol, using 4.8×105 cells/60mm dish. Post transfection, cells were allowed to recover in CD FBS MEM for 3 days before harvesting. Western analysis was performed as previously described (13). Membranes were blocked for 2 hours in 5% non-fat milk and probed with primary antibody overnight at 4°C. The following antibodies were used for western blotting: phospho-SMAD2 (Ser465/467), total SMAD 2/3 (Cell Signaling, Danvers, MA), total TGFβR1 (V-22) and β-actin (Santa Cruz Biotechnology, Santa Cruz, CA).
RESULTS
MicroRNA microarray analysis
A similarity matrix was used to compare microRNA expression using correlation coefficients calculated for each microRNA (Figure 1A). Expression of microRNAs found in clusters A, B and C were more closely correlated to each other than with cluster D, though A-C did inherently differ, as indicated by the branching dendrograms. To further filter significant microRNAs, 1-way ANOVA analysis identified 115 microRNAs as significantly expressed in the resistant cells, using MCF-7aro cells for baseline transformation (Figure 1B). Biological replicates were shown horizontally and based on the heatmap dendrogram, all AI-resistant cells clustered separately from the control MCF-7aro cells, LTEDaro and T+TAM R lines. In addition, microRNAs within each cluster of the heatmap highly correlated with clusters A, B and C from the similarity matrix in Figure 1A, as indicated. The heatmap in Figure 1B also provided microRNA signatures associated with different resistant cell lines. Cluster A included microRNAs that are down-regulated in the AI-resistant lines and inversely regulated in the LTEDaro, T-only and T+TAM R lines. Among the microRNAs identified in cluster A, miR-100/99a, miR-125b, miR-205 and miR-181a/b were selectively down-regulated in the AI-resistant cell lines. Cluster C contained a number of microRNAs up-regulated in the T-only lines, with varying expression in the non-steroidal (letrozole and anastrozole) AI-resistant lines. Cluster B was enriched in microRNAs up-regulated in the steroidal exemestane-resistant lines, including miR-135a, suggesting that microRNA expression in the T+EXE R cells is unique. Detailed microRNA signatures associated with different resistant lines are provided as supplementary material (Table S1).
Figure 1. microRNA microarray analysis of the AI, LTEDaro and tamoxifen-resistant cell lines.
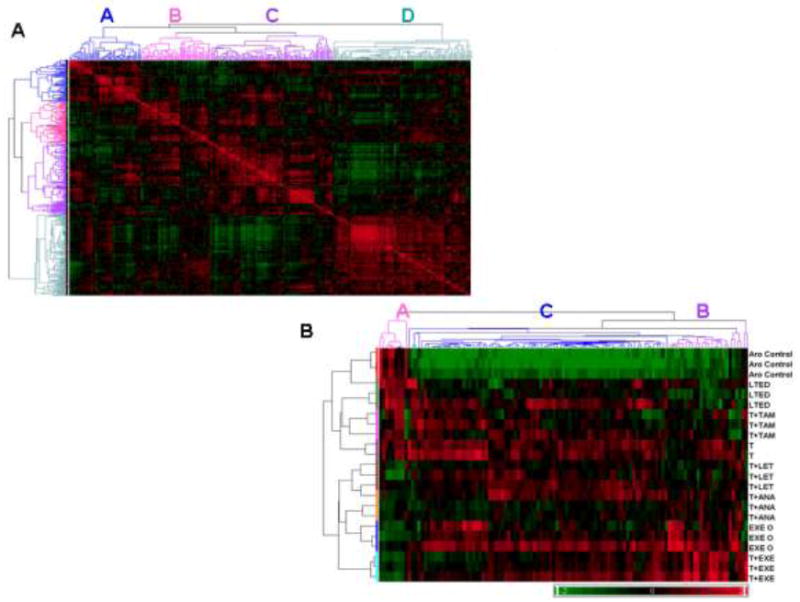
A) Partek Genomics Suite was used to generate the similarity matrix plot that compared expression of 342 RMA normalized microRNAs. Correlation coefficients were calculated using a linear Pearson correlation algorithm and visualized with red and green indicating high (0.9) and low (0.3) correlation, respectively. Unique clusters of microRNAs that share similar expression patterns are labeled A, B, C and D. B) Hierarchical clustering (Pearson’s dissimilarity algorithm) and heat map analysis of 115 significant microRNAs was based on 1-way ANOVA analysis, in comparison to the parental MCF-7aro cells. The false discovery rate (FDR) was set to 1%, with a corresponding p-value of 0.003. Clusters A, B and C corresponded with microRNA clusters visualized with the similarity matrix (Figure 1A), as labeled on the top dendrogram. Biological replicates were shown horizontally and individual microRNAs were clustered vertically, with red indicating up-regulation and green representing down-regulated microRNA expression, according to the color legend.
Of these 115 significant microRNAs, 49 microRNAs were identified as hormone-responsive based on the criteria that their expression was up-regulated in the T-only lines and inversely regulated in both MCF-7aro cells and LTEDaro lines (Figure 2A). The E2-induced expression of several microRNAs, including members of the miR-17-92 cluster, was confirmed by real-time PCR analysis. Similar to the microarray data, the expression of miR-18b, miR-141, miR-128a and miR-17-3p were up-regulated in the T-Only lines and down-regulated in the parental MCF-7aro and LTEDaro lines. The expression of miR-17-3p detected by real-time PCR analysis is shown as representative data (Figure 2B). Likewise, the expression of these microRNAs was induced upon hormone treatment (T or E2) and diminished in the presence of T plus letrozole in the parental MCF-7aro cells (Figure 2C).
