

TO THE EDITOR
The 22q11.2 deletion syndrome (22qDS), also known as DiGeorge or velocardiofacial syndrome, is a multi-system disorder caused by a chromosomal microdeletion most commonly involving a 3 Mb segment on the long arm of chromosome 22. The birth prevalence of 22qDS is estimated at approximately 1 in 4,000 to 1 in 6,000 in multiple population studies [Wilson et al., 1994; Botto et al., 2003; Oskarsdottir et al., 2004] and might be as high as 1 in 2,000 if ascertainment was complete [Shprintzen, 2005]. Although 22qDS is acknowledged as the most common autosomal deletion syndrome, the prevalence is only 1 in 8,000 later in childhood [Oskarsdottir et al., 2004] reflecting significant under-ascertainment, as well as attrition of more severe cases.
The hallmark features of the syndrome are characteristic facial appearance, velopharyngeal insufficiency, conotruncal heart disease, parathyroid and immune dysfunction, developmental delays and learning difficulties. Later onset complications, including recurrent seizures and schizophrenia, are common [Bassett et al., 2005] but little is known about possible neurodegenerative diseases and 22qDS.
Parkinson disease (PD) is a neurodegenerative disorder characterized by tremor, muscle rigidity and bradykinesia. Depression and, later, dementia are common. The lifetime risk for developing PD is 1–2% [Elbaz et al., 2002]. Onset is typically late in life, but onset before age 50 years occurs in about 4% of cases [Van Den Eeden et al., 2003]. In 22qDS, parkinsonian features have been previously described in a case report, possibly related to antipsychotic medication side effects [Krahn et al., 1998]. We report on two patients with both 22qDS and early onset (age <45 years) PD where antipsychotic medications did not complicate the diagnosis. We propose that this co-occurrence in two unrelated patients is unlikely to be coincidental and that PD represents another feature of 22qDS.
Patient 1: This 46-year-old man presented to the Medical Genetics Department in Marshfield, WI at age 42 years because of longstanding hypoparathyroidism combined with a history of learning disabilities, a mildly unusual facial appearance (long face with broad nasal root and bulbous nasal tip, see Fig. 1), and hypernasal speech. He had no history of congenital heart disease or immune deficiency. Chromosome analysis showed a normal male karyotype, but fluorescence in situ hybridization (FISH) confirmed a 22q11.2 deletion.
FIG. 1.
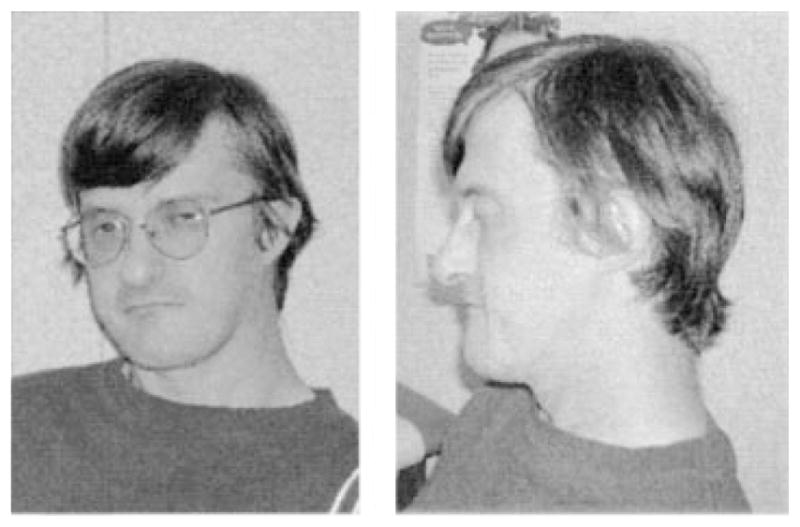
Patient 1 at age 42 years. Note the bulbous nasal tip. Small ear with attached lobule, flat midface and prognathism are evident on the lateral view.
