

Summary
Chronic inflammation is associated with 25% of all cancers. In the inflammation-cancer axis, prostaglandin E2 (PGE2) is one of the major players. PGE2 synthases (PGES) are the enzymes downstream of the cyclooxygenases (COXs) in the PGE2 biosynthesis pathway. Microsomal prostaglandin E2 synthase 1 (mPGES-1) is inducible by pro-inflammatory stimuli and constitutively expressed in a variety of cancers. The potential role for this enzyme in tumorigenesis has been reported and mPGES-1 represents a novel therapeutic target for cancers. In order to identify novel small molecule inhibitors of mPGES-1, we screened the ChemBridge library and identified 13 compounds as potential hits. These compounds were tested for their ability to bind directly to the enzyme using surface plasmon resonance spectroscopy and to decrease cytokine-stimulated PGE2 production in various cancer cell lines. We demonstrate that the compound PGE0001 (ChemBridge ID number 5654455) binds to human mPGES-1 recombinant protein with good affinity (KD = 21.3 ± 7.8 μM). PGE0001 reduces IL-1β-induced PGE2 release in human HCA-7 colon and A549 lung cancer cell lines with EC50 in the submicromolar range. Although PGE0001 may have alternative targets based on the results from in vitro assays, it shows promising effects in vivo. PGE0001 exhibits significant anti-tumor activity in SW837 rectum and A549 lung cancer xenografts in SCID mice. Single injection i.p. of PGE0001 at 100 mg/kg decreases serum PGE2 levels in mice within 5 h. In summary, our data suggest that the identified compound PGE0001 exerts anti-tumor activity via the inhibition of the PGE2 synthesis pathway.
Keywords: prostaglandin E2, drug design, inflammation, cancer, anti-tumor
Introduction
Prostaglandin E2 (PGE2), a key mediator of inflammation, is the most abundant prostanoid with various bioactivities and has been associated with numerous pathologies [1,2]. Thus, inhibition of PGE2 synthesis and its action has been suggested in the treatment of inflammatory-associated diseases, including cancer [2]. PGE2 is synthesized sequentially by the following three enzymatic reactions. Upon the stimulation of IL-1β receptors, for example, membrane-boundand secretory phospholipase A2 (PLA2) isoforms release arachidonic acid (AA) from membrane phospholipids [3]. Next, the cyclooxygenases (COX-1 and COX-2) convert AA into the unstable intermediate, prostaglandin endoperoxide PGH2. Finally, PGE2 synthases (PGESs) isomerizePGH2 into PGE2. Elevated levels of PGE2 and COX-2, which catalyze the rate-limiting step in PGE2 biosynthesis, are often observed in human cancers such as colon cancer [4,5]. Therefore, COX-2 inhibitors have been tested in humans and pre-clinical models for the prevention or treatment of colon cancer [6,7]. However, inhibition of COXs may lead to cardiotoxicity due to the global reduction of other key prostaglandins, and imbalanced production of pro-thrombotic eicosanoids (e.g. increased TxA2) and anti-thrombotic eicosanoids (e.g. decreased PGI2) [8,9]. Therefore, developing inhibitors downstream COXs could represent an alternative therapeutic strategy with potentially less side effects [10,11].
Three PGES isoforms have been cloned [12-14]. The expression and activity of microsomal PGE2 synthase-1, (mPGES-1), is induced by various inflammatory stimuli such as pro-inflammatory cytokines IL-1β and TNF-α [12,15], whereas mPGES-2 (microsomal PGE2 synthase-2) and cPGES (cytosolic PGE2 synthase) are constitutively expressed and active [13,14]. Interestingly, only constitutive over-expression of mPGES-1 has been reported in cancers including colon, lung, gastric, ovarian, pancreatic, and breast cancers [16-22], suggesting its tumorigenic potential. Indeed, the role of mPGES-1 in tumorigenesis has been shown by both transplantation tumor models [18,23,24] and genetic deletion approaches [25,23]. Given the known effects of prostaglandins on cardiovascular function and the recent history of secondary effects associated with high doses of COX-2 specific inhibitors [8,26], there are legitimate concerns regarding the potential cardiotoxicity risks for any new inhibitors of the prostaglandin biosynthesis pathway. However, it was recently reported by Cheng et al. [27] that the deletion of mPGES-1, unlike deletion, disruption, or inhibition of COX-2, did not result in hypertension or a predisposition to thrombosis in normo-lipidemic mice. This important finding suggests that selective mPGES-1 inhibitors would have very low if any cardiotoxic side effects typically associated with COX-2 inhibitors.
Recently, some compounds have been described to inhibit mPGES-1 activity, but none have been developed as anticancer agents in vivo [28]. There are several examples of compounds that were initially developed to target the COX-2 but that were shown later to also inhibit mPGES-1. For example, NS-398 [2-cyclohexyloxy-4-nitrophenyl)-methanesulfonamide], developed in Japan as an arylsulfonamide derivative of the anti-inflammatory agent nimesulide [29], is a COX-2 inhibitor that inhibits mPGES-1 with an IC50 of 20 μM in vitro [30]. In animal models, NS-398 was a potent anti-inflammatory agent [31,32]; however, it had poor bioavailability and produced hepatotoxic metabolites. Thus, NS-398 was not developed into a therapeutic agent. Recently, a series of indole compounds showed selectivity and higher activity against the inducible mPGES-1 with the lowest IC50 value found being 3 nM [33]. However, due to a high degree of protein binding and poor cell permeability, these series of compounds loose potency in cell-based assays and, to our knowledge, have not been tested in vivo. Finally, licofelone and a number of natural compounds were also recently found to inhibit mPGES-1 activity in the low micromolar range [34-37], but many of them also affected COX activity or expression.
