

Abstract
In an effort to probe the structure, mechanism, and biochemical properties of metallo-β-lactamase Bla2 from Bacillus anthracis, the enzyme was over-expressed, purified, and characterized. Metal analyses demonstrated that recombinant Bla2 tightly binds 1 eq of Zn(II). Steady-state kinetic studies showed that mono-Zn(II) Bla2 (1Zn-Bla2) is active, while di-Zn(II) Bla2 (ZnZn-Bla2) was unstable. Catalytically, 1Zn-Bla2 behaves like the related enzymes CcrA and L1. In contrast, di-Co(II) Bla2 (CoCo-Bla2) is substantially more active than the mono-Co(II) analog. Rapid kinetics and UV-Vis, 1H NMR, EPR, and EXAFS spectroscopic studies show that Co(II) binding to Bla2 is distrubuted, while EXAFS shows that Zn(II) binding is sequential. To our knowledge, this is the first documented example of a Zn enzyme that binds Co(II) and Zn(II) via distinct mechanisms, underscoring the need to demonstrate transferability when extrapolating results on Co(II)-substituted proteins to the native Zn(II)-containing forms.
Introduction
Anthrax is a disease that commonly affects cattle and other herbivores, caused by the spore-forming bacterium Bacillus anthracis.1 Anthrax infections in humans can be caused by contact with infected animals, or by ingestion or inhalation of B. anthracis spores. The potential for airborne transmission, together with the known lethality of Anthrax infection,2, 3 has led to the development of anthrax as a potential biological warfare agent,4 and B. anthracis has been labeled as a category A bioterrorism agent by the Centers of Disease Control and Prevention (CDC).3 Currently, the CDC recommends a 60 day antimicrobial regimen of ciprofloxacin, doxycycline, or amoxicillin for anthrax infection.4 Cephalosporins, trimethoprim, and sulfamethoxazole are not prescribed, due to suspected resistance to these drugs. Recently, the Sterne strain of B. anthracis has been reported to produce both a class A (Bla1) and a class B (Bla2) β-lactamase.5
Currently, over 50% of the antibiotics prescribed by physicians are β-lactam-containing compounds, such as penicillins, cephalosporins, and carbapenems.6 However, an increasing number of bacterial pathogens show resistance to β-lactam antibiotics, threatening their efficacy into the future. The most common pathway for resistance is the production of β-lactamases, which hydrolyze the four-membered ring in β-lactam antibiotics, rendering them ineffective.7, 8 To date, over 400 β-lactamases have been described, and these enzymes have been classified into four subgroups (A-D).8 While exhibiting different kinetic and inhibition properties, all group A, C, and D β-lactamases utilize an active site serine to perform nucleophilic attack on the substrate. The serine β-lactamases (SβLs) are the most clinically-significant, and there are some clinical inhibitors that are active towards a majority of the enzymes in these groups.8
In contrast, the group B β-lactamases, often referred to as metallo-β-lactamases or MβLs, require 1-2 Zn(II) ions for catalytic activity, and these enzymes are unaffected by SβL inhibitors.9, 10 Approximately 40 MβLs have been reported, leading to their further classification into 3 subgroups.11 The B1 enzymes require 1-2 Zn(II) ions for full activity, and prefer penicillins as substrates. They bind one Zn(II) in the Zn1 (or 3H) site, which is made up of His116, His118, His196, and a solvent-derived ligand that bridges the two metal ions, and one Zn(II) in the Zn2 (or DCH) site, made up of His263, Asp120, Cys221, the bridging solvent, and a terminally-bound water. The B2 enzymes require only one Zn(II) for full activity, prefer carbapenems as substrates and bind the catalytic Zn(II) in the Zn2 site, while the B3 enzymes, which prefer cephalosporins as substrates, generally require two Zn(II) ions for full activity, binding one Zn(II) in a canonical Zn1 site and one Zn(II) in a modified Zn2 site, where Cys221 is replaced by His121.
Although the Sterne strain of B. anthracis has not yet caused human infection, transfer of the Bla2 gene to a more pathogenic Bacillus is expected to be facile. Bla2 shares 89% sequence identity and 92% sequence homology with the B1 MβL BcII from Bacillus cereus,5, 12 and it would seem that all structural, mechanistic, and computational studies already reported for BcII would be applicable to Bla2. However, steady-state kinetic constants reported for Bla2 and BcII are different,5, 13 and several active site amino acids (IIe39, Thr182 and Gly151) in BcII are not conserved in Bla2. In addition, there are conflicting data on BcII regarding the nature of metal binding and its relation to enzyme mechanism,14-17 and it is not clear which set of data, if either, applies to Bla2.
We report here detailed characterization of recombinant Bla2 from B. anthracis. The metal content of the enzyme was ascertained with ICP-AES, and the steady-state kinetic constants of various analogs of the enzyme, with a range of substrates, were determined. To probe the structure of Bla2, the mono- and di-Co(II) analogs were prepared and characterized by UV-Vis, 1H NMR, EPR, and EXAFS spectroscopies. These studies are complemented by EXAFS of the mono- and di-Zn(II) enzymes. Together the data demonstrate that metal binding by Bla2 follows distinct pathways, dependent on the identity of the metal ion (Co(II) or Zn(II)), and that the reactivity of the Zn- and Co-containing enzymes are quite different.
Experimental Procedures
Steady-State Kinetics
A detailed description of materials, over-expression, protein purification and metal analysis methodology is included in the Supporting Information. Steady-state kinetic studies were conducted at 25° C in 50 mM Hepes, pH 6.5, on a Hewlett-Packard model 5480A diode array spectrophotometer. The molar absorptivities of the substrates used (structures shown in Supporting Information, Figure S2) were Δε485nm = 17,400 M-1cm-1 for nitrocefin, Δε280nm = -6,410 M-1cm-1 for cefaclor, Δε305nm = 7,600 M-1cm-1 for meropenem, and Δε300nm = -9,000 M-1cm-1 for imipenem.18 Substrate concentrations ranged from 10 to 200 μM, and the enzyme concentration was held fixed at ca. 10 nM. Steady-state kinetic constants were determined as described previously.18
UV-Vis Spectrophotometry
Apo-Bla2 (ca. 1 mM) in Chelex-treated 15 mM Hepes, pH 6.5, containing 100 mM NaCl was titrated with CoCl2 (Strem Chemicals, 99.999 %). Samples of Bla2 with 1 or 2 eq of added Co(II) were incubated on ice for 5 min. before being centrifuged (10 minutes at 14,500 × g) to remove precipitated protein. Difference spectra were obtained by subtracting the spectrum of apo-Bla2 from those of the Co(II)-added samples.
Stopped-Flow UV-Vis Studies
Diode array UV-Vis spectra were collected (200 – 700 nm) at 4° C on an Applied Photophysics SX18-MVR stopped-flow spectrophotometer. The buffer used in these experiments was 50 mM Hepes, pH 6.5, and the substrates were nitrocefin, cefaclor, and imipenem. Substrate concentrations ranged from 5 to 100 μM (5, 15, 25, 50, and 100 μM, nitrocefin was also examined at 35 μM), and the concentration of 1Zn-Bla2 was held constant at 20 μM. All experiments were conducted in triplicate, and the data presented here represent the average of these multiple data sets. Stopped-flow absorbance data were converted to concentrations as previously described,19 and the data were corrected for the instrument dead time of 2 ms. The resulting progress curves were fitted to single exponentials, and the resultant values of kobs were plotted against substrate concentration. When using nitrocefin as substrate, these plots were hyperbolic and fitted to kobs = K1[S]k2 / (K1[S] + k-2), as previously described.20 When using cefaclor and imipenem as substrates, these plots were linear and fitted to kobs = k1[S] + k-1. The progress curves were simulated using KINSIM, as previously described. 21, 22 Dynafit was used to verify the simulations,23 and ProK was used to obtain errors for the rate constants, as previously described.21
EPR spectroscopy
EPR spectra were recorded using a Bruker EleXsys E600 EPR spectrometer equipped with an Oxford Instruments ITC4 temperature controller and an ESR-900 helium flow cryostat. Samples were 1 mM protein in 15 mM Hepes, pH 6.5, containing 100 mM NaCl. A Bruker ER-4116DM cavity was used, with a resonant frequency of 9.63 GHz (in perpendicular mode) and 10 G (1 mT) field modulation at 100 kHz was employed. Other recording conditions are given in the legend to Figure 4. Computer simulations of EPR spectra were carried out using the matrix diagonalization program XSophe24 (Bruker Biospin GmbH) assuming a spin Hamiltonian H = βg.B.S + S.D.S, where S = 3/2 and D > 0 corresponds to an MS = |±½〉 ground state Kramers' doublet, as previously described.21, 25, 26
Figure 4.