The patient reported recent deterioration of his gait with right-sided rigidity and generalized bradykinesia. Neurological examination was notable for cogwheel rigidity, bradykinesia, and postural instability leading to a diagnosis of PD. Brain magnetic resonance imaging (MRI) showed only cavum septum pellucidum and non-specific areas of increased white matter signal intensity. He was started on Carbidopa-Levodopa with some initial improvement in his motor symptoms, although he continued disabled due to chronic pain. One year later he reported increasing motor difficulties, as well as dysphagia. During a period of serious physical and financial problems, he presented with a depressed and anxious mood, low energy level, and altered sleeping and eating habits and was started on an antidepressant (escitalopram). He continued to worsen, both physically and psychologically. His wife reported that he attempted suicide. His antidepressant was changed to mirtazapine. Eventually quetiapine, a second generation antipsychotic medication with little tendency for extrapyramidal side effects, resulted in some improvement. With successful treatment of his depression, as well as an increased dose of Carbidopa-Levodopa, his physical condition stabilized. There was no history of psychiatric problems prior to his presentation with PD.
Neuropsychological testing was performed at age 42 years following the diagnosis of PD and 22qDS. Testing of general intellectual skills utilizing the Wechsler Adult Intelligence Scale 3rd edition documented a borderline cognitive disability. He achieved a standard score of 65 on verbal comprehension measures, and perceptual organizational skills yielded a standard score of 74. Academic achievement was consistent with his cognitive ability.
The patient was one of six boys born to apparently healthy, nonconsanguineous parents of Italian ancestry. Two of his five brothers died neonatally, one due to suspected cardiac malformation. Review of family photographs showed facial features suggestive of 22q11.2 deletion in the patient’s father who died from colon cancer at 37 years. There was no known family history of PD, movement disorders, neurological disease, or psychiatric illness. Familial testing for the 22q11.2 deletion was offered but not pursued.
Patient 2: This 56-year-old man presented to the Clinical Genetics Research Program in Toronto at age 52 years with his son to participate in a study of adults with 22qDS. He had previously confirmed diagnoses of PD (onset at 43 years) established by a neurologist and 22qDS by FISH at age 44 following his son’s diagnosis with 22qDS. This case was presented in poster format at an American Society of Human Genetics meeting [Tam et al., 2004].
On physical examination, the patient presented with a bulbous and broad nasal tip (Fig. 2), slightly tapered fingers, mild scoliosis, and hypernasal speech. On cognitive assessment at age 52 years, full-scaled IQ was 94 (verbal IQ = 103, performance IQ = 81) using the Wechsler Adult Intelligence Scale, third edition. On Wechsler Memory Scale-III, delayed verbal memory and immediate and delayed visuospatial memory were within the normal range with immediate verbal memory somewhat worse. Wide Range Achievement Test reading and spelling were consistent with a university education, but arithmetic was at a grade 4 level, similar to other individuals with 22qDS [Chow et al., 2006]. Brain MRI at age 53 years showed nonspecific bright foci in hemispheric white matter and cavum velum interpositum.
FIG. 2.
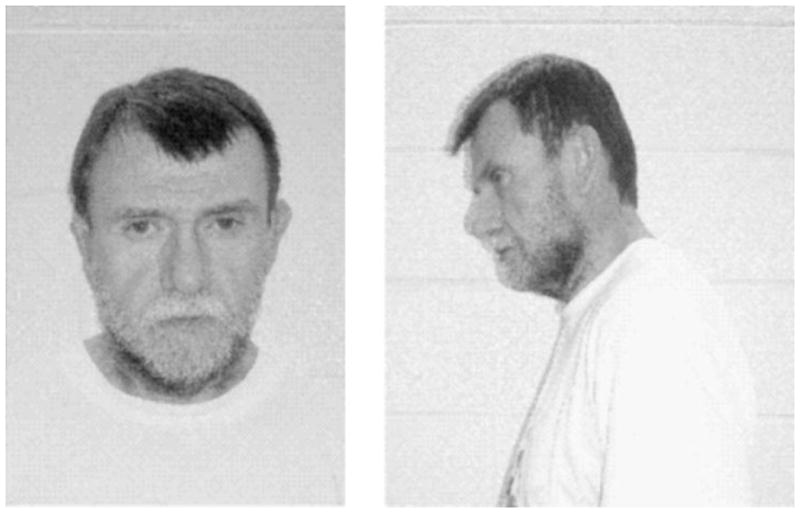
Patient 2 at age 52 years. The general appearance is not dysmorphic, but the nasal tip is somewhat bulbous, and on lateral view the midface is flat.