Figure 2. Hormone-regulated microRNAs.
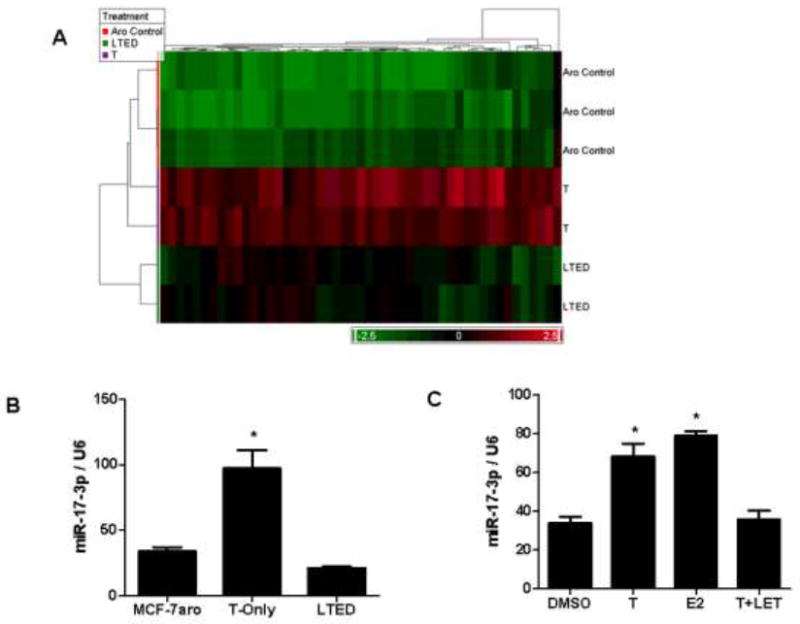
A) Hierarchical clustering of hormone-responsive microRNAs. Based on the 115 significant microRNAs (p-value of 0.003) identified in Figure 1B, average fold-change criteria of 1.2 was used to select microRNAs up-regulated in the T-only lines and inversely regulated in the parental MCF-7aro cells and hormone-deprived LTEDaro lines. B) Hormone-responsive microRNAs were validated by quantitative real-time PCR with miR-17-3p representatively shown. For internal control purposes, microRNA expression was normalized to U6 small RNA expression in the MCF-7aro, T-only and hormone-free LTEDaro cells. C) Expression of hormone-responsive microRNAs was also validated in the parental MCF-7aro cells, with miR-17-3p shown. Treatment conditions compared DMSO vehicle control to hormone (T and E2) and T+letrozole treatment. For statistical analysis, Student’s t-test compared samples to MCF-7aro and DMSO treated controls in 2B and 2C, with * indicating statistical significance at a p-value < 0.05.
To further investigate microRNA regulation of signaling pathways involved in AI-resistance, additional analysis was performed to discern microRNAs differentially regulated in these lines. Selection of microRNAs differentially expressed in resistant cells was based on fold-change criteria from the 115 significant microRNAs previously identified in Figure 1B. It would be expected that microRNAs that are hormone up-regulated in the T-only lines would be down-regulated in the presence of letrozole. Yet, in the T+LET R lines, letrozole does not down-regulate expression of several microRNAs, suggesting a role in resistance. Based on this analysis, 12 microRNAs were found to be up-regulated (1.2 fold) selectively in the T+LET R lines (Figure 3A, Table S1). In addition, 2 microRNAs were up-regulated in the T+ANA R lines and slightly up-regulated in the T+LET R cells (miR-425-3p and miR-191), while 2 microRNAs were up-regulated selectively in the T+ANA R lines (miR-23b and miR-24) (Table S1). This analysis demonstrated inherent differences in microRNA expression profiles between the T+LET R and T+ANA R AI-resistant lines.
Figure 3. Letrozole-resistant microRNAs.
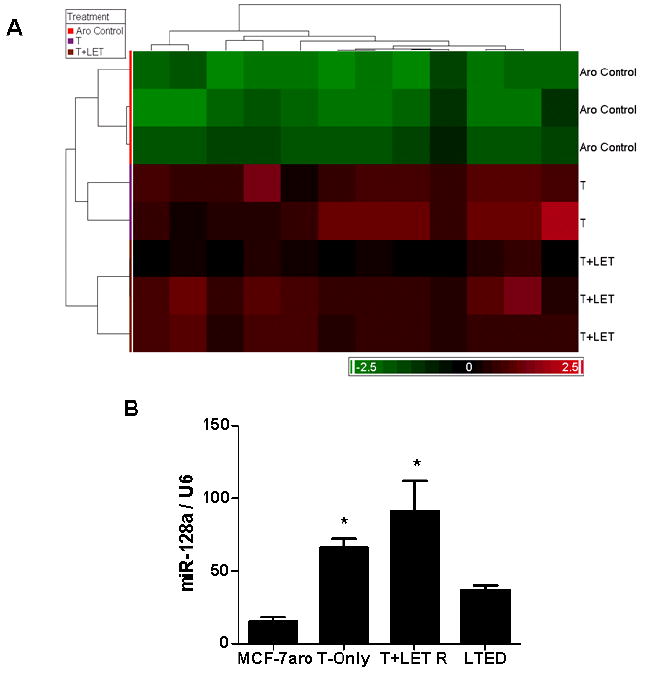
A) Hierarchical clustering and heat map of microRNAs specifically identified in the T+LET R lines. Based on the 115 significant microRNAs (p-value of 0.003) identified in Figure 1B, average fold-change criteria of 1.2 was used to select microRNAs up-regulated in the T+LET R lines. B) Expression of miR-128a was validated using quantitative real- time PCR in the parental MCF-7aro cells, T-only, T+LET R and LTEDaro lines. U6 RNA was used for internal control normalization within samples. For statistical analysis, Student’s t-test compared samples to MCF-7aro control cells, with * indicating statistical significance at a p-value < 0.05.
