

Abstract
Four core structures capable of providing sub-nanomolar inhibitors of anthrax lethal factor (LF) were evaluated by comparing the potential for toxicity, physicochemical properties, in vitro ADME profiles, and relative efficacy in a rat lethal toxin (LT) model of LF intoxication. Poor efficacy in the rat LT model exhibited by the phenoxyacetic acid series (3) correlated with low rat microsome and plasma stability. Specific molecular interactions contributing to the high affinity of inhibitors with a secondary amine in the C2-side chain were revealed by x-ray crystallography.
Keywords: Anthrax, Lethal factor, Inhibitor, in vitro ADME, x-ray crystallography
Anthrax lethal factor (LF) and edema factor (EF) are two proteins secreted by the Gram-positive bacterium Bacillus anthracis to assist in the infection of the host1. LF is a zinc metalloproteinase that disrupts cell signaling pathways by direct cleavage of MEK proteins2, while EF is a Ca2+/calmodulin-dependant adenylate cyclase that increases cytosolic cAMP and also interferes with cell signalling3. When combined with a third protein protective antigen (PA), they are known as lethal toxin (LT) and edema toxin (ET). PA transports EF and LF into cells where they act as potent virulence factors and contribute to the pathogenesis of the disease4. The details of how LF and EF act to suppress the immune system, support dissemination of the bacteria, and contribute to the lethality of the disease is beginning to be revealed5 and suggests that inhibiting the activity of these virulence factors could lead to an increase in survival of the infected host. Of the three forms of the disease, death due to inhalation anthrax is significantly higher when compared to fatalities resulting from cutaneous or gastrointestinal exposure, with fatality rates being >85 % without supportive care6. While not contagious, the potential danger from this disease when used as an of agent bioterrorism was clearly demonstrated by the 2001 U.S. mail attacks, where fatality rates approaching 50% were seen even after aggressive treatment with antibiotics7. Due to the relative ease of production and dispersal of anthrax spores, the potential for mass casualties due to B. anthracis release against an urban or military population is extremely high. As a result, new therapeutics are needed to supplement available methods of treatment and increase the survival rate of patients diagnosed with inhalation anthrax.
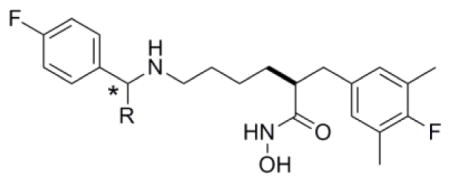
In part 2 of this series8 we disclosed the identification of four core structures (Figure 1) capable of providing anthrax LF inhibitors (LFIs) with Ki values of less than 10 nM and in vivo efficacy in a rat LT model of anthrax. Below we present data from further studies directed towards evaluation of the current core structures, and structural modifications which have led to improved in vivo efficacy in the rat LT model.
Figure 1.

Current lead series of anthrax LFIs (1 to 4).
In a previous report8 we noted the necessity of having a benzylamine fragment located in the C2 side chain to achieve high intrinsic potency with this class of LFIs. We also showed that the replacement of this amine with an oxygen atom (NH to O) to give the corresponding benzyl ether resulted in a >100 fold loss of intrinsic potency, and the all carbon chain analogs (NH to CH2) displayed a greater loss (>1000 fold) in potency. This led to the conclusion that a hydrogen bond or a significant electrostatic interaction was responsible for the observed increase in affinity of the benzylamine analogs to the LF protein. Since then, we have obtained a high resolution x-ray crystal structure of LFI 4 bound to the active site of LF (Figure 2). Consistent with the LF-inhibitor structure obtained by the scientists at Merck9, the two oxygen atoms of hydroxamic acid group were found to chelate the catalytic zinc ion. The orientation and interatomic distances of this group relative to protein atoms is supportive of H-bonds between the NH-group and the backbone carbonyl oxygen of Gly657, the carbonyl oxygen with the hydroxyl group of Tyr728, and the hydroxamate hydroxyl with the catalytic Glu687 residue. This orientation of the hydroxamic acid group and the associated H-bonding network is essentially the same as seen first with hydroxamic acid (HA) based inhibitors thermolysin10 and later with HA inhibitors bound to the matrix metalloproteinases (MMPs)11. In contrast to our expectations based upon the Merck structure we found the C1-C4 axis of the 4-flourophenyl ring present in the core structure of 4 to be at an angle of 57° relative to the axis of the 3-methyl-4-fluorophenyl ring of L915 when bound to LF. This results in a shift of loop residues Lys673, Gly674, and Val 675 away from the catalytic zinc-atom and creates a larger S1-prime subsite (see Fig S1). Early modeling studies and the structure activity relationships (SAR) for these compounds had predicted the core structure of the ligand to bind on the prime side of the catalytic site, however, we were not able to identify a single preferred binding mode for the C2-side chain on the non-prime side with any confidence. It is clear from the present structure that the terminal phenyl ring points directly into the S3 pocket as opposed to the equally accessible S2 subsite12 (see Fig S2). Of particular interest was the presence of a bridging water molecule positioned to help stabilize the complex by H-bonding to both the benzyl amine nitrogen and Glu735. The clear electron density for the oxygen atom of this water molecule placed it 2.38 Å from the N-atom of the benzylamine and 2.58 Å from one of the carboxylate oxygen atoms of Glu735, the other of which is part of the first coordination sphere of the catalytic zinc atom. This H-bonding network combined with the SAR discussed earlier, provides strong support for this interaction as a key contributor to inhibitor potency.
