

1. Introduction
A fundamental challenge in organic chemistry resides in the development of increasingly efficient protocols for carbon–carbon-bond formation. The ideal C–C-bond forming processes should be applicable to both petrochemical and renewable feedstocks and should be aligned with the economic and aesthetic ideals of atom-economy,1 step-economy,2 and Green Chemistry.3 Ultimately, chemical production should be sustainable, that is, it should not compromise human health, the environment, or the economy.
Hydrogen is vastly abundant, constituting roughly 90% of the atoms present in the Universe. Catalytic additions of elemental hydrogen, termed “hydrogenations,” rank among the most significant catalytic transformations in existence in terms of social and economic impact. For example, the catalytic hydrogenation of atmospheric nitrogen to produce ammonia, the Haber-Bosch process,4 is used to produce over 107 metric tons of ammonia annually. Nitrogenous fertilizer obtained from the Haber-Bosch process is estimated to sustain one-third of the Earth’s population.5 The asymmetric hydrogenation of C=X π bonds (X = O, NR) is estimated to account for over half the chiral drugs manufactured industrially, withstanding physical and enzymatic resolution.6
The Fischer-Tropsch reaction7 and alkene hydroformylation8 may be viewed as the prototypical C-C-bond forming hydrogenations. Hydroformylation combines basic feedstocks (α-olefins, carbon monoxide, and hydrogen) with perfect atom-economy, and accounts for the production of over 7 million metric tons of aldehyde annually, making it the largest-volume application of homogeneous metal catalysis.9 Given the impact of hydroformylation, it is surprising that the field of “hydrogenative C-C bond formation” lay fallow for over 70 years.10,11
As described in this account, we find that hydrogenation and transfer hydrogenation may be used to couple diverse π-unsaturated reactants to carbonyl compounds and imines.12 Such hydrogenative C-C couplings define a departure from the use of preformed organometallic reagents in classical C=X (X = O, NR) addition reactions, in many cases enabling completely byproduct-free C=X addition processes. Further, under transfer-hydrogenative coupling conditions, carbonyl addition can be attained from the alcohol or aldehyde oxidation level, thereby circumventing redox manipulations often required to adjust the oxidation level (Scheme 1).
Scheme 1.
Catalytic C-C Coupling via Hydrogenation and Transfer Hydrogenation.
2. Vinylation of Carbonyl Compounds and Imines
Numerous methods exist for the preparation of allylic alcohols and allylic amines.13,14 For example, the metal-catalyzed allylic substitution employing oxygen and nitrogen nucleophiles is a powerful protocol for the synthesis of chiral nonracemic allylic alcohols and allylic amines.15 Another approach, though less developed, involves a catalytic enantioselective aldehyde vinylation.16-19 16,17,18,19 Catalytic enantioselective vinyl transfer to imines had not been achieved prior to our work (vide infra).20,21
Limitations associated with the use of preformed vinyl metal reagents are potentially overcome through direct metal-catalyzed alkyne-carbonyl reductive couplings. The first catalytic process of this type, a rhodium-catalyzed reductive cyclization of acetylenic aldehydes mediated by silane, was reported in 1994 by Ojima et al.22 In 1995, Crowe and Rachita disclosed related silane-mediated titanocene-catalyzed cyclizations.23 The corresponding nickel-catalyzed cyclizations were first reported in 1997 by Montgomery and co-workers.24a-c,e Based on Montgomery’s finding, the nickel-catalyzed intermolecular alkyne-aldehyde reductive coupling was achieved by Jamison in 2000.25 Improved nickel-based catalysts were developed later by Takai26 and Montgomery.24d While reductive couplings of this type signal a departure from the use of preformed organometallic reagents, these methods employ terminal reductants such as hydrosilanes, hydrostannanes, organozinc reagents, organoboron reagents or chromium(II) chloride and, hence, produce molar equivalents of byproducts.
Under hydrogenation conditions, alkynes engage in completely byproduct-free reductive couplings to both carbonyl compounds and imines.12d First-generation catalytic systems based on rhodium promote the highly enantioselective coupling of conjugated alkynes to activated aldehydes and ketones in the form of vicinal dicarbonyl compounds.27a-c 27a,b,c Heterocyclic aromatic aldehydes and ketones couple to conjugated alkynes under closely related conditions, providing access to heteroaryl-substituted carbinols.27d Notably, the diene- and enyne-containing products are not subject to over-reduction under the hydrogenative coupling conditions. Presumably, upon consumption of the electrophile (the limiting reagent) excess alkyne unproductively coordinates rhodium and so impedes the rate of further conventional hydrogenation (Scheme 2).27?
Scheme 2.

Direct, Byproduct-Free Hydrogenative Coupling of Conjugated Alkynes to Activated Carbonyl Compounds and Imines Employing Cationic Rhodium Catalysts. (Ref. 27)
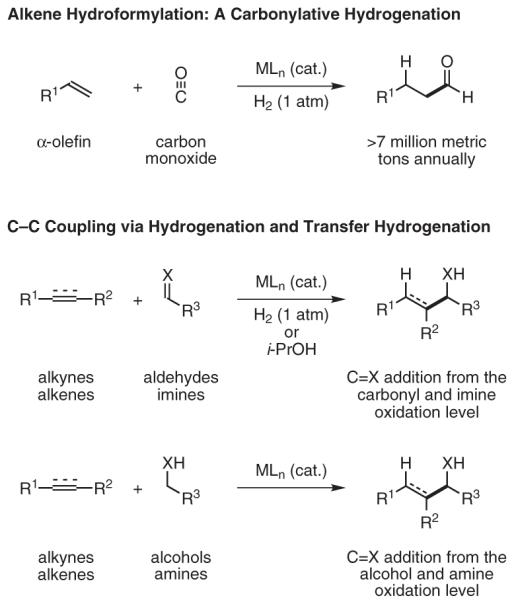
The coupling of conjugated enynes or diynes to ethyl (N-sulfinyl)iminoacetates proceeds efficiently under the conditions of rhodium-catalyzed hydrogenation.28 Using appropriately substituted (N-sulfinyl)iminoacetates, one generates the corresponding β,γ-unsaturated α-amino acid esters as single diastereomers. A second hydrogenation of the unsaturated side chain of the coupling product provides access to β-substituted α-amino acids (Scheme 3).
Scheme 3.
Unnatural α-Amino Acids via C-C-Bond-Forming Hydrogenation. (Ref. 28)
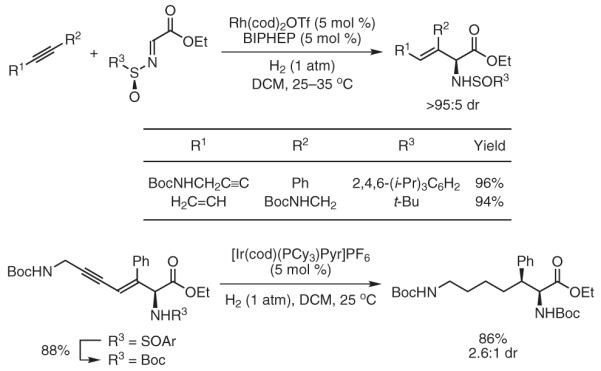
Gaseous acetylene couples to aldehydes and imines under hydrogenation conditions to furnish products of (Z)-butadienylation.29 Using chirally modified rhodium catalysts, allylic alcohols and allylic amines are formed in highly optically enriched form.29,30 These byproduct-free couplings combine acetylene, an abundant feedstock,31 with carbonyl compounds or imines to furnish chiral adducts in the absence of any preformed vinyl metal reagents (Scheme 4).
Scheme 4.