The microRNAs identified as significantly up-regulated in the letrozole-resistant lines were further analyzed for meaningful predicted targets, using TargetScan and miRBase. TGFβ signaling was found to be highly targeted by hsa-miR-128a, which was significantly up-regulated in the T+LET R lines specifically. Our interests were subsequently focused on miR-128a because it is predicted to target both TGFβR1 and SMAD2. Expression levels of miR-128a were confirmed by real-time PCR and in comparison to MCF-7aro and LTEDaro lines, miR-128a was up-regulated in the T-Only as well as T+LET R cells (Figure 3B). The expression of miR-128a was not significantly up-regulated in the other resistant cell lines, including the T+ANA R lines, suggesting functional specificity of miR-128a in letrozole-resistance. Also, miR-128a appeared to be up-regulated by hormone, as its expression was inversely regulated in the parental MCF-7aro cells and the hormone-free LTEDaro lines (Figures 2A and 3B).
Loss of TGFβ signaling in the T+LET R lines
The anti-proliferative effects of TGFβ were investigated using cell proliferation assays to determine if this growth factor could delay the growth of the resistant cells. Dose-response studies were carried out, and though the parental MCF-7aro cells were sensitive to TGFβ1 and demonstrated a dose-dependent decrease in proliferation (Figure 4A), the viability of the T+LET R lines was unaffected by TGFβ1 treatment (Figure 4B). A previous study using hormone-independent MCF-7 cells reported a similar loss of sensitivity to TGFβ (32). Luciferase assays were performed using a SMAD reporter (pGL3-CAGA12 lux) that contains 12 tandem copies of the consensus SMAD 2/3 binding sequence (CAGA). Transient transfection of pGL3-CAGA12 lux reporter resulted in TGFβ1-mediated increase in luciferase activity in the parental MCF-7aro cells, as compared to media treated cells (Figure 4C). In contrast, the T+LET R lines completely lost sensitivity to TGFβ1 treatment by luciferase reporter assays (Figure 4C).
Figure 4. Loss of sensitivity to the growth inhibitory effects of TGFβ in the T+LET R lines.
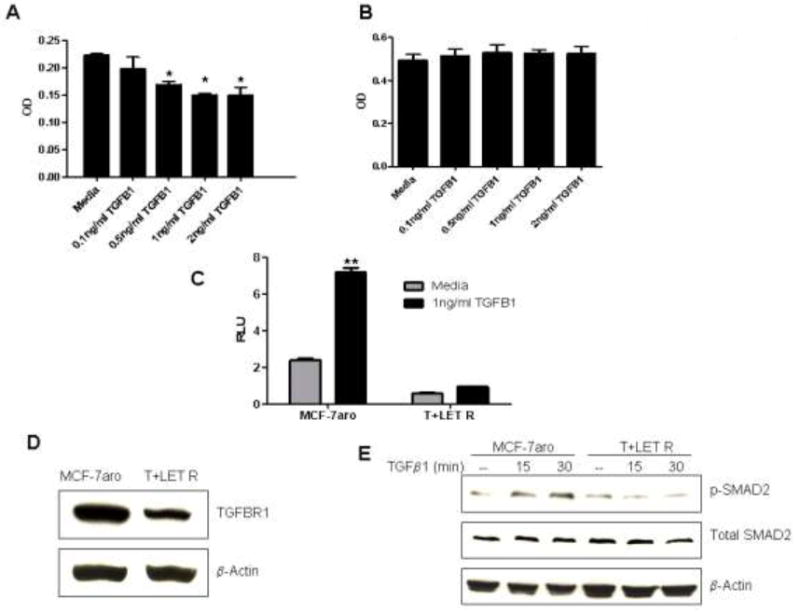
Dose response proliferation studies with TGFβ1 in the parental MCF-7aro (A) and T+LET R lines (B). For statistical analysis, Student’s t-test compared treatment conditions to media control, with * indicating statistical significance at a p-value < 0.05. C) Luciferase assays were performed using a SMAD 2/3 luciferase reporter, containing the CAGA consensus SMAD binding site, co-transfected with a control pRL-CMV renilla reporter. For statistical analysis, Student’s t-test compared treatment conditions to media control, with ** indicating statistical significance at a p-value < 0.01. D) Western analysis was carried out to determine total protein expression of TGFβR1, in comparison to β-actin protein expression in the MCF-7aro and T+LET R lines. E) Activation of SMAD2 by phosphorylation of serine residues 465/467 was detected by western analysis. Levels of phospho-SMAD2 were compared to total SMAD2 and β-actin.