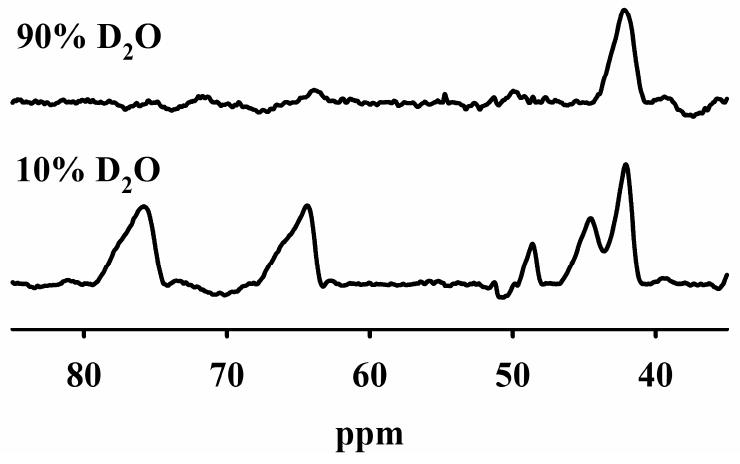
500 MHz 1H NMR spectra of CoCo-Bla2 in 10% D2O and 90% D2O.
NMR Spectroscopy
1H NMR spectra were collected at 298 K on a Bruker Avance 500 spectrometer operating at a proton frequency of 500.13 MHz. Samples of Bla2 in either 10 % or 90 % H2O were 1 mM in Chelex-treated, 15 mM Hepes, pH. 6.5, containing 100 mM NaCl. All samples were incubated for 30 min on ice after D2O addition and centrifuged, before being placed in the NMR tube. Spectra were collected using a presaturation pulse sequence (zgpr) for water suppression, a recycle time of 41 ms, and a sweep width of 400 or 800 ppm. Prior to Fourier transformation, the FID was apodized with an exponential function that resulted in an additional line broadening of 80 Hz.
X-Ray Absorption Spectroscopy
Samples of Bla2 (∼ 1 mM, including 20% (v/v) glycerol added as a glassing agent) were loaded in Lucite cuvettes with 6 μm polypropylene windows and frozen rapidly in liquid nitrogen. X-ray absorption spectra were measured at the National Synchrotron Light Source (NSLS), beamline X3B, with a Si(111) double crystal monochromator; harmonic rejection was accomplished using a Ni focusing mirror. Fluorescence excitation spectra for all samples were measured with a 13-element solid-state Ge detector array. Samples were held at ∼ 15 K in a Displex cryostat during XAS measurements. X-ray energies were calibrated by reference to the absorption spectrum of the appropriate metal foil, measured concurrently with the protein spectra. All of the data shown represent the average of ∼ 12 total scans, from two independently prepared samples each. Data collection and reduction were performed according to published procedures27 with E0 set to 9680 eV for Zn and 7735 eV for Co. The Fourier-filtered EXAFS were fit to Equation 1 using the nonlinear least-squares engine of IFEFFIT that is distributed with SixPack 28, 29
| (1) |
In Eq. 1, Nas is the number of scatterers within a given radius (Ras, ± σas), As(k) is the backscattering amplitude of the absorber-scatterer (as) pair, Sc is a scale factor, φas (k) is the phase shift experienced by the photoelectron, λ is the photoelectron mean free-path, and the sum is taken over all shells of scattering atoms included in the fit. Theoretical amplitude and phase functions, As(k)exp(-2Ras/λ) and φas (k), were calculated using FEFF v. 8.00.30 The scale factor (Sc) and ΔE0 for Zn-N (Sc = 0.78, ΔE0 = -21 eV), Zn-S (0.85, -21 eV), Co-N (0.74, -26 eV) and Co-S (0.85, -26 eV) scattering were determined previously and held fixed throughout this analysis.27, 31 Fits to the current data were obtained for all reasonable integer or half-integer coordination numbers, refining only Ras and σas2 for a given shell. Multiple scattering contributions from histidine ligands were approximated according to published procedures, fixing the number of imidazole ligands per metal ion at half-integral values while varying Ras and σas2 for each of the four combined ms pathways (see Table 4).27, 31 Metal-metal (Co-Co and Zn-Zn) scattering was modeled by fitting calculated amplitude and phase functions to the experimental EXAFS of Co2(salpn)2 and Zn2(sapln)2.
Table 4.
Best fits to Co- and Zn-Bla2 EXAFS. a
| Fit | Sample & Model | M-N/O | M-S | M-His b | M-M | |
|---|---|---|---|---|---|---|
| S1-5 | 1Co-Bla2 | 2.03 (6.9) | 2.27 (1.8) | 2.91 (4.6) | 3.26 (11) | |
| 4 N/O (2 His) + 0.5 S | 4.01 (5.8) | 4.21 (13) | ||||
| S2-5 | CoCo-Bla2 | 2.05 (3.6) | 2.27 (3.5) | 2.91 (3.6) | 3.29 (18) | |
| 4 N/O (2 His) + 0.5 S | 3.85 (10) | 4.17 (17) | ||||
| S3-5 | 1Zn-Bla2 | 2.03 (6.6) | 2.91 (6.6) | 3.16 (9.8) | ||
| 4 N/O (3 His) | 4.11 (19) | 4.42 (23) | ||||
| S4-6 | ZnZn-Bla2 | 2.04 (6.3) | 2.27 (2.6) | 2.90 (7.1) | 3.08 (5.8) | 3.44 (15) |
| 4 N/O (2 His) + 0.5 S + Zn-Zn | 4.08 (18) | 4.43 (25) | ||||
Distances (Å) and disorder parameters (in parentheses, σ2 (10-3 Å2)) shown derive from integer or half-integer coordination number fits to filtered EXAFS data. For detailed fitting results, including ranges in both k- and R-space, fit residuals and fits to unfiltered data, see Figures S1-S6 and Tables S1-S6 in Supporting Information.
Multiple scattering paths represent combined paths, as described previously (see Materials and Methods).
Results
Biochemical Characterization
Steady-State Kinetics of Bla2
Previous kinetic studies by Palzkill and co-workers were conducted in 50 mM Hepes, pH 7.5, containing 50 μM Zn(II) and 20 μg bovine serum albumin (BSA).5 In initial attempts to measure steady-state kinetics under these conditions, we found the enzyme to be unstable. Inclusion of BSA did not stabilize the enzyme in our hands. However, Bla2's predicted pI of 7.6 led us to speculate that lowering the pH from 7.5 would stabilize the protein. Reproducible steady-state kinetic data, summarized in Table 1, were obtained at pH 6.5 in 50 mM Hepes. It is important to bear in mind that the steady-state kinetic buffers contain ∼ 100 nM Zn(II),32 even if the buffers are Chelex-treated, while the assayed enzyme concentration is 1-10 nM. Given this, it is difficult to state with certainty the exact metal content of the Zn-Bla2 analogs in Tables 1 and 2. Within these limits, as-isolated Bla2, which most closely resembles the mono-zinc enzyme, hydrolyzed nitrocefin, imipenem, cefaclor, and meropenem with kcat values ranging from 24 to 92 s-1 and Km values ranging from 25 to 110 μM (Table 1).
Table 1.
Steady-state kinetic parametersa for nitrocefin, imipenem, cefaclor, and meropenem hydrolysis by as-isolated Bla2 containing 1 equivalent of Zn(II).
| Substrate: Nitrocefin | Substrate: Imipenem | Substrate: Cefaclor | Substrate: Meropenem | |
|---|---|---|---|---|
| Km (μM) | 25 ± 4 | 89 ± 35 | 67 ± 6 | 110 ± 42 |
| kcat (s-1) | 41 ± 3 | 92 ± 21 | 24 ± 1 | 49 ± 11 |
Kinetic assays were conducted at 25 °C in 50 mM Hepes, pH 6.5.
Table 2.
Steady-state kinetic parameters with nitrocefin for Bla2 after incubation of the apoenzyme with 1 or 2 eq of Zn(II) or Co(II).
| 1Co-Bla2 | CoCo-Bla2 | 1Zn-Bla2 | ZnZn-Bla2 | |
|---|---|---|---|---|
| Km(μM) | 18 ± 2 | 30 ± 9 | 28 ± 4 | 35 ± 4 |
| kcat (s-1) | 19 ± 1 | 47 ± 5 | 32 ± 2 | 42 ± 2 |
All experiments were conducted at 25 °C in 50 mM Hepes, pH 6.5. The Bla2 analogs were prepared by adding the indicated equivalents of metal to apo-Bla2 before the assays were conducted.
Apo-Bla2 was prepared by dialysis against EDTA, followed by exhaustive dialysis to remove residual EDTA (see Materials and Methods). Apo-Bla2 prepared in this way contained no detectable Zn, Co, Mn, or Fe, as indicated by ICP-AES. The apoenzyme showed ca. 10 – 20 % of the activity of the most active enzyme under steady state conditions, but no detectable activity in stopped-flow. Direct addition of 1 or 2 eq of Zn(II) to the apoenzyme gave the 1Zn- and ZnZn-analogs of Bla2 that were used for metal-dependent steady state kinetic studies (Table 2). 1Zn-Bla2 exhibited a kcat of 32 s-1 and a Km of 28 μM at pH 6.5 using nitrocefin as the substrate. The kcat of this enzyme was ca. 30 % less than that of as-isolated Bla2 (Table 1). Addition of a second eq of Zn(II) (ZnZn-Bla2) resulted in an increase in both kcat and Km, while further additions of Zn(II) led to a steady drop in kcat, consistent with the instability of the dilute enzyme in the presence of excess Zn(II) noted above. The analogous mono- and di-Co(II) enzymes (1Co-Bla2 and CoCo-Bla2, respectively) gave steady-state values for both kcat and Km that were similar to the corresponding Zn(II) enzymes (Table 2).