At age 43 years, he had developed bilateral tremors, mild difficulties with balance, incoordination in his hands, bradykinesia, drooling, and difficulties with speech. Diagnosis of PD was confirmed by a neurologist at age 44 years and treatment with Levodopa started which improved PD symptoms for 2–3 years. Subsequently, problems with tremor, gait, balance, speech, and emotional lability worsened despite increased doses of Levodopa and prevented him from working as a high school teacher.
Within 1 year of the diagnosis of PD, he began experiencing a depressed and anxious mood, low energy level, altered sleeping and eating habits, weight loss, social withdrawal, poor concentration and feelings of worthlessness. There was no previous history of psychiatric problems. Treatment with paroxetine improved these depressive symptoms and the patient discontinued the medication after a few months. About 3 years later, similar sleep and mood disturbances recurred, treated with trazodone, and later zopiclone. At age 54 years, he began gambling which was successfully managed with counseling. There were no signs or symptoms of any other impulse control disorder, mania, or psychosis. At age 56 the patient reported his overall mood to be good, with occasional episodes of irritability. He was maintained on Levodopa, entacapone, ropinirole, and domperidone with good control of PD symptoms.
There was no known family history of PD, other movement or neurological disorders, or major psychiatric illness. The patient’s daughter tested negative for the 22q11 deletion, but testing for other relatives was not pursued.
These two cases are, to our knowledge, the first reports of early onset PD in 22qDS. Occurrence of these two relatively uncommon conditions in two unrelated individuals presenting to different centers is unexpected. The prevalence of clinically recognized 22qDS in older children is 1/8,000 [Oskarsdottir et al., 2004] and the adult prevalence, although not precisely known, is expected to be even lower. The prevalence of PD between ages 40 and 49 is only 1/4,550 [Taba and Asser, 2002].
Although a chance association remains possible, the identification of two such cases, both with fairly typical presentation and response to standard treatment, and with no prior antipsychotic use, suggests a possible etiologic association. The obvious under-ascertainment of 22qDS in adults increases the significance of our observation since both our patients presented with PD in their 40s and the pool of individuals in this age group diagnosed with 22qDS remains quite small compared to the number of younger patients. We therefore propose that early onset PD may be an occasional feature of 22qDS which needs to be explicitly considered in other adult 22qDS patients.
The lack of previous reports of PD in 22qDS may be related to the paucity of information in the medical literature about the natural history of 22qDS and the phenotype in older adults [Bassett et al., 2005]. A report of parkinsonian symptoms in a patient with schizophrenia and 22qDS who had muscle stiffness in infancy and developed multiple extrapyramidal signs as a teenager is complicated by the fact that antipsychotic medications were started concurrently [Krahn et al., 1998]. If accepted as another example of early onset PD in 22qDS, this case would support our conclusion that early onset PD can be a feature of 22qDS. Krahn’s patient, however, presented very differently from ours and we believe antipsychotic medications may have played a significant role. Although there is little published data, neurologic, including parkinsonian, side effects of antipsychotic medications may be common in 22qDS [Bassett and Chow, 2008]. Lynch et al. [1995] reported a young adult with 22qDS and a neurodegenerative disorder with ataxia and parkinsonian features, including bradykinesia. There was no history of exposure to antipsychotic medications. This patient was not diagnosed with PD, presumably because cranial MRI showed marked cerebellar atrophy with only minimal changes in the basal ganglia. Identification of further cases of parkinsonian features in patients with 22qDS, especially those on no antipsychotic medications, will be helpful to confirm an association between this genetic syndrome and PD.