Enantioselective Carbonyl and Imine (Z)-Butadienylation via Rhodium-Catalyzed Hydrogenative Coupling of Acetylene. (Ref. 29,30)
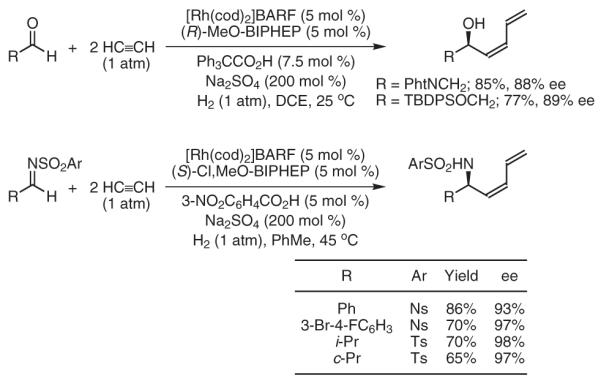
Using second-generation catalysts based on iridium, the highly enantioselective hydrogenative coupling of 1,2-dialkyl-substituted alkynes to N-arylsulfonyl imines is achieved (Scheme 5).32 The trisubstituted allylic amine products are formed with complete levels of E:Z selectivity (≥95:5), and excellent regiocontrol is observed using unsymmetrical alkynes. This byproduct-free coupling provides trisubstituted allylic amines that are not accessible via metal-catalyzed asymmetric allylic alkylation.15
Scheme 5.
Enantioselective Imine Vinylation via Iridium-Catalyzed Hydrogenative Coupling of Unconjugated Alkynes. (Ref. 32)
Finally, intramolecular coupling of alkynes to tethered aldehydes occurs readily in the rhodium-catalyzed hydrogenation. Using chirally modified catalysts, products of reductive carbocyclization are formed with uniformly high levels of optical enrichment.33 Using an achiral rhodium catalyst, chiral racemic acetylenic aldehydes engage in highly syn-diastereoselective reductive cyclizations to furnish cyclic allylic alcohols (Scheme 6).
Scheme 6.
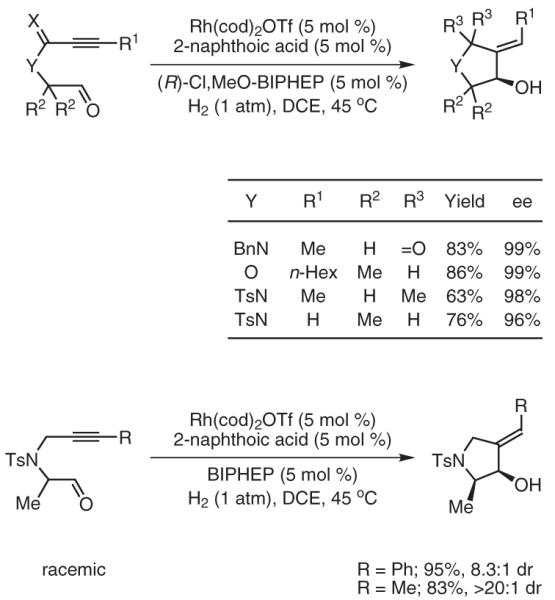
Enantio- and Diastereoselective Carbocyclizations of Acetylenic Aldehydes by the Rhodium-Catalyzed Asymmetric Hydrogenation. (Ref. 33)
3. Allylation and Propargylation of Carbonyl Compounds
Carbonyl allylation is employed routinely in synthetic organic chemistry.34 Asymmetric allylation has been achieved using chirally modified allyl metal reagents,35 chiral Lewis acid catalysts or chiral Lewis base catalysts.36 These methods invariably employ preformed allyl metal reagents, such as allyl stannanes or trichlorosilanes, which generate stoichiometric quantities of metallic byproducts. Other methods for catalytic carbonyl allylation include the reduction of metallo-π-allyls derived from allylic alcohols and allylic carboxylates,37 which require stoichiometric quantities of metal-based terminal reductants for catalytic turnover.38
We find that allyl metal species arising transiently in the course of allene hydrogenation may be captured by exogenous carbonyl electrophiles, thus enabling byproduct-free carbonyl allylation. For example, iridium-catalyzed hydrogenation of dimethylallene in the presence of activated aldehydes or ketones delivers products of reverse prenylation.39a In the iridium-catalyzed transfer hydrogenation employing isopropanol as the terminal reductant, dimethylallene also couples to aldehydes.39b Finally, hydrogen embedded within an alcohol substrate can be redistributed among reactants to generate nucleophile-electrophile pairs, enabling byproduct-free carbonyl reverse prenylation from the alcohol oxidation level (Scheme 7).39b
Scheme 7.

Catalytic Carbonyl Addition via Iridium-Catalyzed Hydrogenative Coupling of Dimethylallene. (Ref. 39)
These results prompted efforts toward general catalytic protocols for alcohol-unsaturate transfer-hydrogenative coupling.40 Under iridium-catalyzed transfer-hydrogenation conditions, 1,3-cyclohexadiene couples readily to aldehydes employing isopropanol as terminal reductant. An identical set of products may be prepared from the corresponding alcohols under nearly identical conditions (Scheme 8).41 In the ruthenium-catalyzed transfer hydrogenation employing RuHCl(CO)(PPh3)3 as precatalyst, simple acyclic dienes (butadiene, isoprene, and 2,3-dimethylbutadiene) couple to diverse alcohols.42 Again, coupling is possible from the alcohol or aldehyde oxidation level. In the latter case, isopropanol or formic acid may be employed as terminal reductants (Scheme 9).
Scheme 8.

Coupling of Dienes to Alcohols or Aldehydes by the Iridium-Catalyzed Transfer Hydrogenation. (Ref. 41)
Scheme 9.

Coupling of Dienes to Alcohols or Aldehydes by the Ruthenium-Catalyzed Transfer Hydrogenation. (Ref. 42)
Under the ruthenium-catalyzed auto-transfer hydrogenation conditions, conjugated enynes couple to benzylic, allylic, and aliphatic alcohols to furnish products of carbonyl propargylation.43-45 43,44,45 Under related transfer-hydrogenation conditions employing isopropanol as terminal reductant, enyne coupling to aldehydes provides identical products of carbonyl propargylation. In this way, carbonyl propargylation is achieved from the alcohol or the aldehyde oxidation level in the absence of preformed allenyl metal reagents. Stereocontrolled variants of these newly developed allene, diene, and enyne couplings are currently under investigation (Scheme 10).
Scheme 10.
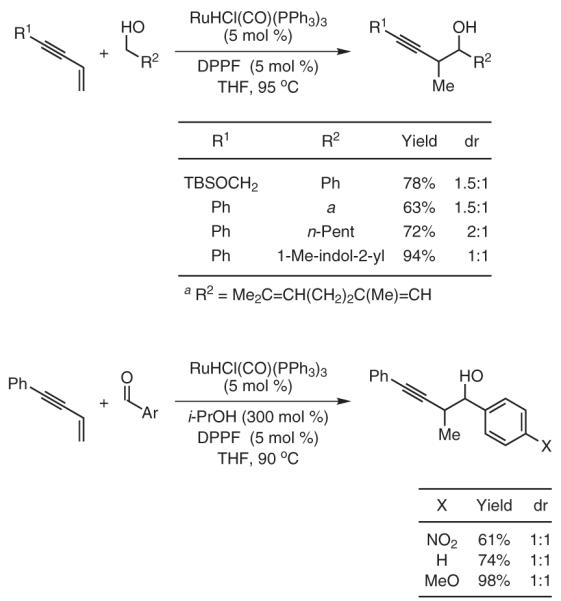
Carbonyl Propargylation from the Alcohol or Aldehyde Oxidation Level via Ruthenium-Catalyzed Transfer-Hydrogenative Coupling of 1,3-Enynes. (Ref. 43-45)
An especially powerful application of transfer hydrogenative C-C coupling involves iridium-catalyzed carbonyl allylation from the aldehyde or alcohol oxidation level employing allyl acetate as the allyl donor.46a Exposure of allyl acetate to benzylic alcohols in the presence of commercially available [Ir(cod)Cl]2 and (R)-BINAP delivers products of C-allylation in good-to-excellent yields and with high levels of asymmetric induction. Allylation from the carbonyl oxidation level is achieved by employing isopropyl alcohol as the terminal reductant. In this case, (-)-TMBTP is used as the chiral phosphine ligand to generate identical allylation adducts with high degrees of enantioselectivity. As such, asymmetric allylation can be achieved from the alcohol or aldehyde oxidation levels with equal facility. More recently, this asymmetric allylation protocol has been extended to allylic alcohols and aliphatic alcohols (Scheme 11).46b
Scheme 11.