Stopped-Flow UV-Vis Kinetic Studies
Stopped-flow kinetic studies were conducted on 1Zn-Bla2 with three substrates (nitrocefin, cefaclor, and imipenem). Similar studies were not performed on ZnZn-Bla2, due to its instability in dilute solution (see Supporting Information). In the reaction of 1Zn-Bla2 (20 μM) with nitrocefin (5-100 μM), only two distinct features are observed: one at 390 nm that decreases with time, corresponding to loss of substrate, and one at 485 nm that increases with time, corresponding to product formation (Figure 1). No transitory absorbance was observed at 665 nm, corresponding to a stable intermediate previously observed in reactions of nitrocefin with CcrA33 and L1,19 but not with BcII.34 Progress curves at 390 and 485 nm using 35 and 50 μM nitrocefin were fitted to single exponentials to obtain kobs (Figure 2A). A plot of kobs vs. substrate concentration was hyperbolic, suggesting a rapid-equilibrium, two-step binding mechanism (Scheme 1).20 The progress curves were simulated (solid lines in Figure 1) using KINSIM, the mechanism in Scheme 1, and the rate constants in Table 3. The King-Altman method35 was used to determine theoretical expressions for kcat and Km, based on the mechanism in Scheme 1, and the resulting steady-state kinetic constants (kcat = 7.4 s-1 and Km = 62 μM) were similar to those determined in steady-state kinetic studies (Table 1).
Figure 1.
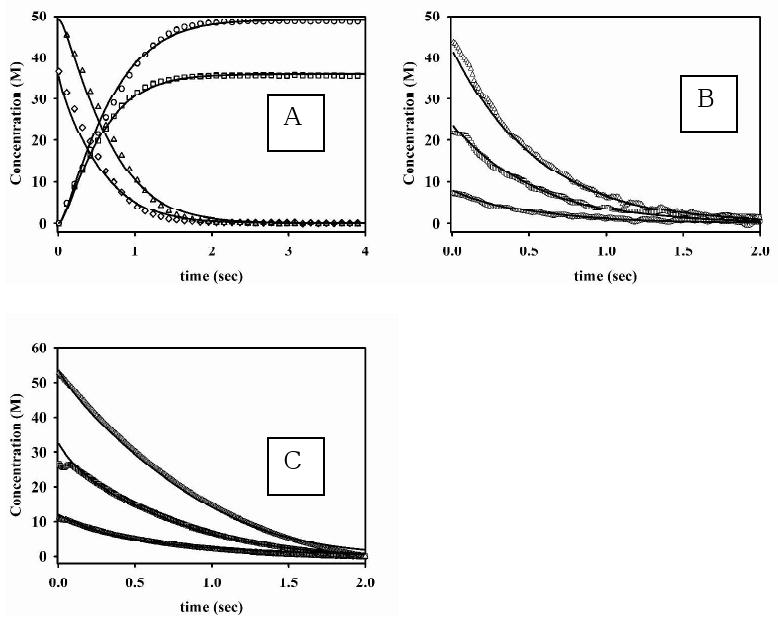
Progress curves for the reaction of 1Zn-Bla2 with nitrocefin (A), imipenem (B) and cefaclor (C). (A) The concentration of product increased over time using 35 (□) and 50 (○) μM nitrocefin, while substrate concentrations decreased over time using 35 (◊) and 50 (△) μM nitrocefin. The solid progress curves were generated by KINSIM, using the mechanism in Scheme 1 and the kinetic constants in Table 3. (B) Progress curves of the reaction of imipenem and Bla2 containing 1 eq. Zn(II) at 4 °C. The concentrations of substrate were (△) 50 μM, (□) 25 μM, and (○) 10 μM, and the concentration of Bla2 was 20 μM. The solid lines were generated by using KINSIM, the mechanism in Scheme 2, and the kinetic constants in Table 3. (C) Progress curves of the reaction of cefaclor and Bla2 containing 1 eq. Zn(II) at 4 °C. The concentrations of substrate were (△) 50 μM, (□) 25 μM, and (○) 10 μM, and the concentration of Bla2 was 20 μM. The solid lines were generated by using KINSIM, the mechanism in Scheme 2, and the kinetic constants in Table 3.
Figure 2.
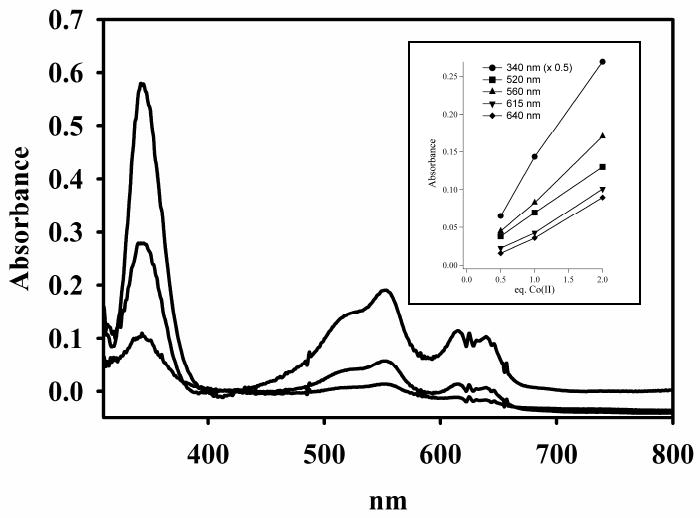
Optical titration of apo-Bla2 with Co(II). The concentration of apo-Bla2 was 1.2 mM apo-Bla2, and the buffer was 15 mM Hepes, pH 6.5, containing 100 mM NaCl. The enzyme was titrated with 0.5, 1.0, and 2.0 equivalents of Co(II) (bottom to top at 550 nm). Inset: Absorbance changes as the equivalents of Co(II) increase.
Scheme 1.

Table 3.
| constant | value used in simulation for nitrocefin | value used in simulation for imipenem | value used in simulation for cefaclor |
|---|---|---|---|
| K1 | 0.12 ± 0.01 | 0.13 ± 0.01 | 2.2 ± 0.1 |
| k2 | 10 ± 1 | 35 ± 1 | 15 ± 1 |
| k-2 | 4 ± 1 | Set to 0 | Set to 0 |
| k3 | 40 ± 1 | - | - |
| k-3 | Set to 0 | - | - |
ProK software (nonlinear Marquardt-Levenberg algorithm) was used to determine the error in the rate constants.
Stopped-flow kinetic studies were also performed on 1Zn-Bla2 with imipenem (Figure 1B) and cefaclor (Figure 1C). In these reactions, only the substrates could be observed. Unlike for nitrocefin, plots of kobs vs. imipenem or cefaclor concentration is linear, suggesting one-step binding mechanisms for both substrates (Scheme 2).35 The resulting progress curves were simulated using KINSIM, the mechanism in Scheme 2, and the rate constants in Table 3. The King-Altman-determined theoretical expressions for kcat and Km give theoretical values for imipenem (kcat = 35 s-1 and Km = 194 μM) and cefaclor (kcat = 15 s-1 and Km = 150 μM) that are similar to those determined by steady-state kinetic studies (Table 1).
Scheme 2.

Metal Binding
Optical Spectroscopy
We first examined Co(II) binding by optical titration of apo-Bla2. The UV-visible difference spectra (Co-bound – apo-Bla2, Figure 2) show the Cys-S to Co(II) charge transfer transition at 340 nm (ε340nm = 608 M-1 cm-1) that is indicative of metal binding at the Zn2 site, even at 0.5 eq of Co(II). Also apparent at 0.5 eq of Co(II) are the ligand field transitions (500 – 650 nm, ε550nm = 218 M-1 cm-1) normally associated with metal binding at the Zn1 site (Figure 2).14, 36 Each of these features increases linearly in absorbance as a function of added Co(II) (Figure 2, inset), suggesting that Co(II) distributes between the Zn1 and Zn2 sites with minimal difference in affinity. We were unable to obtain the optical spectrum of Bla2 containing 3 eq of Co(II) because the protein quickly precipitated, similar to prior observations from Co(II) titrations of CcrA.37
EPR Spectroscopy
EPR spectra from 1Co-Bla2 and CoCo-Bla2 were recorded under non-saturating, saturating, and rapid-passage conditions and are shown in Figure 3. The spectra from 1Co-Bla2 (Figure 3A) and CoCo-Bla2 (Figure 3B) recorded under non-saturating conditions exhibited no resolvable rhombicity or 59Co hyperfine structure, and were characteristic of 5- or 6-coordinate Co(II) with at least one water ligand. Spectra recorded at high power and low temperature (Figure 3D) did not reveal any MS = |± 3/2〉 signals that would indicate tetrahedral Co(II). Slight differences were observed between the non-saturated spectra of 1Co-Bla2 and CoCo-Bla2. The difference spectrum (Figure 3C) revealed the presence of a second Co(II) species in CoCo-Bla2. This component, deconvoluted by direct subtraction, may itself be due to more than one species but its isolation does at least indicate some degree of preference of Co(II) for one binding site over the other. The signal from 1Co-Bla2 could not be satisfactorily simulated as a single species, suggesting that both the spectra of 1Co-Bla2 and of CoCo-Bla2 may be mixtures that differ only in the relative proportions of the species. The best estimate that can be obtained from EPR for the degree of discrimination between sites for Co(II) binding comes from the difference spectrum of Figure 3E, which was generated by subtraction of 1.7 × Trace 3A from Trace 3B. This resulted in a residual that contained no features of either positive or negative amplitude that corresponded to either of the experimental signals, and indicates that 85 % of the Co(II) in CoCo-Bla2 is likely indistinguishable from the Co(II) complement of 1Co-Bla2. The nature of the other 15 % in CoCo-Bla2 is consistent with only a small difference in the affinities of the binding sites.