Western analysis was performed to look at endogenous protein levels of TGFβ signaling pathway members. Based on microRNA target prediction software, TGFβR1 and SMAD2 were believed to be targets of miR-128a. Western analysis demonstrated that expression of endogenous TGFβR1 was decreased in the T+LET R lines versus the parental MCF-7aro cells (Figure 4D). Though SMAD2 is a predicted target of miR-128a, endogenous protein levels of total SMAD2 were unchanged between the MCF-7aro and T+LET R lines (Figure 4E). Activation of downstream factors such as SMADs is critical in mediating TGFβ signaling. Using western blot analysis, levels of phospho-SMAD2 were determined in response to TGFβ1 treatment. Consistent with previous results, phospho-SMAD2 levels were increased by TGFβ1 treatment after 15-30 minutes in the MCF-7aro cells, but in the T+LET R lines, activated SMAD2 could not be detected (Figure 4E). Total protein levels of SMAD2 remained unchanged.
miR-128a targets TGFβR1 in the T+LET R lines
Based on target prediction software, miR-128a was found to regulate SMAD2 and TGFβR1 expression. To identify which targets were regulated by miR-128a, the non-targeting negative control (NC) RNA or pre-miR-128a gain-of-function mimic were transfected into the T+LET R lines, and RT-PCR and western analysis were carried out to determine resulting expression of TGFβR1 and SMAD2. In comparison to the NC transfected cells, pre-miR-128a transfection dramatically increased miR-128a expression (Figure 5A). Subsequent western analysis after microRNA transfection demonstrated that pre-miR-128a did decrease protein levels of TGFβR1, but not SMAD2 (Figure 5C). In addition, though miR-128a did decrease protein levels of TGFβR1, the mRNA levels of TGFβR1 remained unchanged (Figure 5B), as the binding site of miR-128a to the 3’UTR of TGFβR1 gene is an imperfect match.
Figure 5. Human miR-128a targets TGFβR1 at the 3’UTR.
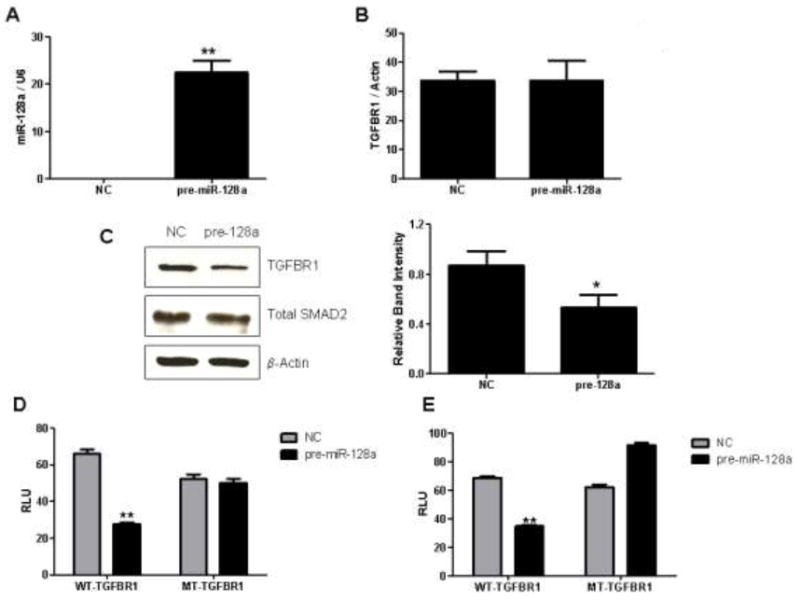
A) Expression of miR-128a was detected by real-time PCR, relative to U6 RNA expression, after pre-miR-128a mimic or negative control (NC) transfection. Student’s t-test compared NC and pre-miR-128a transfected cells, with ** indicating statistical significance at a p-value < 0.01. B) Post NC or pre-miR-128a transient transfection, TGFβR1 mRNA expression was detected by real-time PCR analysis, relative to the β-actin housekeeping gene. C) Transfection of the NC or pre-miR-128a mimic was performed and T+LET R cells were harvested for western analysis to detect total TGFβR1 and total SMAD2 expression levels. Quantified western blots of three independent experiments are shown, with * indicating statistical significance using Student’s t-test at a p-value < 0.05. D-E) WT and MT luciferase constructs containing the putative miR-128a target binding site within the 3’UTR of the TGFβR1 were co-transfected with the NC or pre-miR-128a mimic into MCF-7aro (D) and T+LET R (E) cells. For statistical analysis, Student’s t-test compared pre-miR-128a with NC transfected cells in 5D and 5E, with ** indicating statistical significance at a p-value < 0.01.
TGFβR1 is potentially a target of miR-128a in the T+LET R lines. To further investigate this regulation, the miR-128a target seed sequence within the 3’ UTR of the TGFβR1 gene (5’-CACTGTGA-3’) was cloned into the psiCheck luciferase reporter vector. This miR-128a 8-mer target sequence was also mutated at 4 bases where G became C, T became A and so on (5’-CACTCACT-3’) and inserted into the psiCheck luciferase reporter. Transient transfection of the WT/psiCheck luciferase reporter vector resulted in significantly decreased luciferase activity in the presence of pre-miR-128a, in comparison to the NC transfected cells (Figures 5D and 5E). This decreased luciferase response was observed both in the parental MCF-7aro cells (5D) as well as the T+LET R lines (5E), as the microRNA was overexpressed using the pre-miR-128a mimic. In contrast, transfection of the MT/psiCheck luciferase construct did not result in a loss of luciferase activity, as compared to the NC transfected cells (Figures 5D and 5E). These results further confirm the role of miR-128a in regulating TGFβR1 expression, a process which is mediated by a target binding sequence within the 3’UTR of TGFβR1.