Figure 2.

Representation13 showing the crystallographic water bridging the amine of LFI 4 to Glu735 in the catalytic site of LF.
While clearly important for high binding affinity towards the target, this benzylamine fragment also fits the classic pharmacophore model for CYP2D6 inhbitors14 where the nitrogen is likely to be H-bonded to Glu216 or Asp301 in the CYP2D6 active site. Therefore, we sought a way to limit the potential for our LFIs to bind to this enzyme without sacrificing potency against LF. Since H-bonding is both distance and angle dependant, one potential way to disrupt this interaction is to introduce steric hindrance adjacent to the nitrogen atom. Of the two possible sites for introduction of a branch point on the C2 side chain, the α-benzyl site appeared to be most suitable in terms of synthetic accessibility. It should be noted that at the time these changes in structure were being investigated, the x-ray crystal structure of 4 discussed above was not yet available. At the time, consideration of these structural modifications raised two questions with respect to the potential effect on inhibitor potency; how would the introduction of an R-group at this site, and the creation of a new stereogenic center, affect the intrinsic potency and the in vivo efficacy of this class of LFIs?
To answer these questions our first task was to identify an efficient synthetic route to stereochemically pure α-substituted benzyl amines. Shown in Scheme 1 is the synthesis of the (S)-isobutyl-4-fluorobenzyl amine which is representative of the method used to prepare the needed α-substituted benzylamines. Compound 5, prepared by reaction of the lithium salt of (S)-isopropyl oxazolidinone with the acyl chloride derived from 4-fluorophenylacetic acid, was alkylated with methallyl bromide under standard conditions15. Catalytic hydrogenation using Pd-C provided 6 as a single diastereomer in good overall yield. Removal of the chiral auxiliary with lithium hydroperoxide to give carboxylic acid 7 was followed by a Curtius rearrangement16 using an aqueous acid work up to afford the targeted α-substituted benzylamine 8. To investigate this modification in the C2-side chain, we selected the phenylpropionic acid core series (2) due to their ease of synthesis. These LFIs were then prepared following our previously published procedure involving reductive amination of the benzylamine reagent with the C2-side chain aldehyde connected to the phenylpropionic acid core8.
Scheme 1.

Reagents and conditions: (a) i.) LiHMDS, THF, −78 ºC, ii) methallyl bromide, −78 ºC to rt, (b) H2, Pd-C, EtOH, 3h at rt, (c) LiOOH, THF/H2O, 0 ºC, 1h, (d) i) ClCO2Et, Et3N, acetone, 0 ºC, 15 min, ii) NaN3, H2O, 0 ºC, 30 min, iii) toluene, reflux, 45 min, iv) 8N HCl, 0 ºC to rt.
Inhibitory data for these LFIs against anthrax LF are provided in Table 112. As a first observation, the size of the R-group can have a significant effect on the intrinsic potency of the LFI, with the most potent inhibitor bearing an ethyl group at the benzylic position. Given that this work was conducted prior to obtaining the x-ray structure of 4 bound to LF, we were somewhat surprised to see that introduction of a large substituent such as benzyl (15) can still afford sub-nanomolar inhibitors of LF. By examination of the crystal structure12 of 4, it can be easily seen how a large R-group attached to the α-position can be accommodated without disrupting the interactions essential for high affinity binding (see Fig S3). A second finding was that with the exception of R = methyl, the (R)-isomers were more potent by 3 to 5 fold relative to analogs of the (S)-configuration. When considering both of these parameters, a change in potency of 50-fold can be observed (cf. 11 vs. 16). Perhaps most important, it should be possible to modify the drug-like properties of a given LFI by introduction of various R-groups to this site with minimal concern for size or absolute stereochemistry.