Enantioselective Carbonyl Allylation from the Alcohol or Aldehyde Oxidation Level via Iridium-Catalyzed Transfer-Hydrogenative Coupling of Allyl Acetate. (Ref. 46)
4. Hydrogenative Aldol and Mannich Additions
For well over a century, the aldol reaction has served as a core method in organic synthesis.47 Intensive efforts have led to the realization of aldol addition protocols that enable excellent levels of diastereo- and enantiocontrol.48 A particularly significant advance involves the refinement of methods for the direct asymmetric aldol additions of unmodified ketones employing metallic49 or organic50 catalysts. These byproduct-free processes herald a departure from the use of chiral auxiliaries and preformed enol(ate) derivatives. A significant limitation of these nascent technologies resides in the issue of regiocontrolled enolization. For example, direct catalytic asymmetric aldol additions of unsymmetrical ketones, such as 2-butanone, typically result in coupling at the less substituted enolizable position to furnish linear aldol adducts.51
The challenge of regiocontrolled enolization is overcome via enone reduction. Pioneering work by Stork demonstrates that dissolving metal reduction of enones enables regiospecific generation and capture of enolate isomers that cannot be prepared exclusively under standard conditions for base-mediated deprotonation.52 Subsequently, catalytic reductive couplings of enones to aldehydes emerged.53 To date, myriad metallic catalysts for “reductive aldol coupling” have been devised, including those based on rhodium,54 cobalt,55 iridium,56 ruthenium,57 palladium,58 copper,59,60 nickel,61 and indium.62,63 These protocols invariably employ metallic terminal reductants, such as stannanes, silanes, and organozinc reagents, which mandates the generation of stoichiometric byproducts. Inspired by the prospect of developing completely byproduct-free processes, catalytic reductive aldol additions employing elemental hydrogen as the terminal reductant were investigated.64
Our initial efforts centered on developing intramolecular reductive aldol couplings of tethered enone-aldehydes under hydrogenative conditions (Scheme 12).64a It was found that upon exposure to catalytic quantities of phosphine-modified cationic rhodium complexes under ambient pressures of hydrogen, a range of enone-aldehydes engage in highly diastereoselective cyclization to deliver five- and six-membered-ring products. In a similar fashion, enone-ketones cyclize under nearly identical conditions to furnish syn-aldol adducts as single diastereomers.64b However, in these cases, the diminished electrophilicity of the ketone leads to substantial quantities of simple enone reduction product. Extension of this method to enone-diketone substrates provides a powerful desymmetrization strategy for the stereocontrolled generation of bicyclic frameworks bearing three contiguous stereocenters. The addition of aldehyde enolates to ketones, for which a single stoichiometric variant is known,65 represents a highly challenging class of aldol addition. Under hydrogenative conditions, enal-ketones cyclize with a high degree of efficiency to provide products of aldehyde enolate-ketone addition, although competitive 1,4 reduction is also observed. Specifically, cationic rhodium complexes, in conjunction with catalytic quantities of base, are essential in suppressing conventional reduction of the starting material and enabling the desired coupling (Scheme 13).64c
Scheme 12.

Reductive Aldol Cyclization via Catalytic Hydrogenation. (Ref. 64a,b)
Scheme 13.
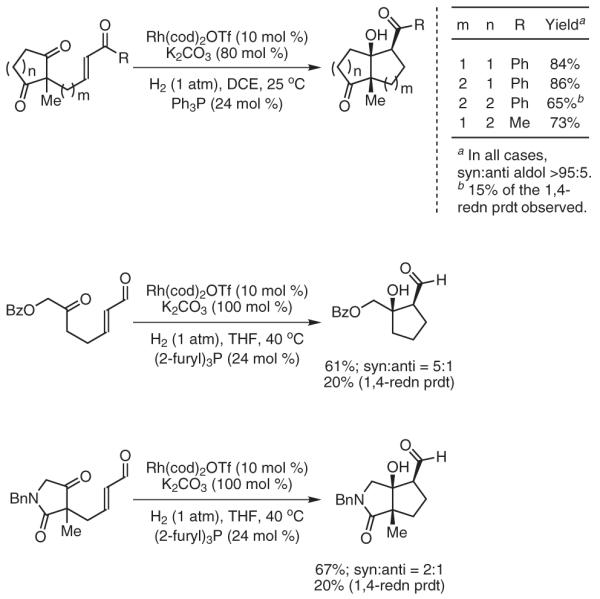
Reductive Aldol Cyclization via Catalytic Hydrogenation. (Ref. 64b,c)
Intermolecular hydrogenative aldol couplings also are possible. Under an atmosphere of hydrogen, cationic rhodium complexes catalyze the coupling of vinyl ketones to diverse aldehydes.64a Whereas the catalyst derived from Rh(cod)2OTf and triphenylphosphine provided aldol adducts as diastereomeric mixtures, high syn-diastereoselectivity may be realized using tri(2-furyl)phosphine as ligand.64e,66 Under these modified conditions, a wide range of aldehydes couple to methyl or ethyl vinyl ketone with exceptional levels of syn-diastereoselectivity. Of note is the wide range of potentially “hydrogen-labile” functionality tolerated, enabling the use of substrates containing alkynes, alkenes, benzylic ethers, nitroarenes, and aryl bromides. Furthermore, functionalized enones also are tolerated, as demonstrated by the employment of crotyl vinyl ketone.64f Remarkably, the mild reaction conditions permit aldol coupling of configurationally sensitive N-Boc-α-aminoaldehydes without racemization. Here, high levels of anti-Felkin-Anh control are achieved by taking advantage of hydrogen-bonded chelates, which arise in reaction media with low dielectric constants (Scheme 14).64g
Scheme 14.