Human miR-128a post-transcriptionally targeted TGFβR1. As a functional consequence of this antagonism, the question was asked whether inhibiting miR-128a would then sensitize the T+LET R cells to the growth inhibitory effect of TGFβ1. To confirm that anti-miR-128a did effectively knockdown endogenous miR-128a expression in the T+LET R lines, total RNA was harvested and used for real-time PCR analysis. Compared to the NC transfected cells, expression of miR-128a was successfully decreased in the anti-miR-128a inhibitor transfected T+LET R lines (Figure 6A). The mRNA levels of TGFβR1 were not significantly changed between the NC versus anti-miR-128a transfected T+LET R cells (Figure 6B). Transfection of 10, 30 and 50nM anti-miR-128a dose-dependently inhibited proliferation of the T+LET R lines in the presence of TGFβ1 (Figure 6C). Anti-miR-128a had no dose-dependent effect on parental MCF-7aro cells. In addition, transfection of another microRNA inhibitor anti-miR-17-5p, which is predicted to target TGFβRII, had no effect on proliferation of T+LET R or MCF-7aro cells (data not shown). These results suggest that inhibiting miR-128a can sensitize the T+LET R lines to TGFβ1-mediated growth inhibition.
Figure 6. Anti-miR-128a transfection sensitizes T+LET R lines to the growth inhibitory effects of TGFβ.
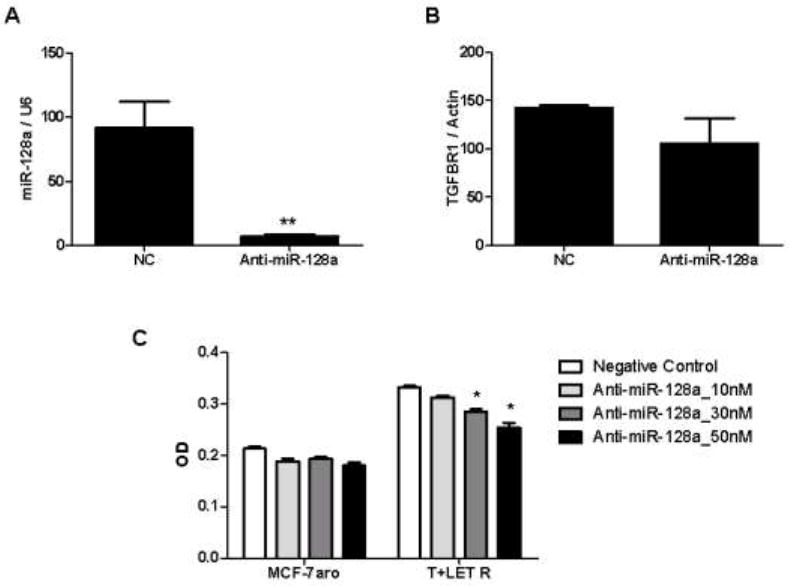
A) Transient transfection of anti-miR-128a inhibitor versus NC was performed to determine expression of miR-128a by real-time PCR, relative to U6 expression. Student’s t-test compared NC and anti-miR-128a transfected cells, with ** indicating statistical significance at a p-value < 0.01. B) Expression of TGFβR1 was quantified using real-time PCR, after transient transfection of NC or anti-miR-128a inhibitor. C) Transient transfection of NC or anti-miR-128a inhibitor was performed at concentrations of 10, 30 and 50nM and all conditions were treated with 1ng/ml TGFβ1 for 5 days. MTT assays for cell viability were performed in the MCF-7aro and T+LET R lines. For statistical analysis, Student’s t-test compared anti-miR-128a with NC transfected cells in 6C, with * indicating statistical significance at a p-value < 0.05.
DISCUSSION
The lack of clinical cross-resistance among the three AIs and tamoxifen has been observed (36), indicating that resistance mechanisms to these endocrine therapy agents are not exactly identical. Our laboratory generated cell lines that have acquired resistance to tamoxifen or each AI (13, 37), and these cells are responsive to second-line inhibitor treatment. These results indicate that our resistant cell lines are valuable tools to study the mechanisms of endocrine resistance, and gene expression profiling of these resistant cells identified unique gene expression signatures (13).
Our laboratory has identified a novel role for hsa-miR-128a in the post-transcriptional regulation of TGFβR1 expression in the T+LET R lines. Based on microarray analysis of microRNA expression, 115 microRNAs were shown to have significant differential expression between the parental MCF-7aro and derivative endocrine therapy resistant cells. As expected, microRNA expression between the AI-resistant lines versus LTEDaro and tamoxifen-resistant cells greatly differed (Figure 1B). Hierarchical clustering of microRNA expression in Figure 1B was used to identify a group of microRNAs selectively down-regulated in all AI-resistant lines, as well as microRNAs that are differentially expressed between AI-resistant lines (Table S1).
Little is known about microRNAs and their role in endocrine therapy resistance. Previous reports have shown miR-221/222 to play a role in tamoxifen-resistance, though these studies implicate two different microRNA targets p27Kip1 (38) and ERα (39) that are negatively regulated at the protein level. Expression of miR-221/222 was not significantly deregulated in comparison to control MCF-7aro expression by our microarray analysis, but several microRNAs were deregulated in our T+TAM R lines (Table S1). Of relevance to the above studies, the T+TAM R cells from our laboratory were generated by long-term selection with hormone plus tamoxifen, generating cells that have acquired resistance to tamoxifen. Cells from the two reported studies were generated either without ligand (38) or are actually tamoxifen-sensitive cells that were rendered resistant to tamoxifen after microRNA overexpression (39).