The genetics of PD is complex. An increased risk to first degree relatives, even in apparently sporadic cases, suggests multifactorial causation of PD. Hereditary forms account for only about 1–3% of all PD cases and tend to have younger age at onset. Mutations in parkin, PINK1 and DJ1 are known to cause autosomal recessive early onset PD and mutations in alpha synuclein, ubiquitin C terminal hydrolase, and LRRK2 can cause autosomal dominant PD with variable age of onset. Genome-wide association studies have yielded various other candidate genes and regions. Most still require replication but it is likely that polymorphisms in multiple genes affect the incidence and age of onset of PD.
Within the commonly deleted 3 Mb 22q11.2 deletion region, which spans over 40 genes, the catechol-O-methyltransferase (COMT) gene is of interest as a possible modifier of PD based on its pharmacogenetic role in Levodopa inhibition. A functional polymorphism at codon 158 in the COMT gene has been extensively studied. The Met allele results in lower COMT activity and, therefore, higher dopamine content in the brain compared to the Val allele. In 22qDS patients, there is a modest effect of the COMT functional allele on frontal lobe measures but none on risk for schizophrenia [Bassett et al., 2007]. There is no previous data regarding 22qDS patients with parkinsonism. COMT genotype data are not available for our patients and would in any case be insufficient for statistical analysis.
PD is associated with low dopamine in the substantia nigra and is treated by dopamine replacement. Met/Met homozygosity may be protective against PD [Bialecka et al., 2005] and may decrease the dosage of Levodopa required for treatment [Bialecka et al., 2004]. Dopamine levels, however, depend upon a complex feedback mechanism. An inverse relationship between prefrontal and striatal dopamine levels has been demonstrated in rodents [Pycock et al., 1980]. Dopamine levels in the prefrontal cortex may be increased in the early stages of PD, even prior to treatment [Rakshi et al., 1999; Kaasinen et al., 2001]. Feedback resulting from extremely high prefrontal dopamine levels theoretically could contribute to low striatal dopamine levels and ultimately to parkinsonian features. Accentuation of the high prefrontal dopamine levels in COMT Met/Met homozygotes is associated with decreased cognitive performance in patients with PD receiving Levodopa [Foltynie et al., 2004; Williams-Gray et al., 2007]. Effects of high prefrontal dopamine are a potential concern for our patients with PD and 22qDS, regardless of COMT genotype, since even Val hemizygotes have lower COMT levels than Val/Met heterozygotes.
In summary, we report early onset PD in two unrelated middle-aged patients with 22qDS. We propose that PD may be an occasional feature of 22qDS. Patients with 22qDS require ongoing surveillance well into adulthood for new complications [Bassett et al., 2005]. Patients with 22qDS and the physicians caring for these individuals should be made aware of this possible association, so they receive prompt diagnosis and treatment for PD, should symptoms develop. Ongoing genetic counseling is important, as is specialty care by providers who are familiar with the adult onset issues that can arise [Bassett et al., 2005]. Furthermore, since pre-existing learning and behavioral problems are relatively common in PD [Bodis-Wollner, 2003; Ishihara and Brayne, 2006], but the majority of such patients have never had genetic evaluation, clinical evaluation for features of 22qDS with genetic testing by FISH when relevant anomalies are identified, should be considered in patients with PD, especially those with early onset. The pathogenesis of early onset PD in patients with 22qDS remains unknown, but if elucidated, may contribute to understanding the etiology of PD and ultimately to prevention and treatment strategies.