syn-Diastereoselective Hydrogen-Mediated Aldol Coupling Employing Cationic Rhodium Catalysts Ligated By Tri(2-furyl)phosphine. (Ref. 64e-g)
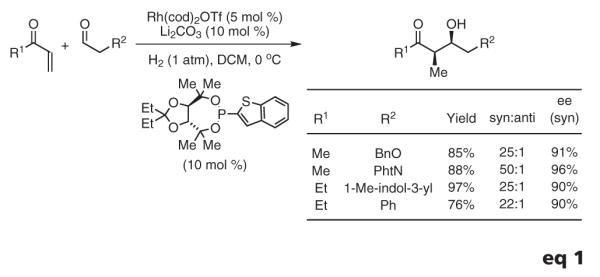
The ability to access syn-aldol adducts relevant to polyketide synthesis inspired further efforts toward enantioselective variants. π-Acidic monodentate phosphine ligands are required to enforce high levels of diastereoselectivity and catalytic turnover. However, commercially available phosphines of this type (e.g., phosphoramidites and BINOL-derived phosphites) give rise to inactive rhodium complexes, suggesting a very narrow window in terms of ligand π acidity. Consequently, the design of an effective, chiral, monodentate, phosphorus-based ligand was undertaken. The versatility of TADDOL-like phosphonites enabled the determination of key structure-selectivity trends, ultimately leading to the design of an effective ligand. Thus, by simply exposing methyl or ethyl vinyl ketone to aldehydes under an atmosphere of gaseous hydrogen in the presence of the rhodium phosphonite complex, aldol addition occurs with high levels of relative and absolute stereocontrol. This method generates optically enriched polyketide substructures and circumvents the stoichiometric generation of byproducts (eq 1).64h
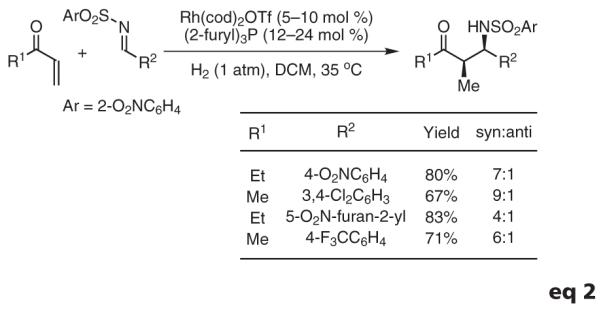
Based on the preceding results, reductive Mannich couplings of vinyl ketones were explored.67 Previously, reductive Mannich couplings had been accomplished using silane,68 the Hantzsch ester,69 or diethylzinc70 as the terminal reductant. Under hydrogenative conditions employing a tri(2-furyl)phosphine-ligated rhodium catalyst, vinyl ketones couple to N-(o-nitrophenyl)sulfonyl aldimines to furnish the desired Mannich addition products with good levels of syn-diastereoselectivity.67 These preliminary studies suggest the feasibility of developing asymmetric variants of this transformation (eq 2).
5. Future Directions
The stereoselective vinylation, allylation, and enolate addition of carbonyl compounds rank among the most broadly utilized methods in organic synthesis. Traditional protocols have relied upon the use of organometallic reagents, which are often basic, moisture sensitive, and give rise to stoichiometric quantities of metallic byproducts. Inspired by alkene hydroformylation and the parent Fischer-Tropsch reaction, hydrogenative variants of these classical carbonyl addition processes are aimed at meeting the environmental, economic, and health and safety ideals set by Green Chemistry. For the hydrogenative protocols, carbonyl and imine addition occurs under essentially neutral conditions simply upon exposure of an unsaturate-electrophile pair to gaseous hydrogen in the presence of a metal catalyst. Accordingly, vinylation, allylation, and enolate addition are achieved without stoichiometric byproduct generation and with stereoselectivities often surpassing traditional methods. The discovery of related transfer-hydrogenative couplings not only evades the stoichiometric generation of metallic byproducts, but also the requirement for substrate oxidation level adjustment. The ability to perform carbonyl addition from either the aldehyde or alcohol oxidation level has broad implications for the field of organic synthesis. These nascent reactivity modes should serve as the basis for innumerable byproduct-free alcohol-unsaturate and amine-unsaturate coupling processes.
 |
 |
6. Acknowledgments
Acknowledgment is made to the Robert A. Welch Foundation, Johnson & Johnson, Eli Lilly, Merck, the NIH-NIGMS (RO1-GM69445), and the ACS-GCI, for partial support of the research described in this account. Dr. Oliver Briel of Umicore is thanked for the generous donation of rhodium and iridium salts.
Biographies

Ryan L. Patman was born in 1982 in Elk City, Oklahoma. In 2006, he received a B.S. degree in chemistry from Oklahoma State University, where he conducted undergraduate research under the supervision of Professor Richard A. Bunce. He is currently a doctoral candidate in the research group of Professor Michael J. Krische at The University of Texas at Austin.

John F. Bower was born in 1980 in Chester, England. In 2003, he obtained an M.Sci. degree in chemistry from the University of Bristol, U.K., where he conducted research under the supervision of Professor Guy C. Lloyd-Jones. He continued doctoral studies at Bristol under the supervision of Professor Timothy Gallagher and, in 2007, received his Ph.D. degree. In May 2007, he joined the research group of Professor Michael J. Krische at The University of Texas at Austin as a postdoctoral research associate.

In Su Kim was born in 1975 in Gapyeong, Republic of Korea. In 2001, he received a B.S. degree from the College of Pharmacy, Sungkyunkwan University, Republic of Korea. He obtained an M.S. degree in 2003 and a Ph.D. degree in 2006, working under the guidance of Professor Young Hoon Jung. In September 2007, he joined the group of Professor Michael J. Krische at the University of Texas at Austin as a postdoctoral fellow of the Korea Research Foundation (KRF).

Michael J. Krische obtained a B.S. degree in chemistry from the University of California at Berkeley, where he performed research under the guidance of Professor Henry Rapoport as a President’s Undergraduate Fellow. After one year of study abroad as a Fulbright Fellow, he initiated graduate research at Stanford University under the mentorship of Professor Barry Trost as a Veatch Graduate Fellow. Following receipt of his Ph.D. degree, he worked with Jean-Marie Lehn at the Université Louis Pasteur as an NIH Post-Doctoral Fellow. In the fall of 1999, he was appointed Assistant Professor at the University of Texas at Austin. He was promoted directly to Full Professor in 2004 and in 2007 he received the Robert A. Welch Chair in Science. Professor Krische’s research program is focused on the development of C-C-bond-forming hydrogenations and transfer hydrogenations. Research from his laboratory demonstrates that hydrogenation and transfer hydrogenation may be used to couple diverse π-unsaturated reactants to carbonyl compounds, imines, and even alcohols offering a byproduct-free alternative to stoichiometrically preformed organometallics in a range of classical C=X (X = O, NR) addition processes. These studies represent the first systematic efforts to exploit hydrogenation in C-C couplings beyond hydroformylation, and define a departure from the use of preformed organometallic reagents in carbonyl and imine additions. His research accomplishments led to the receipt of numerous awards and honors: Novartis Chemistry Lectureship (2008), Presidential Green Chemistry Award (2007), Dowpharma Prize (2007), ACS Elias J. Corey Award (2007), Solvias Ligand Prize (2006), Society of Synthetic Chemistry Japan Lectureship (2005), Johnson & Johnson Focused Giving Award (2005), Dreyfus Teacher Scholar Award (2003), Alfred P. Sloan Research Fellowship (2003), Cottrell Scholar Award (2002), Frasch Foundation Award in Chemistry (2002), Lilly Grantee Award (2002), National Science Foundation-CAREER Award (2000), Maître de Conference, Collége de France (1999), NIH Post-Doctoral Fellow (1997), Veatch Graduate Fellow (1995), Sigma Xi Grantee (1991), and Fulbright Fellow (1990).
7. References
- (1).For reviews, see: Trost BM. Science. 1991;254:1471. doi: 10.1126/science.1962206. Trost BM. Angew. Chem., Int. Ed. Engl. 1995;34:259.
- (2).(a) Wender PA, Miller BL. Org. Synth. Theor. Appl. 1993;2:27. [Google Scholar]; (b) Wender PA, Handy S, Wright DL. Chem. Ind. 1997:767. [Google Scholar]
- (3).(a) Sheldon RA. Chem. Ind. 1997:12. [Google Scholar]; (b) Sheldon RA. Green Chem. 2007;9:1273. [Google Scholar]
- (4).Nobel Foundation . Nobel Lectures, Chemistry, 1901–1921. Elsevier; Amsterdam: 1966. [Google Scholar]
- (5).Smil V. Enriching the Earth: Fritz Haber, Carl Bosch, and the Transformation of World Food Production. MIT Press; Cambridge, MA: 2004. [Google Scholar]
- (6).(a) Thommen M. Spec. Chem. Mag. 2005;25:26. [Google Scholar]; (b) Thayer AM. Chem. Eng. News. 2005;83(36):40. [Google Scholar]; (c) Jäkel C, Paciello R. Chem. Rev. 2006;106:2912. doi: 10.1021/cr040675a. [DOI] [PubMed] [Google Scholar]
- (7).(a) Fischer F, Tropsch H. Brennstoff-Chem. 1923;4:276. Chem. Abstr. 1923, ...., ....... [Google Scholar]; (b) Fischer F, Tropsch H. Chem. Ber. 1923;56B:2428. [Google Scholar]
- (8).Roelen O. Chemische Verwertungsgesellschaft GmbH, Oberhausen. Ger. Patent DE 849,548, 1938; Chem. Abstr. 1944;38:5501.