To date, the TGFβ signaling pathway has not been implicated in AI-resistance, though reports have shown a role for TGFβ in tamoxifen-resistance (33, 34). Our data suggests that the T+LET R lines may have lost sensitivity to the growth inhibitory effect of TGFβ , which remains intact in the parental MCF-7aro cells. This loss of response to TGFβ1 is proposed to be a result of hsa-miR-128a regulation of TGFβR1 expression. This was confirmed by western analysis that showed loss of TGFβR1 expression in response to pre-miR-128a transfection and luciferase assays indicating response when a wild-type miR-128a binding site reporter is used.
Foekens et al. (40) reported 249 microRNAs identified in 38 ER+ lymph node negative breast cancer patient samples. This study was able to identify a subset of microRNAs that were significantly associated with an ER+ luminal signature. Of interest to our study, these ER+ luminal microRNAs were correlated with microRNAs in clusters A, B and C from our bioinformatic analysis (Figure 1), suggesting the microRNAs identified by our in vitro analysis also correlate with microRNAs observed in breast cancer samples. Foekens et al. also determined that four microRNAs are associated with breast cancer aggressiveness, miR-7, miR-210, miR-516-3p and miR-128a, as seen by Kaplan-Meier survival plots (40). With the exception of miR-516-3p, the three other microRNAs were found in cluster C of our data (Figure 1), and we have implicated miR-128a in letrozole-resistance. This correlation further substantiates our in vitro analysis and possibly suggests that up-regulation of ‘aggressive’ miR-128a expression in hormone-dependent breast cancer could accelerate letrozole-resistance/estrogen-independence. This idea requires further confirmation in breast tumor specimens, though we are currently limited to confirming our results with published studies regarding miR-128a in “aggressive” breast cancers. Information regarding AI-resistance mechanisms is still limited to cell line and animal models, due to a lack of “acquired” AI-resistant breast cancer patient tumors.
Blenkiron et al. (41) reported on 133 microRNAs identified in 93 primary breast tumors, 5 normal breast samples and 21 breast cancer cell lines. This study elegantly correlated microRNA expression with breast cancer subtypes, luminal A/B, basal-like, HER2+ and normal-like. An ER+ (luminal A and B) microRNA signature was published in this study (41), and similar to Foekens et al., the ER+ luminal microRNAs are found only in clusters A, B and C of our analysis. We could not correlate cluster D from our study with an ER+ luminal mRNA signature from the Foekens (40) or Blenkiron et al. (41) studies. Closer examination of this group of microRNAs is needed, as they may play a very unique role in endocrine therapy resistance, which may be independent of ER signaling.
Analysis of clusters A, B and C from our study was expanded, which subsequently identified 86% of the hormone-responsive microRNAs within cluster C, and the remaining 14% within cluster B. Of the microRNAs identified by our microarray analysis, the miR-17-92 cluster was identified to be hormone-responsive. Similar to our study, the miR-17-92 cluster was also recently reported to be regulated by estrogen (42). The hormone-responsive microRNAs identified by our microarray analysis were correlated and matched with 30% of predicted hormone-responsive microRNAs reported by Creighton et al. (43). A study was recently published by Bhat-Nakshatri et al. that compared microRNA expression in the MCF-7 vector transfected cells and MCF-7/AKT overexpressing lines treated with E2. Though this system is not identical to our own, we were able to correlate 5 of the 21 E2-induced microRNAs to our own hormone-responsive microRNA list, let-7g, let-7i, miR-200a, mir-103, miR-107 (44).
It is believed that hormone-dependent breast cancer cells utilize the transcriptional program of ER for transactivation of genes involved in survival and proliferation. It may be possible that in addition to alterations in estrogen-responsive gene expression, microRNAs could simultaneously achieve a secondary level of post-transcriptional regulation of pathways implicated in breast cancer and resistance to endocrine therapy. This study identified a number of hormone-responsive microRNAs as well as microRNAs significantly expressed between the AI, LTEDaro and tamoxifen-resistant lines. Also, miR-128a was identified to play an important role in regulating TGFβR1 expression, leading to compromised growth inhibitory effects of TGFβ in letrozole-resistant breast cancer cells. We believe a number of the microRNAs identified in this study could play important roles in modulating acquired resistance to endocrine therapy.
Supplementary Material
Acknowledgments
The authors would like to thank Dr. Xiwei Wu and Dr. Min (Sierra) Li for help with microarray processing and statistical analysis.
Support: NIH pre-doctoral training fellowship CA123691 to S. Masri and NIH grant CA044735 to S. Chen.
Footnotes
Microarray data in this publication has been deposited in NCBIs Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) and is accessible through GEO Series accession number GSE17775.