Figure 3. EPR spectra from Co(II)-containing Bla2.
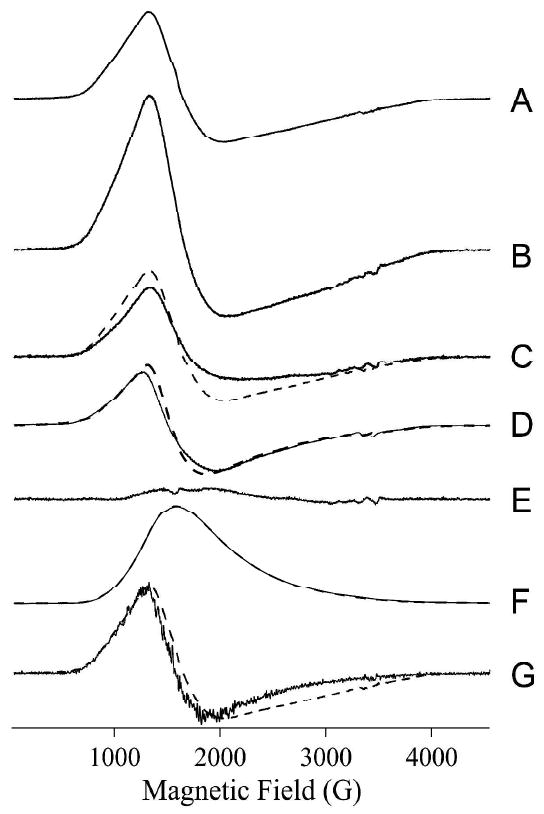
(A) 1Co-Bla2, 2 mW, 11 K; (B) CoCo-Bla2, 2 mW, 11 K; (C, solid line) = (B) − (A); (C, dashed line) 1Co-Bla2, 2 mW, 11 K; (D, solid line) CoCo-Bla2 × 0.5, 50 mW, 7 K; (D, dashed line) 1Co-Bla2, 50 mW, 7 K; (E) (B) - (1.7 × A); (F, solid line) CoCo-Bla2 × 0.5, 100 mW, 7 K, rapid passage; (F, dashed line) 1Co-Bla2, 100 mW, 7 K, rapid passage; (G, solid line) derivative of rapid passage spectrum of CoCo-Bla2; (G, dashed line) 1Co-Bla2, 2 mW, 11 K. Rapid passage spectra were recorded using second-derivative quadrature phase-sensitive detection. The intensities of spectra shown in A – C and E are correct relative to each other. Intensities of pairs of spectra in D, F and G are arbitrary, but within each pair the intensities are correct when the multiplication factors given are taken into account.
The rapid passage spectra of 1Co-Bla2 and CoCo-Bla2 (Figure 3F) were indistinguishable in form and differed only in intensity, by a factor of 2. This indicates that the species that is responsible for this spectrum is the same in both 1Co-Bla2 and CoCo-Bla2, and that twice as much of it is present in CoCo-Bla2 than in 1Co-Bla2. Comparison of the normal, non-saturated spectrum of 1Co-Bla2 and the (CoCo-Bla2 - 1Co-Bla2) difference spectrum (Figure 3C) shows that the spectrum of 1Co-Bla2 is sharper in the g⊥ region, around 2000 G (200 mT). The derivative of the rapid passage spectrum (of either 1Co-Bla2 or CoCo-Bla2) is, in turn, sharper still than that of 1Co-Bla2; the former more likely represents a single chemical species and provides some direct evidence that the signal from 1Co-Bla2 contains more than one species.
Taken overall, then, the EPR data indicate that the signal from 1Co-Bla2 is likely due to two components, one of which can be isolated by recording the spectrum under rapid passage conditions for that signal, and another that is similar in line shape, but could not be deconvoluted. Some 85 % of the signal from CoCo-Bla2 consists of Co(II) in the same environments, with the same distribution among those environments, as in 1Co-Bla2. These data indicate a binding mechanism that is largely either random or cooperative. A minor fraction of the Co(II) spin density in CoCo-Bla2, 15 %, appears either to (i) indicate Co(II) that resides in a third, distinct environment, or (ii) represent a slight difference in the distribution of Co(II) between two binding sites, or else (iii) is indicative of a broadening mechanism due to weak spin-coupling between Co(II) ions that is more pronounced in CoCo-Bla2 than in 1Co-Bla2. While the latter possibility would suggest random binding to sites with similar affinities, rather than cooperative binding, there was no additional evidence from parallel mode EPR (B0‖B1, not shown) for weak magnetic coupling between the two Co(II) ions in CoCo-Bla2.
NMR Spectroscopy
The 1H NMR spectrum of CoCo-Bla2 in 10 % D2O shows five downfield shifted resonances between 35 and 85 ppm (Figure 4). The spectrum is nearly identical in appearance to that previously reported for di-Co BcII,14 where each peak was assigned to a single proton, despite significant differences in linewidth and intensity. The 1H NMR spectrum of Bla2 containing one eq of Co(II) is identical to that of CoCo-Bla2, although of lower signal-to-noise (not shown). In 90 % D2O, four of the resonances of CoCo-Bla2 (78, 65, 49, and 44 ppm) disappear. With analogy to the crystal structure of BcII, we assign these four resonances to the NH protons of His116, His118 and His196 from the Zn1 site and His263 of the Zn2 site. The resonance at 42 ppm is not solvent-exchangeable and can be assigned to the meta proton of His118, which binds metal through the δN in BcII. This is consistent with the multiple Co(II) binding sites identified by EPR, and the observation of both the S6Co(II) CT band and the d-d bands, both with one eq of Co(II), indicating that the first eq of Co(II) distributes between the two metal binding sites.
The NMR spectra of Bla2 are very similar to those previously reported for BcII,14 with the notable exception of the absence of a resonance shifted beyond 100 ppm assignable to the β-CH2 protons of a Co(II)-coordinated cysteinate (spectra were recorded to 400 ppm). One possibility is that our procedure to prepare the 1H NMR samples resulted in oxidation of Cys221 to the sulfoxide. Similar oxidation has been suggested for CcrA,31 and an oxidized Cys221 has been observed in crystal structures of BcII38 and VIM-2.39
Co K-Edge XAS
Comparing the EXAFS of 1Co-Bla2 and CoCo-Bla2 (Figure 5) shows little quantifiable difference, consistent with the optical, NMR and EPR studies described above. That the data are indeed different favors independent, distributed binding to two sites of comparable affinity, rather than cooperative binding. The major difference is in the height of the main peak, reflective of either higher coordination number or lower overall disorder in the first shell on addition of the second eq of Co(II). The shape, particularly the width, of the main feature is largely unchanged, and curve-fitting results suggest the latter interpretation (lower disorder) better reflects the apparent difference. Best fits for 1Co- and CoCo-Bla2 are shown in Figure 5, and summarized in Table 4. Detailed fitting results, including fits to unfiltered EXAFS, are presented in Supporting Information, Figure S2 and Table S1 for 1Co-Bla2, and Figure S3 and Table S2 for CoCo-Bla2.
Figure 5.
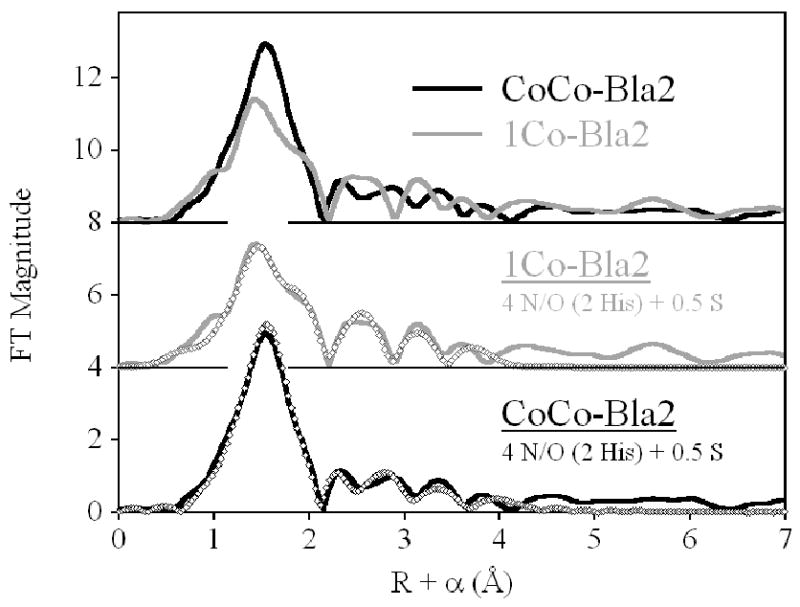
Fourier transformed EXAFS spectra of Co(II)-substituted Bla2. (Top) Direct comparison of 1Co- (gray line) and CoCo-Bla2 (black line). (Center) Best fit (open symbols) for 1Co-Bla2. (Bottom) Best fit (open symbols) for CoCo-Bla2. See Table 4 and Supporting Information for fit details.