- (9).(a) Frohning CD, Kohlpaintner CW. In: Applied Homogeneous Catalysis with Organometallic Compounds. Cornils B, Herrmann WA, editors. Vol. 1. Wiley-Vch; Weinheim: 1996. pp. 29–104. [Google Scholar]; (b) Van Leeuwen PWNM. Homogeneous Catalysis: Understanding the Art. Kluwer; Dordrecht: 2004. [Google Scholar]
- (10).Prior to our systematic studies, only two isolated reports of hydrogenative C–C coupling had appeared in the literature: Molander GA, Hoberg JO. J. Am. Chem. Soc. 1992;114:3123. Kokubo K, Miura M, Nomura M. Organometallics. 1995;14:4521.
- (11).On rare occasions, side products of reductive C–C-bond formation have been observed in catalytic hydrogenations: Moyes RB, Walker DW, Wells PB, Whan DA, Irvine EA. In: Catalysis and Surface Characterisation (Special Publication) Dines TJ, Rochester CH, Thomson J, editors. Vol. 114. Royal Society of Chemistry; City, U.K.: 1992. pp. 207–212. Bianchini C, Meli A, Peruzzini M, Vizza F, Zanobini F, Frediani P. Organometallics. 1989;8:2080.
- (12).For recent reviews on hydrogen-mediated C–C couplings, see: Ngai M-Y, Krische MJ. Chim. Oggi/Chemistry Today. 2006;24(4) (Chiral technologies supplement), 12. Iida H, Krische MJ. Top. Curr. Chem. 2007;279:77. Ngai M-Y, Kong J-R, Krische MJ. J. Org. Chem. 2007;72:1063. doi: 10.1021/jo061895m. Skucas E, Ngai M-Y, Komanduri V, Krische MJ. Acc. Chem. Res. 2007;40:1394. doi: 10.1021/ar7001123.
- (13).For reviews encompassing the synthesis of allylic alcohols, see: Seidel A, editor. Kirk-Othmer’s Encyclopedia of Chemical Technology. 5th ed. Vol. 2. Wiley; Hoboken, NJ: 2004. pp. 234–249. Banerjee AK, Poon PS, Laya MS, Vera WJ. Russ. Chem. Rev. 2004;73:621.
- (14).For reviews encompassing the synthesis of allylic amines, see: Cheikh RB, Chaabouni R, Laurent A, Mison P, Nafti A. Synthesis. 1983:685. Laurent A, Mison P, Nafti A, Cheikh RB, Chaabouni RJ. Chem. Res. 1984:354. Johannsen M, Jørgensen KA. Chem. Rev. 1998;98:1689. doi: 10.1021/cr970343o.
- (15).For reviews on the metal-catalyzed allylic amination and alkoxylation, see: Acemoglu L, Williams JMJ. In: Handbook of Organopalladium Chemistry for Organic Synthesis. Negishi E, de Meijere A, editors. Vol. 2. Wiley; New York: 2002. pp. 1689–1705. Trost BM, Crawley ML. Chem. Rev. 2003;103:2921. doi: 10.1021/cr020027w. Trost BM. J. Org. Chem. 2004;69:5813. doi: 10.1021/jo0491004. Miyabe H, Takemoto Y. Synlett. 2005:1641. Takeuchi R, Kezuka S. Synthesis. 2006:3349.
- (16).For enantioselective catalytic additions of vinylzinc reagents to aldehydes, see: Oppolzer W, Radinov RN. Helv. Chim. Acta. 1992;75:170. Oppolzer W, Radinov RN. J. Am. Chem. Soc. 1993;115:1593. Soai K, Takahashi K. Chem. Soc., Perkin Trans. 1. 1994:1257. Wipf P, Xu W. Tetrahedron Lett. 1994;35:5197. Oppolzer W, Radinov RN, De Brabander J. Tetrahedron Lett. 1995;36:2607. Wipf P, Ribe S. J. Org. Chem. 1998;63:6454. Oppolzer W, Radinov RN, El-Sayed E. J. Org. Chem. 2001;66:4766. doi: 10.1021/jo000463n. Dahmen S, Bräse S. Org. Lett. 2001;3:4119. doi: 10.1021/ol016954r. Chen YK, Lurain AE, Walsh PJ. J. Am. Chem. Soc. 2002;124:12225. doi: 10.1021/ja027271p. Ji J-X, Qiu L-Q, Yip CW, Chan ASC. J. Org. Chem. 2003;68:1589. doi: 10.1021/jo026551k. Lurain AE, Walsh PJ. J. Am. Chem. Soc. 2003;125:10677. doi: 10.1021/ja035213d. Ko D-H, Kang S-W, Kim KH, Chung Y, Ha D-C. Bull. Korean. Chem. Soc. 2004;25:35. Sprout CM, Richmond ML, Seto CT. J. Org. Chem. 2004;69:6666. doi: 10.1021/jo049160+. Jeon S-J, Chen YK, Walsh PJ. Org. Lett. 2005;7:1729. doi: 10.1021/ol050255n. Lauterwasser F, Gall J, Höfener S, Bräse S. Adv. Synth. Catal. 2006;348:2068. Jeon S-J, Fisher EL, Carroll PJ, Walsh PJ. J. Am. Chem. Soc. 2006;128:9618. doi: 10.1021/ja061973n. Salvi L, Jeon S-J, Fisher EL, Carroll PJ, Walsh PJ. J. Am. Chem. Soc. 2007;129:16119. doi: 10.1021/ja0762285. Wu H-L, Wu P-Y, Uang B-J. J. Org. Chem. 2007;72:5935. doi: 10.1021/jo0709403.
- (17).For reviews encompassing catalytic enantioselective aldehyde vinylations using organozinc reagents, see: Wipf P, Kendall C. Chem.—Eur. J. 2002;8:1778. doi: 10.1002/1521-3765(20020415)8:8<1778::aid-chem1778>3.0.co;2-h. Wipf P, Nunes RL. Tetrahedron. 2004;60:1269.
- (18).For catalytic enantioselective ketone vinylation using organozinc reagents, see: Li H, Walsh PJ. J. Am. Chem. Soc. 2004;126:6538. doi: 10.1021/ja049206g. Li H, Walsh PJ. J. Am. Chem. Soc. 2005;127:8355. doi: 10.1021/ja0425740. Jeon S-J, Li H, García C, LaRochelle LK, Walsh PJ. J. Org. Chem. 2005;70:448. doi: 10.1021/jo048683e.
- (19).Schmidt F, Rudolph J, Bolm C. Synthesis. 2006:3625. [Google Scholar]
- (20).The catalyzed addition of vinylzirconocenes to imines is known, but enantioselective variants have not been developed: Kakuuchi A, Taguchi T, Hanzawa Y. Tetrahedron Lett. 2003;44:923. Wipf P, Kendall C, Stephenson CRJ. J. Am. Chem. Soc. 2003;125:761. doi: 10.1021/ja028092a.