References
- 1.Yager JD, Liehr JG. Molecular mechanisms of estrogen carcinogenesis. Annu Rev Pharmacol Toxicol. 1996;36:203–32. doi: 10.1146/annurev.pa.36.040196.001223. [DOI] [PubMed] [Google Scholar]
- 2.Yue W, Santen RJ, Wang JP, et al. Genotoxic metabolites of estradiol in breast: potential mechanism of estradiol induced carcinogenesis. J Steroid Biochem Mol Biol. 2003;86(3-5):477–86. doi: 10.1016/s0960-0760(03)00377-7. [DOI] [PubMed] [Google Scholar]
- 3.Santner SJ, Chen S, Zhou D, Korsunsky Z, Martel J, Santen RJ. Effect of androstenedione on growth of untransfected and aromatase-transfected MCF-7 cells in culture. J Steroid Biochem Mol Biol. 1993;44(4-6):611–6. doi: 10.1016/0960-0760(93)90267-z. [DOI] [PubMed] [Google Scholar]
- 4.Yue W, Wang JP, Hamilton CJ, Demers LM, Santen RJ. In situ aromatization enhances breast tumor estradiol levels and cellular proliferation. Cancer Res. 1998;58(5):927–32. [PubMed] [Google Scholar]
- 5.Tamoxifen for early breast cancer: an overview of the randomised trials. Early Breast Cancer Trialists’ Collaborative Group. Lancet. 1998;351(9114):1451–67. [PubMed] [Google Scholar]
- 6.Coombes RC, Hall E, Gibson LJ, et al. A randomized trial of exemestane after two to three years of tamoxifen therapy in postmenopausal women with primary breast cancer. N Engl J Med. 2004;350(11):1081–92. doi: 10.1056/NEJMoa040331. [DOI] [PubMed] [Google Scholar]
- 7.Goss PE, Ingle JN, Martino S, et al. A randomized trial of letrozole in postmenopausal women after five years of tamoxifen therapy for early-stage breast cancer. N Engl J Med. 2003;349(19):1793–802. doi: 10.1056/NEJMoa032312. [DOI] [PubMed] [Google Scholar]
- 8.Howell A, Cuzick J, Baum M, et al. Results of the ATAC (Arimidex, Tamoxifen, Alone or in Combination) trial after completion of 5 years’ adjuvant treatment for breast cancer. Lancet. 2005;365(9453):60–2. doi: 10.1016/S0140-6736(04)17666-6. [DOI] [PubMed] [Google Scholar]
- 9.Britton DJ, Hutcheson IR, Knowlden JM, et al. Bidirectional cross talk between ERalpha and EGFR signalling pathways regulates tamoxifen-resistant growth. Breast Cancer Res Treat. 2006;96(2):131–46. doi: 10.1007/s10549-005-9070-2. [DOI] [PubMed] [Google Scholar]
- 10.Massarweh S, Osborne CK, Creighton CJ, et al. Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Cancer Res. 2008;68(3):826–33. doi: 10.1158/0008-5472.CAN-07-2707. [DOI] [PubMed] [Google Scholar]
- 11.Jelovac D, Sabnis G, Long BJ, Macedo L, Goloubeva OG, Brodie AM. Activation of mitogen-activated protein kinase in xenografts and cells during prolonged treatment with aromatase inhibitor letrozole. Cancer Res. 2005;65(12):5380–9. doi: 10.1158/0008-5472.CAN-04-4502. [DOI] [PubMed] [Google Scholar]
- 12.Santen RJ, Song RX, Zhang Z, Yue W, Kumar R. Adaptive hypersensitivity to estrogen: mechanism for sequential responses to hormonal therapy in breast cancer. Clin Cancer Res. 2004;10(1 Pt 2):337S–45S. doi: 10.1158/1078-0432.ccr-031207. [DOI] [PubMed] [Google Scholar]
- 13.Masri S, Phung S, Wang X, et al. Genome-wide analysis of aromatase inhibitor-resistant, tamoxifen-resistant, and long-term estrogen-deprived cells reveals a role for estrogen receptor. Cancer Res. 2008;68(12):4910–8. doi: 10.1158/0008-5472.CAN-08-0303. [DOI] [PubMed] [Google Scholar]
- 14.Chan CM, Martin LA, Johnston SR, Ali S, Dowsett M. Molecular changes associated with the acquisition of oestrogen hypersensitivity in MCF-7 breast cancer cells on long-term oestrogen deprivation. J Steroid Biochem Mol Biol. 2002;81(4-5):333–41. doi: 10.1016/s0960-0760(02)00074-2. [DOI] [PubMed] [Google Scholar]
- 15.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–97. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 16.Llave C, Xie Z, Kasschau KD, Carrington JC. Cleavage of Scarecrow-like mRNA targets directed by a class of Arabidopsis miRNA. Science. 2002;297(5589):2053–6. doi: 10.1126/science.1076311. [DOI] [PubMed] [Google Scholar]
- 17.Palatnik JF, Allen E, Wu X, et al. Control of leaf morphogenesis by microRNAs. Nature. 2003;425(6955):257–63. doi: 10.1038/nature01958. [DOI] [PubMed] [Google Scholar]
- 18.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75(5):843–54. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- 19.Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993;75(5):855–62. doi: 10.1016/0092-8674(93)90530-4. [DOI] [PubMed] [Google Scholar]
- 20.Esquela-Kerscher A, Slack FJ. Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer. 2006;6(4):259–69. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- 21.Cimmino A, Calin GA, Fabbri M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A. 2005;102(39):13944–9. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson SM, Grosshans H, Shingara J, et al. RAS is regulated by the let-7 microRNA family. Cell. 2005;120(5):635–47. doi: 10.1016/j.cell.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 23.O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435(7043):839–43. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- 24.Huang Q, Gumireddy K, Schrier M, et al. The microRNAs miR-373 and miR-520c promote tumour invasion and metastasis. Nature cell biology. 2008;10(2):202–10. doi: 10.1038/ncb1681. [DOI] [PubMed] [Google Scholar]