The distributed binding indicated above leads to the expectation that both 1Co-and CoCo-Bla2 would reflect an average of the Zn1 and Zn2 coordination spheres, with 4 N/O and 0.5 S comprising the first shell. In both cases, inclusion of a partial sulfur scatterer dramatically improves the quality of the first shell fits. For 1Co-Bla2, the average first shell bond length is reflective of 4- or 5-coordination (Fit S1-1). While the fit residual decreases nearly 3-fold (63 %) when the fit includes a mixed first shell of nitrogen and oxygen (compare Fits S1-1 and S1-2), the difference in refined distances (0.15 Å) is at the resolution of the data (0.14 Å). An even greater improvement is seen on addition of a sulfur scatterer (70 %, compare Fits S1-1 and S1-3). The three shell fit, including resolved N, O and S shells, results in an overall 83 % improvement from the single scattering fit (Fit S1-1 vs. S1-4), and 43 % over the N/O + S fit (Fit S1-1 vs. S1-4), although the refined N and O distances are outside the resolution of the data. For this reason, multiple scattering fits were conducted using a first shell of 4 N/O + 0.5 S and indicate an average of 2 His ligands per Co ion (Fit S1-5). Inclusion of a Co-Co vector in this fit leads to only 8 % improvement in the fit residual, suggesting a bridged bimetallic cluster is not present (Fit S1-6).
Similar results are obtained for CoCo-Bla2 (Fig. 5, Bottom, and Figure S3 and Table S2). The average first shell bond length is unchanged from 1Co-Bla2 (compare Fits S2-1 and S1-1). A mixed first shell of N and O leads to a 49 % improvement in fit residual with N and O distances at the resolution of the data (Fits S2-1 and S2-2) and inclusion of a partial sulfur scatterer leads to a 70 % improvement in the fit residual. Multiple scattering fits, using 4 N/O + 0.5 S to model the first shell, again indicate an average of 2 His ligands per Co ion. The appearance of the outer shell scattering would suggest some rearrangement on addition of the second Co(II) ion, but this information is outside the scope of the current analysis. Inclusion of a Co-Co vector in this fit leads to a 20 % improvement in fit residual, while increasing the number of variable parameters to 14 from 12 (a 17 % increase). The refined Co-Co distance is shorter than the Zn-Zn separation in similar systems, including this one (below), while it is expected, if present to be slightly longer due to the modestly larger size of the Co(II) ion. We take this as evidence that the aquo bridge is replaced by one solvent on each metal ion in fully loaded CoCo-Bla2, in accord with our previous studies of CcrA,31 and consistent with the lack of both resolved 59Co hyperfine structure and a parallel mode signal in the EPR.
Zn K-Edge XAS
In contrast to the distributed binding observed with Co, the binding of Zn by Bla2 appears to be sequential. The Fourier-transformed EXAFS for 1Zn-Bla2 shows much higher outer shell intensity than its ZnZn counterpart, which shows much better resolution in its outer shell scattering (Figure 6, Top). Qualitatively, this is consistent with binding of the first equivalent of Zn(II) in the Zn1 site, bearing three His ligands. Population of the Zn2 site would then be expected to (i) broaden the first shell with addition of the sulfur ligand, (ii) lower the overall outer shell scattering, as the average number of His ligands is reduced from 3 to 2 and (iii) sharpen the outer shell features as the dinuclear site becomes better organized. This description is fully consistent with the Fourier transforms presented in Figure 6.
Figure 6.
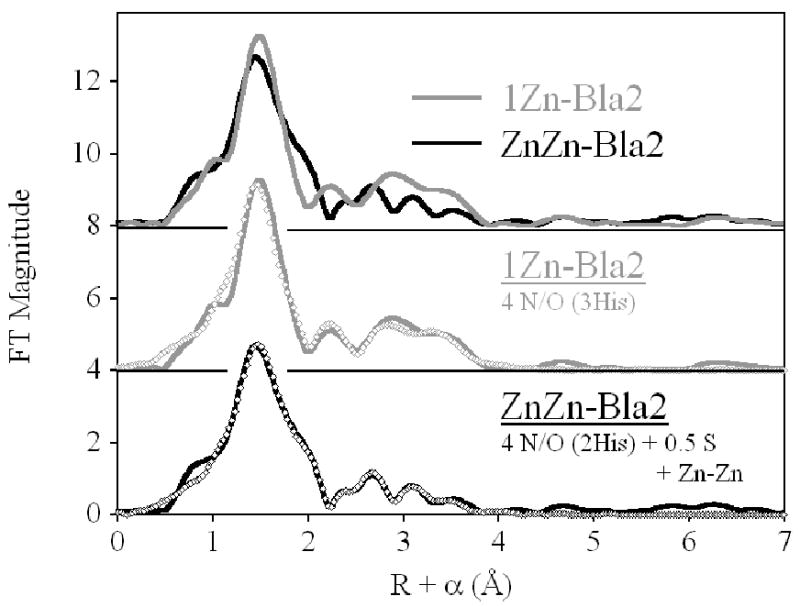
Fourier transformed EXAFS spectra of Zn(II)-Bla2. (Top) Direct comparison of 1Zn- (gray line) and ZnZn-Bla2 (black line). (Center) Best fit (open symbols) for 1Zn-Bla2. (Bottom) Best fit (open symbols) for ZnZn-Bla2. See Table 4 and Supporting Information for fit details.
The qualitative description presented above is also supported by the curve fitting results (best fits are shown in Figure 6 and Table 4, detailed results are shown Figure S4 and Table S3 for 1Zn-Bla2, and Figure S5 and Table S4 for ZnZn-Bla2), which show the Zn in 1Zn-Bla2 is coordinated by 4 N/O donors, including 3 His ligands. Inclusion of a partial sulfur scatterer does not significantly improve the fit (< 30 %), while resulting in an unreasonably short Zn-S distance (2.23 Å, compare Fits S3-1 and S3-2). Similarly, a mixed first shell of N and O donors leads to a minimal improvement in the fit residual, only slightly larger than simply increasing the number of variable parameters (compare Fits S3-3 and S3-4 to Fit S3-1), with a separation in Zn-N and Zn-O distances that is less than the resolution of the data. Multiple scattering fits indicate an average of 3 His ligands per Zn, and inclusion of a Zn-Zn vector in this fit results in only 18 % improvement in fit residual with a relatively short refined Zn-Zn distance of 3.36 Å. Together, we take this as evidence that there is no population of the Zn2 site in 1Zn-Bla2, and that there are not sub-populations of di-Zn and apo-Bla2.
In contrast, the coordination sphere of the average Zn(II) in ZnZn-Bla2 clearly includes 0.5 S donors, as well as 4 N/O. The average first shell bond length (in N/O only fits) increases by 0.05 Å over 1Zn-Bla2 (compare Fits S4-1 and S3-1), suggesting an increase in average coordination number and/or the presence of a larger scatterer. A mixed first shell of resolved Zn-N and Zn-O leads to a 70 % decrease in the fit residual, though the distances are right at the resolution of the data (Fits S4-1 and S4-2). Inclusion of 0.5 S with 4 unresolved N/O donors leads to an 88 % improvement in the fit residual (compare Fits S4-3 and S4-1), and a three-component N + O + S fit results in an overall 93 % reduction in residual. For simplicity, multiple-scattering fits were restricted to a first shell of 4 N/O + 0.5 S donors. The fits are consistent with the average number of imidazoles being reduced from 3 to 2 on addition of the second eq of Zn(II). Inclusion of a Zn-Zn interaction leads to a 43 % improvement in the fit residual, while increasing the number of variable parameters from 12 to 14. The refined Zn-Zn distance of 3.44 Å is the same as that seen in other resting state dinuclear MβLs by both XAS40 and diffraction,41-43 but very different from the 3.6-3.7 Å EXAFS-derived separation reported earlier for BcII.44
Stopped-Flow of 1Zn- and 1Co-Bla2
The distributed binding of Co(II) by Bla2, as demonstrated above, cannot distinguish between potential populations of mono-Co(II) Bla2 (loaded randomly in either the Zn1 or the Zn2 site) and a mixture of apo- and fully-loaded di-Co(II) Bla2, and indeed all variants may coexist in the absence of substrate. As can be seen in Figure 7, while 1Zn-Bla2 does not stabilize an intermediate with nitrocefin, 1Co-Bla2 clearly does. The lack of accurate values of Km and kcat for 1Co-Bla2 preclude the simulation of these data. However, in recent work with the dinuclear B3 MβL L1, we showed that occupation of the Zn1 site was sufficient for catalysis, but both metal ions were needed to stabilize the nitrocefin-derived intermediate.45 Thus, the data in Fig. 7 indicate that there is a significant population of CoCo-Bla2 present at 1 eq Co(II) per protein, at least under turnover conditions. The apparent concentration of the intermediate suggests that substrate may shift the metal binding equilibrium in favor of the dinuclear enzyme. This is consistent with previous work by Page and co-workers on BcII.15, 46
Figure 7.