Acknowledgments
We thank both patients for giving permission to publish their case histories. We also would like to acknowledge Bradley Hiner, MD and Fred Theye, PhD for their neurological and neuropsychological evaluations of case 1 and Mark Watson, MA and Don Young, PhD for the neuropsychological evaluations of case 2. The authors also thank Marshfield Clinic Research Foundation for its support through the assistance of Jennifer Hayes and Alice Stargardt in the preparation of this manuscript.
References
- Bassett AS, Chow EW. Schizophrenia and 22q11.2 deletion syndrome. Curr Psychiatry Rep. 2008;10:148–157. doi: 10.1007/s11920-008-0026-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett AS, Chow EW, Husted J, Weksberg R, Caluseriu O, Webb GD, Gatzoulis MA. Clinical features of 78 adults with 22q11 deletion syndrome. Am J Med Genet Part A. 2005;138A:307–313. doi: 10.1002/ajmg.a.30984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett AS, Caluseriu O, Weksberg R, Young D, Chow EW. Catechol-O-methyl transferase and expression of schizophrenia in 73 adults with 22q11 deletion syndrome. Biol Psychiatry. 2007;61:1135–1140. doi: 10.1016/j.biopsych.2006.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bialecka M, Drozdzik M, Klodowska-Duda G, Honczarenko K, Gawronska-Szklarz B, Opala G, Stankiewicz J. The effect of monoamine oxidase B (MAOB) and catechol-O-methyltransferase (COMT) polymorphisms on levodopa therapy in patients with sporadic Parkinson’s disease. Acta Neurol Scand. 2004;110:260–266. doi: 10.1111/j.1600-0404.2004.00315.x. [DOI] [PubMed] [Google Scholar]
- Bialecka M, Drozdzik M, Honczarenko K, Gawronska-Szklarz B, Stankiewicz J, Dabrowska E, Kubisiak M, Klodowska-Duda G, Opala G. Catechol-O-methyltransferase and monoamine oxidase B genes and susceptibility to sporadic Parkinson’s disease in a Polish population. Eur Neurol. 2005;53:68–73. doi: 10.1159/000084302. [DOI] [PubMed] [Google Scholar]
- Bodis-Wollner I. Neuropsychological and perceptual defects in Parkinson’s disease. Parkinsonism Relat Disord. 2003;9:S83–S89. doi: 10.1016/s1353-8020(03)00022-1. [DOI] [PubMed] [Google Scholar]
- Botto LD, May K, Fernhoff PM, Correa A, Coleman K, Rasmussen SA, Merritt RK, O’Leary LA, Wong LY, Elixson EM, Mahle WT, Campbell RM. A population-based study of the 22q11.2 deletion: Phenotype, incidence, and contribution to major birth defects in the population. Pediatrics. 2003;112:101–107. doi: 10.1542/peds.112.1.101. [DOI] [PubMed] [Google Scholar]
- Chow EW, Watson M, Young DA, Bassett AS. Neurocognitive profile in 22q11 deletion syndrome and schizophrenia. Schizophr Res. 2006;87:270–278. doi: 10.1016/j.schres.2006.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbaz A, Bower JH, Maraganore DM, McDonnell SK, Peterson BJ, Ahlskog JE, Schaid DJ, Rocca WA. Risk tables for parkinsonism and Parkinson’s disease. J Clin Epidemiol. 2002;55:25–31. doi: 10.1016/s0895-4356(01)00425-5. [DOI] [PubMed] [Google Scholar]
- Foltynie T, Goldberg TE, Lewis SG, Blackwell AD, Kolachana BS, Weinberger DR, Robbins TW, Barker RA. Planning ability in Parkinson’s disease is influenced by the COMT val158met polymorphism. Mov Disord. 2004;19:885–891. doi: 10.1002/mds.20118. [DOI] [PubMed] [Google Scholar]