- 25.Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007;449(7163):682–8. doi: 10.1038/nature06174. [DOI] [PubMed] [Google Scholar]
- 26.Wrana JL, Attisano L, Wieser R, Ventura F, Massague J. Mechanism of activation of the TGF-beta receptor. Nature. 1994;370(6488):341–7. doi: 10.1038/370341a0. [DOI] [PubMed] [Google Scholar]
- 27.Feng XH, Derynck R. Specificity and versatility in tgf-beta signaling through Smads. Annual review of cell and developmental biology. 2005;21:659–93. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- 28.Buck MB, Knabbe C. TGF-beta signaling in breast cancer. Ann N Y Acad Sci. 2006;1089:119–26. doi: 10.1196/annals.1386.024. [DOI] [PubMed] [Google Scholar]
- 29.Siegel PM, Shu W, Cardiff RD, Muller WJ, Massague J. Transforming growth factor beta signaling impairs Neu- induced mammary tumorigenesis while promoting pulmonary metastasis. Proc Natl Acad Sci U S A. 2003;100(14):8430–5. doi: 10.1073/pnas.0932636100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muraoka RS, Koh Y, Roebuck LR, et al. Increased malignancy of Neu- induced mammary tumors overexpressing active transforming growth factor beta1. Mol Cell Biol. 2003;23(23):8691–703. doi: 10.1128/MCB.23.23.8691-8703.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tucker RF, Shipley GD, Moses HL, Holley RW. Growth inhib itor from BSC-1 cells closely related to platelet type beta transforming growth factor. Science. 1984;226(4675):705–7. doi: 10.1126/science.6093254. [DOI] [PubMed] [Google Scholar]
- 32.Kalkhoven E, Beraldi E, Panno ML, De Winter JP, Thijssen JH, Van Der Burg B. Growth inhibition by anti-estrogens and progestins in TGF-beta-resistant and -sensitive breast-tumor cells. International journal of cancer. 1996;65(5):682–7. doi: 10.1002/(SICI)1097-0215(19960301)65:5<682::AID-IJC20>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 33.Arteaga CL, Koli KM, Dugger TC, Clarke R. Reversal of tamoxifen resistance of human breast carcinomas in vivo by neutralizing antibodies to transforming growth factor-beta. J Natl Cancer Inst. 1999;91(1):46–53. doi: 10.1093/jnci/91.1.46. [DOI] [PubMed] [Google Scholar]
- 34.Yoo YA, Kim YH, Kim JS, Seo JH. The functional implications of Akt activity and TGF-beta signaling in tamoxifen-resistant breast cancer. Biochim Biophys Acta. 2008;1783(3):438–47. doi: 10.1016/j.bbamcr.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 35.Sun XZ, Zhou D, Chen S. Autocrine and paracrine actions of breast tumor aromatase. A three-dimensional cell culture study involving aromatase transfected MCF-7 and T-47D cells. J Steroid Biochem Mol Biol. 1997;63(1-3):29–36. doi: 10.1016/s0960-0760(97)00068-x. [DOI] [PubMed] [Google Scholar]
- 36.Lonning PE. Lack of complete cross-resistance between different aromatase inhibitors; a real finding in search for an explanation? Eur J Cancer. 2009;45(4):527–35. doi: 10.1016/j.ejca.2008.10.019. [DOI] [PubMed] [Google Scholar]
- 37.Chen S, Masri S, Hong Y, et al. New experimental models for aromatase inhibitor resistance. J Steroid Biochem Mol Biol. 2007;106(1-5):8–15. doi: 10.1016/j.jsbmb.2007.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miller TE, Ghoshal K, Ramaswamy B, et al. MicroRNA-221/222 confers tamoxifen resistance in breast cancer by targeting p27Kip1. J Biol Chem. 2008;283(44):29897–903. doi: 10.1074/jbc.M804612200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao JJ, Lin J, Yang H, et al. MicroRNA-221/222 negatively regulates estrogen receptor alpha and is associated with tamoxifen resistance in breast cancer. J Biol Chem. 2008;283(45):31079–86. doi: 10.1074/jbc.M806041200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 40.Foekens JA, Sieuwerts AM, Smid M, et al. Four miRNAs associated with aggressiveness of lymph node-negative, estrogen receptor-positive human breast cancer. Proc Natl Acad Sci U S A. 2008;105(35):13021–6. doi: 10.1073/pnas.0803304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blenkiron C, Goldstein LD, Thorne NP, et al. MicroRNA expression profiling of human breast cancer identifies new markers of tumor subtype. Genome biology. 2007;8(10):R214. doi: 10.1186/gb-2007-8-10-r214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Castellano L, Giamas G, Jacob J, et al. The estrogen receptor-{alpha}-induced microRNA signature regulates itself and its transcriptional response. Proc Natl Acad Sci U S A. 2009 doi: 10.1073/pnas.0906947106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Creighton CJ, Nagaraja AK, Hanash SM, Matzuk MM, Gunaratne PH. A bioinformatics tool for linking gene expression profiling results with public databases of microRNA target predictions. RNA (New York, NY. 2008;14(11):2290–6. doi: 10.1261/rna.1188208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bhat-Nakshatri P, Wang G, Collins NR, et al. Estradiol-regulated microRNAs control estradiol response in breast cancer cells. Nucleic acids research. 2009 doi: 10.1093/nar/gkp500. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.