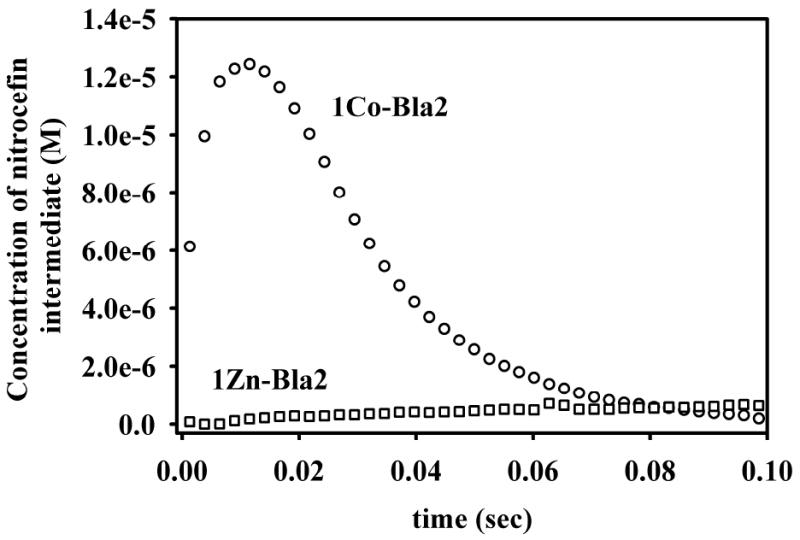
Stopped-flow kinetic traces for the hydrolysis of nitrocefin by 1Co- and 1Zn-Bla2, monitored at 665 nm.
Discussion
Biochemical Characterization
Steady-state kinetic studies on as-isolated Bla2 containing 1 equivalent of Zn(II), using several different substrates, show that the as-isolated enzyme is catalytically-active (Table 1). Previously, Palzkill and co-workers reported that recombinant Bla2 (Zn(II) content unknown) exhibited a Km of 75 ± 5 μM and a kcat 313 ± 21 s-1, when using nitrocefin as substrate.5 In our hands, recombinant Bla2 containing 1 eq of Zn(II) exhibited a Km of 25 ± 4 μM and a kcat of 41 ± 3 s-1 with nitrocefin as substrate. These values compare favorably with previous steady-state kinetic data on BcII, which exhibits a Km of 9 μM and a kcat of 43 s-1 with nitrocefin.47 The stark difference in steady-state kinetic behavior of the two recombinant Bla2 preparations may be due to differing metal content and/or different assay conditions, such as pH. Our assays were conducted at 25° C, while the preceding studies were conducted at 30° C,5 although this should result in only a 1.25-fold difference in kcat.
Metal Binding
Steady-state kinetic studies revealed that Bla2 containing 1 equivalent of Co(II) is less than ½ as catalytically active as CoCo-Bla2 (Table 2), suggesting that (i) Co(II) binding to Bla2 is random and that both metal ions bind with similar KD values or (ii) that Bla2 exhibits positive cooperative binding of Co(II). A distributed binding model is supported by optical studies, which show both the d-d bands (Zn1) and the Scys6Co(II) LMCT (Zn2), even at a loading of only 0.5 eq of Co(II), whose intensity maximizes above 2 eq of Co(II) per protein (Figure 2). The EPR spectra of 1Co-and CoCo-Bla2 are nearly superimposable, and suggest 5- or 6-coordination for both, with at least one solvent ligand (Figure 3). The 1H NMR spectra of 1Co- and CoCo-Bla2 are nearly identical, showing the same five paramagnetically-shifted resonances. Four of these resonances are solvent exchangeable (Fig. 4), indicating a total of four coordinated histidines with only 1 eq of Co(II) added. The Co K-edge EXAFS are also consistent with a distributed binding model, with only an apparent decrease in disorder accompanying addition of the second eq of Co(II).
In sum, the spectroscopic studies of Co(II)-binding by Bla2 overwhelmingly indicate distributed binding of Co(II) by Bla2 in the absence of substrate. In addition, while the electronic absorption, EPR and EXAFS data all indicate that the average Co(II) environment in 1Co-Bla2 and CoCo-Bla2 bulk samples is substantially similar, the EPR and EXAFS nevertheless detected differences between them. The appearance of an ill-defined broadening in the EPR and of lower overall disorder in the EXAFS, upon binding a second Co(II), argue strongly against a strictly cooperative binding mechanism and, therefore, favor independent, distributed binding of Co(II) to sites with comparable affinities. Samples of 1Co-Bla2 would then be expected to contain a statistical distribution of [Co_(Bla2)], [_Co(Bla2)], [CoCo(Bla2)] and apo-Bla2, and steady state kinetics are consistent with this.
In contrast to Co(II), the binding of Zn(II) by Bla2 appears to be sequential. EXAFS of 1Zn-Bla2 show the first eq of Zn(II) clearly binds to the Zn1 site. Addition of a 2nd eq of Zn(II) to form ZnZn-Bla2 leads to the observation of a Zn-S interaction, consistent with Cys221 coordination (and metal bound at the Zn2 site), and a clear Zn-Zn interaction at ∼ 3.4 Å. The EXAFS are consistent with the steady-state kinetics, which showed that ZnZn-Bla2 is not twice as active as 1Zn-Bla2, suggesting that the second Zn(II) ion does not greatly affect catalysis and that the first site of Zn(II) binding, Zn1 in this case, is more important kinetically.14, 44, 48 To our knowledge this is the first documented example of a naturally-occurring Zn(II) enzyme that follows distinct metal binding pathways, dependent on whether the metal is Zn(II) or Co(II), and strongly underscores the need to demonstrate transferability when studying Co(II)-substituted Zn(II) enzymes.
The stopped-flow kinetics studies are consistent with the difference in metal binding mechanisms, and they suggest that substrate binding may alter the metal-binding equilibrium for Co(II), but not for Zn(II). 1Zn-Bla2 does not stabilize the nitrocefin-derived reaction intermediate. The lack of an intermediate suggests that the situation described for resting 1Zn-Bla2 by EXAFS, a homogeneous population of mono-Zn enzyme, with Zn(II) bound in the canonical Zn1 site, is preserved during turnover. In contrast, the appearance of an intermediate is clear in 1Co-Bla2-catalyzed nitrocefin hydrolysis. Within the accuracy of ε665nm for the intermediate, the limiting concentration of the Co-bound intermediate (ca. 12 μM) suggests that, under turnover conditions, 1Co-Bla2 may, in fact, contain nearly equal populations of apo- and CoCo-Bla2, while all of the resting state studies presented clearly indicate a statistical distribution. At the current level of the data we can only speculate on this point.
Comparison with Other B1 MβLs
The metal-binding properties of two other B1 MβLs have been studied in detail. A set of enzymological, optical, X-ray absorption and magnetic resonance studies of Co(II) binding, similar to those presented here, by the B1 MβL CcrA indicated sequential binding, with the first equivalent of Co(II) bound exclusively at the Zn1 site.31 Both EPR and XAS of 1Co-CcrA and CoCo-CcrA were suggestive of 5/6-coordination, rather than the expected 4/5. Neither XAS nor EPR showed evidence of a covalent bridge between the Co ions.
The EPR studies presented here also suggest 5/6-coordination for both Co(II) ions in Bla2, with little or no evidence of a bridging interaction. However, the XAS-derived average first shell bond lengths for 1Co- and 1Zn-Bla2 differ by only 0.02 Å, while those of 1Co-31 and 1Zn-CcrA (see Supporting Information) differ by 0.12 Å. A similar trend is apparent comparing the di-Co and di-Zn enzymes. The average first shell bond lengths for CoCo- and ZnZn-Bla2 differ by only 0.01 Å, while those of CoCo-31 and ZnZn-CcrA differ by 0.12 Å. These data clearly indicate that Co(II)-substitution better preserves the coordination number of the native Zn(II) in Bla2 than it does in CcrA, which clearly indicates an increase in coordination number for both metal ions.