Table 1.
Anthrax LF inhibitory data for LFIs bearing α-benzyl substituents on the C2-side chain.
| |||
|---|---|---|---|
| LFI | (*) | R | LF (FRET)
|
| KinM) | |||
| 9 | R | Me | 0.30 |
| 10 | S | Me | 0.49 |
| 11 | R | Et | 0.04 |
| 12 | S | Et | 0.11 |
| 13 | R | iso-butyl | 0.23 |
| 14 | S | iso-butyl | 1.30 |
| 15 | R | -CH2Ph | 0.60 |
| 16 | S | -CH2Ph | 2.00 |
Values are the average of two experiments; sd < 15%
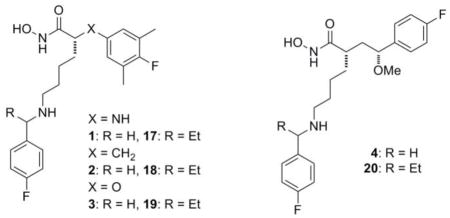
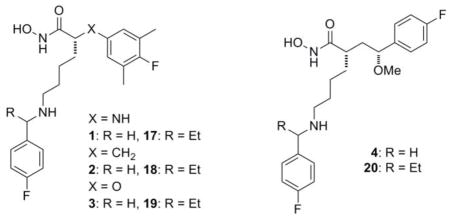
Having identified structural features which provided sub-nanomolar LFIs in four core structures, we set out to see if one or more of these structural classes would be preferred for the lead optimization phase of this work. Towards this end we chose to compare in vitro potency, toxicity, physicochemical (PC) properties, and in vivo efficacy of matched pairs of LFIs representing each core structure bearing an unsubstituted or α-substituted benzylamine on the C2-side chain. Based on the results presented in Table 1, and to ease the burden of synthesis, we selected the racemic α-ethylbenzylamine analogs for our comparison of C2-side chain effects. The data collected from these studies is provided in Tables 2 and 3 below.
Table 2.
The in vitro potency, potential for toxicity, and PC properties for match pairs of LFIs representing each of the four core structures.
| |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LFI | MW | Kia (nM) | CC50a (μM) | ClogPb | PSAc (Å2) | PAMPAd (log Pe) | MMP Ki (μM)a
|
CYP2D6a (μM) | hERGa (μM) | ||||
| −1 | −3 | −9 | −12 | −14 | |||||||||
| 1 | 391.2 | 0.24 | 61 | 3.7 | 92.6 | −5.9 | >50 | >50 | >50 | >50 | >50 | 1.35 | 1.23 |
| 17 | 419.2 | 0.17 | 32 | 4.6 | 88.1 | −5.1 | >50 | >50 | >50 | >50 | >50 | 0.24 | 0.95 |
| 2 | 390.2 | 0.13 | 16 | 4.0 | 88.9 | −4.8 | >50 | >50 | >50 | >50 | >50 | 0.02 | 4.65 |
| 18 | 418.2 | 0.31 | 15 | 4.8 | 74.6 | −4.1 | >50 | >50 | >50 | >50 | >50 | 0.08 | 13.3 |
| 3 | 392.2 | 1.20 | 26 | 3.8 | 86.9 | −5.2 | >50 | >50 | >50 | >50 | >50 | 0.23 | 0.79 |
| 19 | 420.2 | 1.00 | 21 | 4.7 | 83.9 | −4.4 | >50 | >50 | >50 | >50 | >50 | 0.34 | 2.7 |
| 4 | 406.2 | 0.58 | 55 | 3.2 | 81.9 | −5.8 | 33 | >50 | 21 | 2.3 | >50 | 1.26 | 2.0 |
| 20 | 434.3 | 0.27 | 35 | 4.0 | 80.1 | −5.1 | 47 | 31 | 11 | 12 | >50 | 0.16 | 10.0 |
see SI for experimental procedures; IC50 values given for CYP2D6 and hERG,
ClogP calculated with ChemDraw v12.0,
PSA calculated using PCModel v. 8.5,
PAMPA determined by the method of Wohnsland17 using Mllipore multiscreen 96-well plates
Table 3.