- (21).The enantioselective Ni-catalyzed alkyne, imine, and triethylborane three-component coupling has been reported, but modest selectivities (51–89% ee’s) are observed. In this method, vinylation is accompanied by ethyl transfer: Patel SJ, Jamison TF. Angew. Chem., Int. Ed. 2004;432:3941. doi: 10.1002/anie.200460044.
- (22).Ojima I, Tzamarioudaki M, Tsai C-Y. J. Am. Chem. Soc. 1994;116:3643. [Google Scholar]
- (23).(a) Crowe WE, Rachita MJ. J. Am. Chem. Soc. 1995;117:6787. [Google Scholar]; Kablaoui NM, Buchwald SL. J. Am. Chem. Soc. 1995;117:6785. [Google Scholar]
- (24).(a) Oblinger E, Montgomery J. J. Am. Chem. Soc. 1997;119:9065. [Google Scholar]; (b) Tang X-Q, Montgomery J. J. Am. Chem. Soc. 1999;121:6098. [Google Scholar]; (c) Tang X-Q, Montgomery J. J. Am. Chem. Soc. 2000;122:6950. [Google Scholar]; (d) Mahandru GM, Liu G, Montgomery J. J. Am. Chem. Soc. 2004;126:3698. doi: 10.1021/ja049644n. [DOI] [PubMed] [Google Scholar]; (e) Knapp-Reed B, Mahandru GM, Montgomery J. J. Am. Chem. Soc. 2005;127:13156. doi: 10.1021/ja054590i. [DOI] [PubMed] [Google Scholar]
- (25).(a) Huang W-S, Chan J, Jamison TF. Org. Lett. 2000;2:4221. doi: 10.1021/ol006781q. [DOI] [PubMed] [Google Scholar]; (b) Miller KM, Huang W-S, Jamison TF. J. Am. Chem. Soc. 2003;125:3442. doi: 10.1021/ja034366y. [DOI] [PubMed] [Google Scholar]; (c) Miller KM, Jamison TF. Org. Lett. 2005;7:3077. doi: 10.1021/ol051075g. [DOI] [PubMed] [Google Scholar]
- (26).Takai K, Sakamoto S, Isshiki T. Org. Lett. 2003;5:653. doi: 10.1021/ol0272996. [DOI] [PubMed] [Google Scholar]
- (27).(a) Kong J-R, Ngai M-Y, Krische MJ. J. Am. Chem. Soc. 2006;128:718. doi: 10.1021/ja056474l. [DOI] [PubMed] [Google Scholar]; (b) Cho C-W, Krische M. J. Org. Lett. 2006;8:3873. doi: 10.1021/ol061485k. [DOI] [PubMed] [Google Scholar]; (c) Hong Y-T, Cho C-W, Skucas E, Krische M. J. Org. Lett. 2007;9:3745. doi: 10.1021/ol7015548. [DOI] [PubMed] [Google Scholar]; (d) Komanduri V, Krische MJ. J. Am. Chem. Soc. 2006;128:16448. doi: 10.1021/ja0673027. [DOI] [PubMed] [Google Scholar]
- (28).Kong J-R, Cho C-W, Krische MJ. J. Am. Chem. Soc. 2005;127:11269. doi: 10.1021/ja051104i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Kong J-R, Krische MJ. J. Am. Chem. Soc. 2006;128:16040. doi: 10.1021/ja0664786. [DOI] [PubMed] [Google Scholar]
- (30).Skucas E, Kong J-R, Krische MJ. J. Am. Chem. Soc. 2007;129:7242. doi: 10.1021/ja0715896. [DOI] [PubMed] [Google Scholar]
- (31).Seidel A, editor. Kirk-Othmer's Encyclopedia of Chemical Technology. 5th ed. Vol. 1. Wiley; Hoboken, NJ: 2004. pp. 216–217. [Google Scholar]
- (32).(a) Barchuk A, Ngai M-Y, Krische MJ. J. Am. Chem. Soc. 2007;129:8432. doi: 10.1021/ja073018j. [DOI] [PubMed] [Google Scholar]; (b) Ngai M-Y, Barchuk A, Krische MJ. J. Am. Chem. Soc. 2007;129:12644. doi: 10.1021/ja075438e. [DOI] [PubMed] [Google Scholar]
- (33).Rhee J-U, Krische MJ. J. Am. Chem. Soc. 2006;128:10674. doi: 10.1021/ja0637954. [DOI] [PubMed] [Google Scholar]
- (34).For reviews on enantioselective carbonyl allylations, see: Ramachandran PV. Aldrichimica Acta. 2002;35:23. Kennedy JWJ, Hall DG. Angew. Chem., Int. Ed. 2003;42:4732. doi: 10.1002/anie.200301632. Denmark SE, Fu J. Chem. Rev. 2003;103:2763. doi: 10.1021/cr020050h. Yu C-M, Youn J, Jung H-K. Bull. Korean Chem. Soc. 2006;27:463. Marek I, Sklute G. Chem. Commun. 2007:1683. doi: 10.1039/b615042j. Hall DG. Synlett. 2007:1644.
- (35).Chirally modified allyl metal reagents: Herold T, Hoffmann RW. Angew. Chem., Int. Ed. Engl. 1978;17:768. Hoffmann RW, Herold T. Chem. Ber. 1981;114:375. Hayashi T, Konishi M, Kumada M. J. Am. Chem. Soc. 1982;104:4963. Brown HC, Jadhav PK. J. Am. Chem. Soc. 1983;105:2092. Roush WR, Walts AE, Hoong LK. J. Am. Chem. Soc. 1985;107:8186. Reetz M. Pure Appl. Chem. 1988;60:1607. Short RP, Masamune S. J. Am. Chem. Soc. 1989;111:1892. Corey EJ, Yu C-M, Kim SS. J. Am. Chem. Soc. 1989;111:5495. Seebach D, Beck AK, Imwinkeiried R, Roggo S, Wonnacott A. Helv. Chim. Acta. 1987;70:954. Riediker M, Duthaler RO. Angew. Chem., Int. Ed. Engl. 1989;28:494. Panek JS, Yang M. J. Am. Chem. Soc. 1991;113:6594. Kinnaird JWA, Ng PY, Kubota K, Wang X, Leighton JL. J. Am. Chem. Soc. 2002;124:7920. doi: 10.1021/ja0264908.
- (36).Catalytic asymmetric carbonyl allylation: Costa AL, Piazza MG, Tagliavini E, Trombini C, Umani-Ronchi A. J. Am. Chem Soc. 1993;115:7001. Keck GE, Tarbet KH, Geraci LS. J. Am. Chem. Soc. 1993;115:8467. Denmark SE, Coe DM, Pratt NE, Griedel BD. J. Org. Chem. 1994;59:6161. doi: 10.1021/jo052202p. Denmark SE, Fu J. J. Am. Chem. Soc. 2001;123:9488. doi: 10.1021/ja016552e.
- (37).For selected examples of reactions involving nucleophilic π-allyls, see: Tabuchi T, Inanaga J, Yamaguchi M. Tetrahedron Lett. 1986;27:1195. Palladium. Takahara JP, Masuyama Y, Kurusu Y. J. Am. Chem. Soc. 1992;114:2577. Kimura M, Ogawa Y, Shimizu M, Sueishi M, Tanaka S, Tamaru Y. Tetrahedron Lett. 1998;39:6903. Kimura M, Shimizu M, Shibata K, Tazoe M, Tamaru Y. Angew. Chem., Int. Ed. 2003;42:3392. doi: 10.1002/anie.200351182. Zanoni G, Gladiali S, Marchetti A, Piccinini P, Tredici I, Vidari G. Angew. Chem., Int. Ed. 2004;43:846. doi: 10.1002/anie.200352743. Rhodium. Masuyama Y, Kaneko Y, Kurusu Y. Tetrahedron Lett. 2004;45:8969. Ruthenium. Tsuji Y, Mukai T, Kondo T, Watanabe Y. J. Organomet. Chem. 1989;369:C51. Kondo T, Ono H, Satake N, Mitsudo T.-a., Watanabe Y. Organometallics. 1995;14:1945.