Recent studies have shown that BcII shows a very modest preference for binding Co(II) at the Zn1 site, but evidence of a di-Co(II) population is apparent at very low (∼0.6 eq) Co(II) stoichiometries.14 The present studies on Bla2 are not inconsistent with this model, though they offer no direct evidence as to the speciation of resting 1Co-Bla2. Freeze quench spectroscopic studies of di-Co(II) BcII indicate the presence of a covalent bridge in the resting enzyme, based on the observation of a parallel mode EPR signal.49 The presence of the bridge appears, at present, unique to di-Co BcII, as in both CcrA and Bla2, we see no concrete evidence for Co-Co interactions in the Co K-edge XAS or significant magnetic coupling in their EPR.31
The distributed binding of Co(II) by BcII14 is consistent with an earlier EXAFS study of Zn(II)-binding by the same enzyme.44 Using optical competition and fluorescence spectroscopy, de Seny, et al suggested that the first equivalent of Zn(II) distributes between the two sites in BcII. In contrast, both CcrA and Bla2 have been shown here to bind Zn(II) sequentially. To the greater issue of Co(II)-substitution, it appears at present that the substitution is completely faithful in BcII; it reproduces the order of addition in CcrA, but not coordination number, without the formation of a bridged cluster; and it fails for Bla2 where distinct metal-dependent mechanisms are observed.
Co(II) substitution has long been used to study zinc enzymes and often furnishes catalytically active forms of zinc enzymes.50 Tetrahedral Zn(II) active centers can be substituted with Co(II) and, in some cases, Co(II) directly adopts the original Zn(II) coordination.51-53 In other zinc-dependent metalloenzymes, four-coordinate Zn(II) is replaced by five-coordinate,52, 54, 55 or even six-coordinate56 Co(II). The general propensity of Co(II) to adopt higher coordination in a hitherto four-coordinate Zn(II) site has perhaps been underappreciated,57 but the affinity of Co(II) for an extra solvent ligand appears to have little effect on catalytic activity, metal ion affinity, the mechanism of binding, or gross structural features.58 Indeed, the present case of Bla2 is the first that we know of where Co(II) binding and Zn(II) binding clearly differ, and renewed vigilance should be exercised in interpreting studies on Co(II)-substituted zinc enzymes.
Summary
Taken together, the present data demonstrate that 1Zn-Bla2 is catalytically-active and EXAFS data on this analog demonstrate that Zn(II) binds in the consensus Zn1 site. The EXAFS data also show that Zn(II) binding is sequential, as in CcrA31 and L1.40 In contrast, all of the spectroscopic and steady-state kinetic data on Co(II)-containing Bla2 indicate that Co(II)-binding is distributed in the absence of substrate. The stopped-flow data suggest that substrate binding may affect the Co(II)-binding equilibrium. A summary scheme showing how Zn(II) and Co(II) bind to Bla2 is shown in Figure 8. Further studies are required to address why Zn(II) and Co(II) behave differently in Bla2, and X-ray crystal structures of both analogs may yield information about the different metal binding properties. We believe this is the first documented example of a Zn enzyme that binds Co(II) and Zn(II) via distinct mechanisms, underscoring the importance of validating transferability when extrapolating results on Co(II)-substituted proteins to the native Zn(II)-containing forms.
Figure 8.
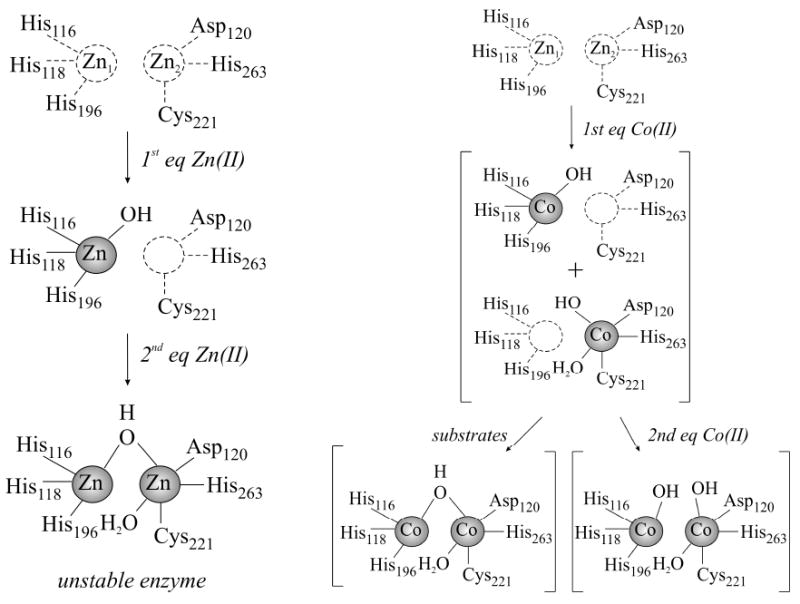
The proposed active site of Bla2 after the addition of 1 or 2 equivalents of Zn(II) (left) or Co(II) (right) to apo-Bla2.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (AI056231 to BB; EB001980 to the Medical College of Wisconsin; RR16480 from the BRIN/INBRE Program of the National Center for Research Resources to DLT), the Volwiler Professorship at Miami University (to MWC) and the National Science Foundation (CHE0809985 to DLT). Funds to purchase the 500 MHz NMR were provided by the Hayes Investment Fund (Ohio Board of Regents). The National Synchrotron Light Source is supported by the US Department of Energy.
Abbreviations
- ε
extinction coefficient
- EDTA
ethylenediaminetetraacetic acid
- EPR
electron paramagnetic resonance
- EXAFS
extended X-ray absorption fine structure
- FPLC
Fast Protein Liquid Chromatography
- Hepes
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- ICP-AES
Inductively Coupled Plasma spectrometer with atomic emission spectroscopy detection
- IPTG
isopropyl-β-D-thiogalactoside
- MβLs
metallo-β-lactamases
- NMR
nuclear magnetic resonance
Footnotes
Supporting Information Available. A description of materials, over-expression and protein purification is provided, along with ten Figures and six Tables, including SDS-PAGE gels of Bla2 purification (Figure S1), structures of the substrates used in kinetic studies (Figure S2), detailed EXAFS fitting results for 1Co-Bla2 (Figure S3 and Table S1), CoCo-Bla2 (Figure S4 and Table S2), 1Zn-Bla2 (Figure S5 and Table S3), ZnZn-Bla2 (Figure S6 and Table S4). Zn K-edge XAS of 1Zn- and ZnZn-CcrA are also presented and discussed (Figures S7-S10 and Tables S5 and S6). This information is available free of charge at http://pubs.acs.org.
References
- 1.Mock R, Fouet A. Annu Rev Microbiol. 2001;55:647–671. doi: 10.1146/annurev.micro.55.1.647. [DOI] [PubMed] [Google Scholar]
- 2.Meselson M, Guillemin J, Hugh-Jones M, Langmuir A, Popova I, Shelokov A, Yampolskaya O. Science. 1994;266:1202–1208. doi: 10.1126/science.7973702. [DOI] [PubMed] [Google Scholar]
- 3.Jernigan JA, Stephens DS, Ashford DA, Omenaca C, Topiel MS, Galbraith M, Tapper M, Fisk TL, Zaki S, Popovic T, Meyer RF, Quinn CP, Harper SA, Fridkin SK, Sejvar JJ, Shepard CW, McConnell M, Guarner J, Shieh WJ, Malecki JM, Gerberding JL, Hughes JM, Perkins BA. Emerging Infect Dis. 2001;7:933–944. doi: 10.3201/eid0706.010604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Inglesby TV, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Friedlander AM, Hauer J, McDade J, Osterholm MT, O'Toole T, Parker G, Perl TM, Russell PK, Tonat K Working Group on Civilian Biodefense. JAMA. 1999;281:1735–1745. doi: 10.1001/jama.281.18.1735. [DOI] [PubMed] [Google Scholar]
- 5.Materon IC, Queenan AM, Koehler TM, Bush K, Palzkill T. Antimicrob Agents Chemother. 2003;47:2040–2042. doi: 10.1128/AAC.47.6.2040-2042.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neu HC. Science. 1992;257:1064–1073. doi: 10.1126/science.257.5073.1064. [DOI] [PubMed] [Google Scholar]
- 7.Rice LB, Bonomo RA. Drug Resist Updates. 2000;3:178–189. doi: 10.1054/drup.2000.0144. [DOI] [PubMed] [Google Scholar]
- 8.Perez F, Endimiani A, Hujer KM, Bonomo RA. Curr Opin Pharmacol. 2007;7:459–469. doi: 10.1016/j.coph.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walsh CT, Wright GD. Chem Rev. 2005;105:391–393. doi: 10.1021/cr030100y. [DOI] [PubMed] [Google Scholar]
- 10.Crowder MW, Spencer J, Vila AJ. Acc Chem Res. 2006;39:721–728. doi: 10.1021/ar0400241. [DOI] [PubMed] [Google Scholar]