The in vitro ADME and rat LT challenge assay data for match pairs of LFIs representing each of the four core structures.
| |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LFI | Kia | CC50a | ClogPb | ElogDc | In Vitro ADMEd | 5.0 mg/kge | 2.5 mg/kge | 1.25 mg/kge | |||||
|
|
|
||||||||||||
| (nM) | (μM) | M | P | U | surv | rMST | surv | rMST | surv | rMST | |||
| 1 | 0.24 | 61 | 3.7 | 2.3 | 60 | 90 | 38 | 3/3 | - | 3/5 | 16.8 | nt | - |
| 17 | 0.17 | 32 | 4.6 | 3.3 | 75 | 84 | 74 | 3/3 | - | 3/3 | - | 0/3 | 2.4 |
| 2 | 0.13 | 16 | 4.0 | 2.2 | 18 | 100 | 79 | 3/3 | - | 1/3 | 19.3 | nt | - |
| 18 | 0.31 | 15 | 4.8 | 2.9 | 55 | 95 | 100 | 3/3 | - | 3/3 | - | 5/6 | 4.2 |
| 3 | 1.20 | 26 | 3.8 | 3.1 | 3 | 61 | 76 | 0/3 | 16.0 | nt | - | nt | - |
| 19 | 1.00 | 21 | 4.7 | 3.3 | 7 | 38 | 75 | 1/3 | 16.0 | nt | - | nt | - |
| 4 | 0.58 | 55 | 3.2 | 2.4 | 91 | 98 | 99 | 2/3 | 16.0 | nt | - | nt | - |
| 20 | 0.27 | 35 | 4.0 | 2.9 | 75 | 94 | 100 | 3/3 | - | 2/5 | 16.8 | nt | - |
see SI for experimental procedure;
ClogP calculated with ChemDraw v;
ElogD measure using the method of Lombardo15.
Values given indicate percent of compound remaining at t = 30 min for M = miocrosome stability, U = UGT assays and the percent remaining at 3 hours for P = plasma stability;
Fischer 344 rats were dosed IV with LFIs follwed 20–30 minutes later by 10 μg of LT (10 μg of PA + 10 μg of LF); surv. =Number of survivors per group. rMST = MST(LFI)/MST(controls) where vehicle treated controls (MST = 74.8 ± 7.2 minutes (n=11; when less than 50% deaths occurred the average survival time was used: rMST = aveST(LFI)/MST(controls); nt = not tested, (−) = not applicable. Survival curves for all LFIs at each dose showed statistical significance (P<0.05) relative to controls.
The in vitro potency, potential for toxicity, PC properties, and the activity of these compounds versus possible anti-targets (MMPs, CYP2D6, and hERG) is provided in Table 2. In terms of intrinsic potency, the analogs based upon the phenoxyacetic acid core structure are the least potent, though they still possess Ki values <2 nM. Cytoxicity is very low for all of the analogs with the observed CC50 values being 20K fold greater than their respective Ki values. The PC properties for all of the core structures are very similar. Molecular mass is well below 500 Da for each series and differs by < 50 mass units. ClogP values are on the high side of the preferred range with all being > 3.0 but still less than 5.0 while the calculated polar surface areas are at the lower end of the preferred range of 75Å <PSA<125Å. Determination of relative permeability using a hexadecane based PAMPA assay17 show all of the LFIs to have fair to good permeability with log Pe values being > −6.0. While no apparent trend emerges with respect to the specific core structures, better permeability is seen with the α-ethyl analogs 17 to 20. Selectivity for LF versus various MMPs was excellent, being >50K fold for the one atom linking group series (X= NH, CH2, O) and > 4000 fold for the γ-ether series represented by LFI 4 and 20. Consistent with the current pharmacophore models for binding to CYP2D6, many of these compounds were found to be strong inhibitors of this enzyme. Further, when comparing analogs where R = H versus R = ethyl, introduction of steric hindrance by a simple alkyl group at the benzylic position did not decrease CYP2D6 inhibitory activity. Activity against hERG was considerably lower compared to CYP2D6, but still significant (< 10 μM) for all but two analogs. Taken together, these data did not show a bias towards any one core structure in terms their suitability for the lead optimization process, however, the potential for each series to significantly inhibit CYP2D6 and hERG was made clear.
Continuing our investigation of the four core structures, the same set of eight LFIs were tested in various in vitro ADME assays and finally in the rat LT challenge assay18. A concurrent goal of these studies was to determine if the data obtained from the in vitro ADME assays would correlate with survival outcomes observed in the rat LT challenge experiments and aid in selection of future candidates for in vivo studies. The results of this work are provided in Table 3. We also determined the ElogD values for these compounds which show good correlation with logD7.4 values when applied to neutral or basic compounds19. In this case, the data shows these LFIs to have the potential for better drug-like properties at physiologic pH when compared to evaluation of the ClogP values alone.