- Ishihara L, Brayne C. What is the evidence for a premorbid parkinsonian personality: A systematic review. Mov Disord. 2006;21:1006–1072. doi: 10.1002/mds.20980. [DOI] [PubMed] [Google Scholar]
- Kaasinen V, Nurmi E, Bruck A, Eskola O, Bergman J, Solin O, Rinne JO. Increased frontal [(18)F]fluorodopa uptake in early Parkinson’s disease: Sex differences in the prefrontal cortex. Brain. 2001;124:1125–1130. doi: 10.1093/brain/124.6.1125. [DOI] [PubMed] [Google Scholar]
- Krahn LE, Maraganore DM, Michels VV. Childhood-onset schizophrenia associated with parkinsonism in a patient with a microdeletion of chromosome 22. Mayo Clin Proc. 1998;73:956–959. doi: 10.4065/73.10.956. [DOI] [PubMed] [Google Scholar]
- Lynch DR, McDonald-McGinn DM, Zackei EH, Emanuel BS, Driscoll DA, Whitaker LA, Fishbeck KH. Cerebellar atrophy in a patient with velocardiofacial syndrome. J Med Genet. 1995;32:561–563. doi: 10.1136/jmg.32.7.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oskarsdottir S, Vujic M, Fasth A. Incidence and prevalence of the 22q11 deletion syndrome: A population-based study in Western Sweden. Arch Dis Child. 2004;89:148–151. doi: 10.1136/adc.2003.026880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pycock CJ, Carter CJ, Kerwin RW. Effect of 6-hydroxydopamine lesions of the medial prefrontal cortex on neurotransmitter systems in subcortical sites in the rat. J Neurochem. 1980;34:91–99. doi: 10.1111/j.1471-4159.1980.tb04625.x. [DOI] [PubMed] [Google Scholar]
- Rakshi JS, Uema T, Ito K, Bailey DL, Morrish PK, Ashburner J, Dagher A, Jenkins IH, Friston KJ, Brooks DJ. Frontal, midbrain and striatal dopaminergic function in early and advanced Parkinson’s disease A 3D [(18)F]dopa-PET study. Brain. 1999;122:1637–1650. doi: 10.1093/brain/122.9.1637. [DOI] [PubMed] [Google Scholar]
- Shprintzen R. Velo-cardio-facial syndrome. In: Cassidy SB, Allanson JE, editors. Management of genetic syndromes. 2. New York: Wiley-Liss; 2005. pp. 615–631. [Google Scholar]
- Taba P, Asser T. Prevalence of Parkinson’s disease in Estonia. Acta Neurol Scand. 2002;106:276–281. doi: 10.1034/j.1600-0404.2002.01286.x. [DOI] [PubMed] [Google Scholar]
- Tam K, Bassett A, Chow E. The co-occurrence of 22q11 deletion syndrome and Parkinson disease: A new neurological feature?. American Society of Human Genetics Meeting; Toronto. 2004; 2004. [accessed August 7, 2008]. Available at: http://www.ashg.org/genetics/abstracts/abs04/f769.htm. [Google Scholar]
- Van Den Eeden SK, Tanner CM, Bernstein AL, Fross RD, Leimpeter A, Bloch DA, Nelson LM. Incidence of Parkinson’s disease: Variation by age, gender, and race/ethnicity. Am J Epidemiol. 2003;157:1015–1022. doi: 10.1093/aje/kwg068. [DOI] [PubMed] [Google Scholar]
- Williams-Gray CH, Hampshire A, Robbins TW, Owen AM, Barker RA. Catechol O-methyltransferase val158Met genotype influences frontoparietal activity during planning in patients with Parkinson’s disease. J Neurosci. 2007;27:4832–4838. doi: 10.1523/JNEUROSCI.0774-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson DI, Cross IE, Wren C, Scambler PJ, Burn J, Goodship J. Minimum prevalence of chromosome 22q11 deletions. Am J Hum Genet. 1994;55:A169. [Google Scholar]