- (38).For selected reviews covering carbonyl allylation via umpolung of π-allyls, see: Tamaru Y. In: Handbook of Organopalladium Chemistry for Organic Synthesis. Negishi E, de Meijere A, editors. Vol. 2. Wiley; New York: 2002. pp. 1917–1943. Tamaru Y. In: Perspectives in Organopalladium Chemistry for the XXI Century. Tsuji J, editor. Elsevier; Amsterdam: 1999. pp. 215–231. Kondo T, Mitsudo T-A. Curr. Org. Chem. 2002;6:1163.
- (39).(a) Skucas E, Bower JF, Krische MJ. J. Am. Chem. Soc. 2007;129:12678. doi: 10.1021/ja075971u. [DOI] [PubMed] [Google Scholar]; (b) Bower JF, Skucas E, Patman RL, Krische MJ. J. Am. Chem. Soc. 2007;129:15134. doi: 10.1021/ja077389b. [DOI] [PubMed] [Google Scholar]
- (40).The alcohol-unsaturate couplings developed in our laboratory provide products of carbonyl addition. To date, all other reported hydrogen auto-transfer processes provide products of oxidation–condensation–reduction, resulting in formal substitution of the alcohol. For recent reviews, see: Guillena G, Ramón DJ, Yus M. Angew. Chem., Int. Ed. 2007;46:2358. doi: 10.1002/anie.200603794. Hamid MHSA, Slatford PA, Williams JM. J. Adv. Synth. Catal. 2007;349:1555.
- (41).Bower JF, Patman RL, Krische M. J. Org. Lett. 2008;10:1033. doi: 10.1021/ol800159w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Shibahara F, Bower JF, Krische MJ. J. Am. Chem. Soc. 2008;130:6338. doi: 10.1021/ja801213x. Allenes also couple to carbonyl electrophiles under ruthenium-catalyzed transfer-hydrogenative conditions: Ngai M-Y, Skucas E, Krische MJ. Org. Lett. 2008;10 doi: 10.1021/ol800836v. in press.
- (43).Patman RL, Williams VM, Bower JF, Krische M. J. Angew. Chem., Int. Ed. 2008;47 doi: 10.1002/anie.200801359. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).For reviews that encompass carbonyl propargylation employing allenyl metal reagents, see: Moreau J-L. In: The Chemistry of Ketenes, Allenes and Related Compounds. Patai S, editor. Wiley; New York: 1980. pp. 363–413. Marshall JA. Chem. Rev. 1996;96:31. doi: 10.1021/cr950037f. Gung BW. Org. React. 2004;64:1. Marshall JA, Gung BW, Grachan ML. In: Modern Allene Chemistry. Krause N, Hashmi ASK, editors. Wiley-VCH; Weinheim, Germany: 2004. pp. 493–592. Marshall JA. J. Org. Chem. 2007;72:8153. doi: 10.1021/jo070787c.
- (45).For selected milestones in carbonyl propargylation, see: Prévost C, Gaudemar M, Honigberg J. Compt. Rend. 1950;230:1186. Wotiz JH. J. Am. Chem. Soc. 1950;72:1639. Karila M, Capmau ML, Chodkiewicz W. Compt. Rend. 1969;269:342. Lequan M, Guillerm G. J. Organomet. Chem. 1973;54:153. Mukaiyama T, Harada T. Chem. Lett. 1981:621. Favre E, Gaudemar M. Compt. Rend. 1966;263:1543. Danheiser RL, Carini DJ. J. Org. Chem. 1980;45:3925. Haruta R, Ishiguro M, Ikeda N, Yamamoto H. J. Am. Chem. Soc. 1982;104:7667. Corey EJ, Yu C-M, Lee D-H. J. Am. Chem. Soc. 1990;112:878. Minowa N, Mukaiyama T. Bull. Chem. Soc. Jpn. 1987;60:3697. Marshall JA, Wang X-J. J. Org. Chem. 1991;56:3211. Marshall JA, Maxson K. J. Org. Chem. 2000;65:630. doi: 10.1021/jo991543y. Matsumoto Y, Naito M, Uozumi Y, Hayashi T. J. Chem. Soc., Chem. Commun. 1993:1468. Keck GE, Krishnamurthy D, Chen X. Tetrahedron Lett. 1994;35:8323. Denmark SE, Wynn T. J. Am. Chem. Soc. 2001;123:6199. doi: 10.1021/ja016017e.
- (46).(a) Kim IS, Ngai M-Y, Krische MJ. J. Am. Chem. Soc. 2008;130:6340. doi: 10.1021/ja802001b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kim IS, Ngai M-Y, Krische MJ. University of Texas; Austin, TX: Unpublished work, 200X. [Google Scholar]
- (47).Though largely attributed to Würtz, the aldol reaction was reported first by Borodin: Von Richter V. Ber. Deut. Chem. Ges. 1869;2:552. Borodin's earliest results are cited in this article. Würtz A. Bull. Soc. Chim. Fr. 1872;17:436. Borodin A. Ber. Dtsch. Chem. Ges. 1873;6:982. Kane R. Ann. Phys. Chem., Ser. 2. 1838;44:475.
- (48).For selected reviews on stereoselective aldol additions, see: Heathcock CH. Science. 1981;214:395. doi: 10.1126/science.214.4519.395. Heathcock CH. ACS Symp. Ser. 1982;185:55–72. Evans DA, Nelson JV, Taber TR. Top. Stereochem. 1982;13:1. Machajewski TD, Wong C-H. Angew. Chem., Int. Ed. 2000;39:1352. doi: 10.1002/(sici)1521-3773(20000417)39:8<1352::aid-anie1352>3.0.co;2-j. Palomo C, Oiarbide M, García JM. Chem. Soc. Rev. 2004;33:65. doi: 10.1039/b202901d.
- (49).For a recent review on the use of metallic catalysts for direct enantioselective aldol additions, see: Shibasaki M, Matsunaga S, Kumagai N. In: Modern Aldol Reactions. Mahrwald R, editor. Vol. 2. Wiley-VCH; Weinheim: 2004. pp. 197–227.
- (50).For recent reviews on the use of organic catalysts for direct enantioselective aldol addition, see: List B. In: Modern Aldol Reactions. Mahrwald R, editor. Vol. 1. Wiley-VCH; Weinheim: 2004. pp. 161–200. Notz W, Tanaka F, Barbas C., III Acc. Chem. Res. 2004;37:580–591. doi: 10.1021/ar0300468.
- (51).A notable exception involves the direct asymmetric catalytic aldol additions to deliver glycolate aldol adducts. For example, see: Notz W, List B. J. Am. Chem. Soc. 2000;122:7386. Yoshikawa N, Kumagai N, Matsunaga S, Moll G, Ohshima T, Suzuki T, Shibasaki M. J. Am. Chem. Soc. 2001;123:2466. doi: 10.1021/ja015580u. Trost BM, Ito H, Silcoff ER. J. Am. Chem. Soc. 2001;123:3367. doi: 10.1021/ja003871h.
- (52).(a) Stork G, Rosen P, Goldman NL. J. Am. Chem. Soc. 1961;83:2965. [Google Scholar]; (b) Stork G, Rosen P, Goldman N, Coombs RV, Tsuji J. J. Am. Chem. Soc. 1965;87:275. [Google Scholar]
- (53).For recent reviews on the reductive aldol reaction, see: Nishiyama H, Shiomi T. Top. Curr. Chem. 2007;279:105. Garner SA, Han SB, Krische MJ. In: Modern Reductions. Andersson P, Munslow I, editors. Wiley-VCH; Weinheim: 2008. pp. 387–408.