As in our previously studies, the rat LT challenge study was conducted by dosing Fischer 344 rats (IV) with the LFI followed 20 to 30 minutes later by a 10 μg dose (IV) of LT (10 μg PA + 10 μg LF). When administered at a dose of 5.0 mg/kg (Table 3), five of the eight LFIs provided 100% protection, one animal was lost in the group treated with LFI 4, but only a single animal survived from the two groups treated with the LFIs based on the phenoxy acetic acid core structure (3 and 19). This finding was qualitatively consistent with the observed in vitro ADME data where 3 and 19 displayed the lowest stability in the microsome and plasma stability assays among all of the compounds tested. Of particular significance was the low metabolic stability for these compounds in the rat liver microsome assay where < 10% of either compound remained after incubation for 30 minutes. This observation combined with the fact that the animals were challenged with lethal toxin 20 to 30 minutes after receiving the LFI is consistent with the inability of 3 and 19 to provide significant protection from the lethal effects of LF in this experiment. In an attempt to probe further into possible differences in efficacy due to the core structure, the LFIs which provided full protection at 5.0 mg/kg were then tested at 2.5 mg/kg. In this case, two of the five compounds tested (17 and 18) were still able to protect all of the animals in their respective groups. The finding that the LFIs which perform best were α-substituted benzylamine analogs, combined with a similar trend seen with the γ-ether analogs 4 (R=H) and 20 (R=Et), indicates that α-substitution appears to benefit survival in this model of anthrax LT intoxication. In a final experiment LFIs 17 and 18 were tested at 1.25 mg/kg. At this dose, neither LFI was able to fully protect the animals from death due to LF, however, the phenylpropionic acid derivative 18 appeared to be more effective (5/6 survivors) compared to the aniline based analog 17 (0/3 survivors).
The pathogenesis of inhalation anthrax observed in the rabbit is considered to be a good representation of the disease course in humans20. In preparation for these studies we determined the PK profile of LFI 20 in NZW rabbits. Since the current protocol call for the administration of the drug over several days, and may require multiple doses of the test compound per day, we selected the subcutaneous (SC) route of administration for this initial study. The PK data for LFI 20 shown in Table 4 indicates that this compound is rapidly absorbed when dosed at 10 mg/kg via the SC route and results in a good peak plasma concentration (Cmax = 1.3 μM). In addition, the terminal half-life (t1/2β) and AUC values suggest a good residence time for this compound. Finally, the mean plasma concentration of 20 at the 12 hour time point indicates that the compound is present at levels more than 290-fold above the Ki value which would support a twice a day dosing schedule in a future efficacy study.
Table 4.
PK data for LFI 20 after single dose of 10 mg/kg (SC; DMSO:PEG400:pH6 citrate buffer, 2:10:88) in NZW rabbits.
| Ki | 0.27 nM |
| Dose (SC) | 10 mg/kg |
| Tmax | 0.3 h |
| Cmax | 561.3 ng/mL (1.3μM) |
| T1/2(β) | 5.3 h |
| AUC∞ | 2637.6 ng-h/mL |
| [20]12 h | 79 nM |
In summary, evaluation of four core structures capable of providing sub-nanomolar inhibitors of anthrax lethal factor was accomplished by comparing the potential for toxicity, PC properties, in vitro ADME profiles, and relative efficacy in a rat LT model of LF intoxication. Based upon these results, the phenoxyacetic acid core structure appears to be inferior to the other three tested. All of the core structures provided analogs which had significant inhibitory activity against CYP2D6 and hERG indicating that new analogs should be tested against these anti-targets as part of the lead candidate selection process. Addition of an R-group at the benzylic position of the C2-side chain was well tolerated with respect to intrinsic potency, was consistent with the x-ray structure of LFI 4 bound to the catalytic site of LF, but did not decrease inhibitor potency against CYP2D6. In addition, this modification appeared to provide analogs with improved protection profiles in the rat LT challenge model. Compound 20 was also found to have a good PK profile when administered SC to NZW rabbits and supports the potential of these inhibitors to provide protection in a rabbit spore challenge model of inhalation anthrax.