- 11.Heinz U, Adolph HW. CMLS, Cell Mol Life Sci. 2004;61:2827–2839. doi: 10.1007/s00018-004-4214-9. [DOI] [PubMed] [Google Scholar]
- 12.Chen Y, Tenover FC, Koehler TM. Antimicrob Agents Chemother. 2004;48:4873–4877. doi: 10.1128/AAC.48.12.4873-4877.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Felici A, Amicosante G, Oratore A, Strom R, Ledent P, Joris B, Fanuel L, Frere JM. Biochem J. 1993;291:151–155. doi: 10.1042/bj2910151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Llarrull LI, Tioni MF, Kowalski J, Bennett B, Vila AJ. J Biol Chem. 2007;282:30586–30595. doi: 10.1074/jbc.M704613200. [DOI] [PubMed] [Google Scholar]
- 15.Badarau A, Damblon C, Page MI. Biochem J. 2007;401:197–203. doi: 10.1042/BJ20061002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Badarau A, Page MI. Biochemistry. 2006;45:10654–10666. doi: 10.1021/bi060934l. [DOI] [PubMed] [Google Scholar]
- 17.Gonzalez JM, Medrano Martin FJ, Costello AL, Tierney DL, Vila AJ. J Mol Biol. 2007;373:1141–1156. doi: 10.1016/j.jmb.2007.08.031. [DOI] [PubMed] [Google Scholar]
- 18.Crowder MW, Walsh TR, Banovic L, Pettit M, Spencer J. Antimicrob Agents Chemother. 1998;42:921–926. doi: 10.1128/aac.42.4.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McManus-Munoz S, Crowder MW. Biochemistry. 1999;38:1547–1553. doi: 10.1021/bi9826512. [DOI] [PubMed] [Google Scholar]
- 20.Spencer J, Clark AR, Walsh TR. J Biol Chem. 2001;276:33638–33644. doi: 10.1074/jbc.M105550200. [DOI] [PubMed] [Google Scholar]
- 21.Sharma NP, Hajdin C, Chandrasekar S, Bennett B, Yang KW, Crowder MW. Biochemistry. 2006;45:10729–10738. doi: 10.1021/bi060893t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matthews ML, Periyannan G, Hajdin C, Sidgel TK, Bennett B, Crowder MW. J Am Chem Soc. 2006;128:13050–13051. doi: 10.1021/ja0627343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuzmic P. Analytical Biochemistry. 1996;237:260–273. doi: 10.1006/abio.1996.0238. [DOI] [PubMed] [Google Scholar]
- 24.Hanson GR, Gates KE, Noble CJ, Mitchell A, Benson S, Griffin M, Burrage K. XSophe - Sophe - XeprViewA computer simulation software suite for the analysis of continuous wave EPR spectra. In: Shiotani M, Lund A, editors. EPR of Free Radicals in Solids: Trends in Methods and Applications. Kluwer Press; New York: 2003. pp. 197–237. [Google Scholar]
- 25.Bennett B, Holz RC. Biochemistry. 1997;36:9837–9846. doi: 10.1021/bi970735p. [DOI] [PubMed] [Google Scholar]
- 26.Bennett B. Curr Topics Biophys. 2002;26:49–57. [Google Scholar]
- 27.Thomas PW, Stone EM, Costello A, Tierney DL, Fast W. Biochemistry. 2005;44:7559–7569. doi: 10.1021/bi050050m. [DOI] [PubMed] [Google Scholar]
- 28.Sixpack is available free of charge from its author at http://www-ssrl.slac.stanford.edu/∼swebb/sixpack.htm
- 29.IFEFFIT is open source software available from http://cars9.uchicago.edu/ifeffit
- 30.Ankudinov AL, Ravel B, Rehr JJ, Conradson SD. Phys Rev B. 1998;58:7565–7576. [Google Scholar]
- 31.Periyannan G, Costello AL, Tierney DL, Yang KW, Bennett B, Crowder MW. Biochemistry. 2006;45:1313–1320. doi: 10.1021/bi051105n. [DOI] [PubMed] [Google Scholar]
- 32.Outten CE, O'Halloran TV. Science. 2001;292:2488–2492. doi: 10.1126/science.1060331. [DOI] [PubMed] [Google Scholar]
- 33.Wang Z, Fast W, Benkovic SJ. J Am Chem Soc. 1998;120:10788. [Google Scholar]
- 34.Rasia RM, Vila AJ. ARKIVOC. 2003;3:507–516. [Google Scholar]
- 35.Segel IH. Enzyme Kinetics. John Wiley and Sons, Inc.; New York: 1993. p. 955. [Google Scholar]
- 36.Llarrull LI, Tioni MF, Vila AJ. J Am Chem Soc. 2008;130:15842–15851. doi: 10.1021/ja801168r. [DOI] [PubMed] [Google Scholar]
- 37.Wang Z, Benkovic SJ. J Biol Chem. 1998;273:22402–22408. doi: 10.1074/jbc.273.35.22402. [DOI] [PubMed] [Google Scholar]
- 38.Fabiane SM, Sohi MK, Wan T, Payne DJ, Bateson JH, Mitchell T, Sutton BJ. Biochemistry. 1998;37:12404–12411. doi: 10.1021/bi980506i. [DOI] [PubMed] [Google Scholar]
- 39.Garcia-Saez I, Docquier JD, Rossolini GM, Dideberg O. J Mol Biol. 2008;375:604–611. doi: 10.1016/j.jmb.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 40.Costello A, Periyannan G, Yang KW, Crowder MW, Tierney DL. J Biol Inorg Chem. 2006;11:351–358. doi: 10.1007/s00775-006-0083-z. [DOI] [PubMed] [Google Scholar]
- 41.Concha NO, Rasmussen BA, Bush K, Herzberg O. Structure. 1996;4:823–836. doi: 10.1016/s0969-2126(96)00089-5. [DOI] [PubMed] [Google Scholar]
- 42.Ullah JH, Walsh TR, Taylor IA, Emery DC, Verma CS, Gamblin SJ, Spencer J. J Mol Biol. 1998;284:125–136. doi: 10.1006/jmbi.1998.2148. [DOI] [PubMed] [Google Scholar]
- 43.Concha NO, Janson CA, Rowling P, Pearson S, Cheever CA, Clarke BP, Lewis C, Galleni M, Frere JM, Payne DJ, Bateson JH, Abdel-Meguid SS. Biochemistry. 2000;39:4288–4298. doi: 10.1021/bi992569m. [DOI] [PubMed] [Google Scholar]
- 44.de Seny D, Heinz U, Wommer S, Kiefer M, Meyer-Klaucke W, Galleni M, Frere JM, Bauer R, Adolph HW. J Biol Chem. 2001;276:45065–45078. doi: 10.1074/jbc.M106447200. [DOI] [PubMed] [Google Scholar]
- 45.Hu Z, Periyannan G, Bennett B, Crowder MW. J Am Chem Soc. 2008;130:14207–14216. doi: 10.1021/ja8035916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Badarau A, Page MI. J Biol Inorg Chem. 2008;13:919–928. doi: 10.1007/s00775-008-0379-2. [DOI] [PubMed] [Google Scholar]
- 47.de Seny D, Prosperi-Meys C, Bebrone C, Rossolini GM, Page MI, Noel P, Frere JM, Galleni M. Biochem J. 2002;363:687–696. doi: 10.1042/0264-6021:3630687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rasia RM, Vila AJ. Biochemistry. 2002;41:1853–1860. doi: 10.1021/bi010933n. [DOI] [PubMed] [Google Scholar]
- 49.Tioni MF, Llarrull LI, Poeylaut-Palena AA, Marti MA, Saggu M, Periyannan GR, Mata EG, Bennett B, Murgida DH, Vila AJ. J Am Chem Soc. 2008;130:15852–15863. doi: 10.1021/ja801169j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maret W, Vallee BL. Methods in Enzymology. Vol. 226. Academic Press; New York: 1993. Cobalt as Probe and Label of Proteins; pp. 52–71. [DOI] [PubMed] [Google Scholar]
- 51.Werth MT, Tang SF, Formicka G, Zeppezauer M, Johnson MK. Inorg Chem. 1995;34:218–228. [Google Scholar]
- 52.Crawford PA, Yang KW, Sharma N, Bennett B, Crowder MW. Biochemistry. 2005;44:5168–5176. doi: 10.1021/bi047463s. [DOI] [PubMed] [Google Scholar]
- 53.Wang J, Stieglitz A, Kantrowitz ER. Biochemistry. 2005;44:8378–8386. doi: 10.1021/bi050155p. [DOI] [PubMed] [Google Scholar]
- 54.Bennett B, Holz RC. J Am Chem Soc. 1997;119:1923–1933. [Google Scholar]
- 55.Garrity JD, Bennett B, Crowder MW. Biochemistry. 2005;44:1078–1087. doi: 10.1021/bi048385b. [DOI] [PubMed] [Google Scholar]
- 56.Kleifeld O, Rulisek L, Bogin O, Frenkel A, Havlas Z, Burstein Y, Sagi I. Biochemistry. 2004;43:7151–7161. doi: 10.1021/bi0302696. [DOI] [PubMed] [Google Scholar]
- 57.Kremer-Aach A, Kläui W, Bell R, Strerath A, Wunderlich H, Mootz D. Inorg Chem. 1997;36:1552–1563. doi: 10.1021/ic961434r. [DOI] [PubMed] [Google Scholar]
- 58.Munih P, Moulin A, Stamper CC, Bennett B, Ringe D, Petsko GA, Holz RC. J Inorg Biochem. 2007;101:1099–1107. doi: 10.1016/j.jinorgbio.2007.03.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.