- (54).For rhodium-catalyzed reductive aldol reactions mediated by silane, see: Revis A, Hilty TK. Tetrahedron Lett. 1987;28:4809. Matsuda I, Takahashi K, Sato S. Tetrahedron Lett. 1990;31:5331. Taylor SJ, Morken JP. J. Am. Chem. Soc. 1999;121:12202. Taylor SJ, Duffey MO, Morken JP. J. Am. Chem. Soc. 2000;122:4528. Zhao C-X, Bass J, Morken JP. Org Lett. 2001;3:2839. doi: 10.1021/ol016279l. Emiabata-Smith D, McKillop A, Mills C, Motherwell WB, Whitehead AJ. Synlett. 2001:1302. Freiría M, Whitehead AJ, Tocher DA, Motherwell WB. Tetrahedron. 2004;60:2673. Nishiyama H, Shiomi T, Tsuchiya Y, Matsuda I. J. Am. Chem. Soc. 2005;127:6972. doi: 10.1021/ja050698m. Willis MC, Woodward RL. J. Am. Chem. Soc. 2005;127:18012. doi: 10.1021/ja056130v. Fuller NO, Morken JP. Synlett. 2005:1459. Ito J-I, Shiomi T, Nishiyama H. Adv. Synth. Catal. 2006;348:1235. Shiomi T, Ito J-I, Yamamoto Y, Nishiyama H. Eur. J. Org. Chem. 2006:5594. Shiomi T, Nishiyama H. Org Lett. 2007;9:1651. doi: 10.1021/ol070251d.
- (55).For cobalt-catalyzed reductive aldol reactions, see: Isayama S, Mukaiyama T. Chem. Lett. 1989:2005. Baik TG, Luis A. l., Wang LC, Krische MJ. J. Am. Chem. Soc. 2001;123:5112. doi: 10.1021/ja0040971. Wang LC, Jang H-Y, Roh Y, Lynch V, Schultz AJ, Wang X, Krische MJ. J. Am. Chem. Soc. 2002;124:9448. doi: 10.1021/ja020223k. Lam HW, Joensuu PM, Murray GJ, Fordyce EAF, Prieto O, Luebbers T. Org. Lett. 2006;8:3729. doi: 10.1021/ol061329d. Lumby RJR, Joensuu PM, Lam HW. Org. Lett. 2007;9:4367. doi: 10.1021/ol701980e.
- (56).For iridium-catalyzed reductive aldol reactions, see: Zhao CX, Duffey MO, Taylor SJ, Morken JP. Org. Lett. 2001;3:1829. doi: 10.1021/ol015859f.
- (57).For ruthenium-catalyzed reductive aldol reactions, see: Doi T, Fukuyama T, Minamino S, Ryu I. Synlett. 2006;18:3013.
- (58).Kiyooka SI, Shimizu A, Torii S. Tetrahedron Lett. 1998;39:5237. [Google Scholar]
- (59).For copper-promoted reductive aldol reactions, see: Chiu P, Chen B, Cheng KF. Tetrahedron Lett. 1998;39:9229. Chiu P. Synthesis. 2004:2210. Chung WK, Chiu P. Synlett. 2005;1:55. Chiu P, Leung SK. Chem. Commun. 2004:2308. doi: 10.1039/b407842j.
- (60).For copper-catalyzed reductive aldol reactions, see: Ooi T, Doda K, Sakai D, Maruoka K. Tetrahedron Lett. 1999;40:2133. Lam HW, Joensuu PMA. Org. Lett. 2005;7:4225. doi: 10.1021/ol051649h. Lam HW, Murray GJ, Firth JD. Org. Lett. 2005;7:5743. doi: 10.1021/ol052599j. Deschamp J, Chuzel O, Hannedouche J, Riant O. Angew. Chem., Int. Ed. 2006;45:1292. doi: 10.1002/anie.200503791. Chuzel O, Deschamp J, Chauster C, Riant O. Org. Lett. 2006;8:5943. doi: 10.1021/ol062398v. Zhao D, Oisaki K, Kanai M, Shibasaki M. Tetrahedron Lett. 2006;47:1403. Zhao D, Oisaki K, Kanai M, Shibasaki M. J. Am. Chem. Soc. 2006;128:14440. doi: 10.1021/ja0652565. Welle A, Díez-González S, Tinant B, Nolan SP, Riant O. Org. Lett. 2006;8:6059. doi: 10.1021/ol062495o.
- (61).For nickel-catalyzed reductive aldol reactions, see Chrovian CC, Montgomery J. Org Lett. 2007;9:537. doi: 10.1021/ol063028+.
- (62).For a reductive aldol coupling employing stoichiometric quantities of indium reagent, see Inoue K, Ishida T, Shibata I, Baba A. Adv. Synth. Catal. 2002;344:283.
- (63).For indium-catalyzed reductive aldol reactions, see: Shibata I, Kato H, Ishida T, Yasuda M, Baba A. Angew. Chem., Int. Ed. 2004;43:711. doi: 10.1002/anie.200352738. Miura K, Yamada Y, Tomita M, Hosomi A. Synlett. 2004:1985.
- (64).For rhodium-catalyzed reductive aldol reactions mediated by hydrogen, see: Jang H-Y, Huddleston RR, Krische MJ. J. Am. Chem. Soc. 2002;124:15156. doi: 10.1021/ja021163l. Huddleston RR, Krische MJ. Org. Lett. 2003;5:1143. doi: 10.1021/ol0300219. Koech PK, Krische MJ. Org. Lett. 2004;6:691. doi: 10.1021/ol030136c. Marriner GA, Garner SA, Jang H-Y, Krische MJ. J. Org. Chem. 2004;69:1380. doi: 10.1021/jo030310a. Jung C-K, Garner SA, Krische MJ. Org. Lett. 2006;8:519. doi: 10.1021/ol052859x. Han SB, Krische MJ. Org. Lett. 2006;8:5657. doi: 10.1021/ol0624023. Jung C-K, Krische MJ. J. Am. Chem. Soc. 2006;128:17051. doi: 10.1021/ja066198q. Bee C, Han SB, Hassan A, Iida H, Krische MJ. J. Am. Chem. Soc. 2008;130:2746. doi: 10.1021/ja710862u.
- (65).Yachi K, Shinokubo H, Oshima K. J. Am. Chem. Soc. 1999;121:9465. [Google Scholar]
- (66).For tri(2-furyl)phosphine and triphenylarsine effects in metal- catalyzed reactions, see: Farina V, Krishnan B. J. Am. Chem. Soc. 1991;113:9585. Farina V. Pure Appl. Chem. 1996;68:73. Anderson NG, Keay BA. Chem. Rev. 2001;101:997. doi: 10.1021/cr000024o.
- (67).Garner SA, Krische MJ. J. Org. Chem. 2007;72:5843. doi: 10.1021/jo070779w. [DOI] [PubMed] [Google Scholar]
- (68).For metal-catalyzed reductive Mannich couplings mediated by silane, see: Muraoka T, Kamiya S-I, Matsuda I, Itoh K. Chem. Commun. 2002:1284. doi: 10.1039/b202773a. Townes JA, Evans MA, Queffelec J, Taylor SJ, Morken JP. Org. Lett. 2002;4:2537. doi: 10.1021/ol020106u.
- (69).For secondary-amine-catalyzed reductive Mannich coupling of enal to imines mediated by Hantzsch ester, see Zhao G-L, Cordova A. Tetrahedron Lett. 2006;47:7417.
- (70).Prieto O, Lam HW. Org. Biomol. Chem. 2008;6:55. doi: 10.1039/b715839d. [DOI] [PubMed] [Google Scholar]