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health for their support of this work with grants R44 AI052587 and U01 AI078067. The rat LT challenge studies were supported by the Intramural Research Program of the NIH, National Institute of Allergy and Infectious Diseases. The PK study was conducted by Covance Laboratories, Inc. (Madison, WI). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIAID or the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Tournier JN, Paccani SR, Quesnel-Hellmann A, Baldari CT. Mol Aspects Med. 2009;30:456. doi: 10.1016/j.mam.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 2.(a) Duesbury NS, Webb CP, Leppla SH, Gordon VM, Copeland TD, Ahn NG, Oskarsson MK, Fukasawa K, Paull KD, Vande Woude GF. Science. 1998;280:734. doi: 10.1126/science.280.5364.734. [DOI] [PubMed] [Google Scholar]; (b) Vitale G, Bernardi L, Napolitani G, Mock M, Montecucco C. Biochem J. 2000;352:739. [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Leppla SH. Proc Natl Acad Sci USA. 1982;79:3162. doi: 10.1073/pnas.79.10.3162. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tang WJ, Guo Q. Mol Aspects Med. 2009;30:423. doi: 10.1016/j.mam.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Collier RJ, Young JA. Annu Rev Cell Dev Bio. 2003;19:45. doi: 10.1146/annurev.cellbio.19.111301.140655. [DOI] [PubMed] [Google Scholar]
- 5.Moayeri M, Leppla SH. Mol Aspects Med. 2009;30:439. doi: 10.1016/j.mam.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holty JEC, Bravata DM, Liu H, Olshen RA, McDonald KM, Owens DK. Ann Intern Med. 2006;144:270. doi: 10.7326/0003-4819-144-4-200602210-00009. [DOI] [PubMed] [Google Scholar]
- 7.Jernigan JA, Stephens DS, Ashford DA, Omenaca C, Topiel MS, Galbraith M, et al. Emerg Infect Diseases. 2001;7:933. doi: 10.3201/eid0706.010604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim S, Jiao GS, Moayeri M, Crown D, Cregar-Hernandez L, McKasson L, Margosiak SS, Leppla SH, Johnson AT. Bioorg Med Chem Lett. 2011;21:2030. doi: 10.1016/j.bmcl.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shoop WL, Xiong Y, Wiltsie J, Woods A, et al. Proc Natl Acad Sci USA. 2005;102:7958. doi: 10.1073/pnas.0502159102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holmes MA, Matthews BW. Biochemistry. 1981;20:6912. doi: 10.1021/bi00527a026. [DOI] [PubMed] [Google Scholar]
- 11.Skiles JW, Gonnella NC, Jeng AY. Curr Med Chem. 2001;8:425. doi: 10.2174/0929867013373417. [DOI] [PubMed] [Google Scholar]
- 12.See supplementary data for details on experimental procedures and additional figures from the crystallographic study.
- 13.This image was made with VMD which was developed with NIH support by the Theoretical and Computational Biophysics group at the Beckman Institute, University of Illinois at Urbana-Champaign. http://www.ks.uiuc.edu/Research/vmd/
- 14.Smith DA, van de Waterbeemd H, Walker DK. In: Methods and Principles in medicinal Chemistry. Mannhold R, Kubinyi H, Timmerman H, editors. Vol. 13. Wiley-VCH Verlag GmbH; Weinheim: 2001. pp. 78–80. [Google Scholar]
- 15.Evans DA, Ennis MD, Mathre DJ. J Am Chem Soc. 1982;104:1737. [Google Scholar]
- 16.Kaiser C, Weinstock J. Org Synth. 1971;51:48. [Google Scholar]
- 17.Wohnsland F, Faller B. J Med Chem. 2001;44:923. doi: 10.1021/jm001020e. [DOI] [PubMed] [Google Scholar]
- 18.Gupta PK, Moayeri M, Crown D, Fattah RJ, Leppla SH. PLoS ONE. 2008;3:e3130. doi: 10.1371/journal.pone.0003130. note: this assay was not designed to model inhalation anthrax in humans but to demonstrate that a small molecule LFI can prevent death due to anthrax lethal factor in an in vivo model. The protocol used provides for a reproducible assay with the resolution necessary to rank order the in vivo efficacy of LFIs in an animal model where death results specifically from the action of anthrax LF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lombardo F, Shalaeva MY, Tupper KA, Gao F. J Med Chem. 2001;44:2490. doi: 10.1021/jm0100990. [DOI] [PubMed] [Google Scholar]
- 20.Goossens PL. Mol Aspects Med. 2009;30:467. doi: 10.1016/j.mam.2009.07.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.