Herein, we generated a pharmacophore query using the structure of triclosan, an anti-inflammatory compound sharing pharmacophore features with NS-398. The anti-inflammatory property of triclosan has been attributed in part to the inhibition of PGE2 biosynthesis. The molecular docking model of triclosan within the mPGES-1 active site has also been described [28]. This query was used to perform Unity-based three dimensional searches on the ChemBridge diversity library to identify thirteen compounds. Using surface plasmon resonance (SPR) spectroscopy, we confirmed the binding of several compounds to human recombinant mPGES-1 which correlated with the inhibition of IL-1β-induced PGE2 production in colon and lung cancer cells. In this report, we show that one of these compounds, ChemBridge 5654455 (hereafter referred to as “PGE0001”) exhibited good cellular activity in colorectal and lung cancer cells and promising anti-tumor activities in their corresponding subcutaneous xenograft mouse models with appropriate pharmacokinetic properties.
Materials and methods
Compounds and reagents
Compounds ID 5654455, 5933870, 6795274, 7384071, 7418129, 7786927, 7882458, 5662444, 5807166, 5935487, 5724933, 6239316, and 5862295 were purchased from ChemBridge Corp. (San Diego, CA). Anti-COX-2 monoclonal antibody (mAb) (clone CX229), anti-mPGES-1 mAb (clone C6C), anti-mPGES-2 polyclonal antibody (pAb), and anti-cPGES pAb were all purchased from Cayman Chemical (Ann Arbor, MI). Anti-β-actin mAb was purchased from Sigma-Aldrich (St. Louis, MO). Reduced L-glutamine (GSH) was purchased from Sigma-Aldrich. Recombinant human mPGES-1 was purchased from Cayman Chemical. Recombinant Human IL-1β was purchased from R&D Systems (Minneapolis, MN). Prostaglandin H2 was purchased from Enzo Life Sciences (Plymouth Meeting, PA). Sphingosine kinase (SPHK-1) inhibitor 2 and compound MK-886 were both purchased from Cayman Chemical. Celecoxib was purchased from LKT Laboratories (St. Paul, MN).
Molecular modeling procedure
Docking was performed using the Sybyl 8.0 modeling software package from Tripos Inc (St Louis, MS). The crystal structure of mPGES-1 (PDB code: 3dww) was used for all docking protocols. The protein structure along with the active site and different ligands were used as inputs. The structures of the small molecules were constructed in Sybyl 8.0 and minimized using Tripos forcefield and Gasteiger-Huckel charges. FlexiDock within Sybyl 8.0 generates 20 different docking orientations of the ligand within the active site. These docking orientations were analyzed on the basis of the FlexiDock score. LogD values were calculated using ACD/PhysChem database version 12.00 (Advanced Chemistry Development, Inc., Toronto, ON, Canada, www.acdlabs.com, 2008).
Bacterial expression of human mPGES-1
The 6xHis-tagged human mPGES-1 was expressed from the pET30(b) vector in E. coli BL21(DE3) cells. An overnight culture of BL21(DE3) cells in LB broth containing kanamycin (50 μg/ml) was diluted 1:100 into LB broth containing kanamycin. The culture was grown at 37°C with shaking (250 rpm) until the A600 nm was around 0.6. Expression of 6xHis-mPGES-1 was then induced by the addition of 0.5 mM IPTG, and the culture was grown for another 3 h at 37°C with shaking. The cell pellets were harvested by centrifugation (5,000 ×g, 10 min at 4°C) and stored at –20°C for further purification.
Bacterial membrane preparation and purification of 6xHis-mPGES-1
Preparation of membranes was performed by following the procedure from Thoren et al. [38]. The supernatant of the membrane preparation was loaded onto a Ni-NTA (Qiagen, Valencia, CA) chromatography column equilibrated with binding buffer containing 15 mM Tris-HCl, pH 7.4, 150 mM NaCl, 10% glycerol, 0.2% Triton X-100, and 1 mM GSH, then washed with washing buffer (60 mM imidazole in binding buffer). The bound protein was then eluted with elution buffer (200 mM imidazole in binding buffer). The eluted peak was immediately desalted into 20 mM sodium phosphate buffer, pH 7.5, 50 mM NaCl, 2.5 mM GSH, and 0.2% reduced Triton X-100, using Zeba Spin Desalting Columns, 7K MWCO (Thermo Scientific, Rockford, IL).
Surface plasmon resonance (SPR) spectroscopy binding assays
All interaction analyses were performed with a Biacore 2000, Biacore 2000 Control Software v. 3.2, and BIAevaluation v. 4.1 analysis software (Biacore, Piscataway, NJ) as already described in reference [39]. His-tagged mPGES-1 fusion protein was immobilized on a CM5 sensorchip (Biacore BR-1000-12) using Biacore's Amine Coupling Kit (Biacore BR-1000-50) to a level of 10,000 Response units (RUs). Small molecule analytes at concentrations ranging from one tenth to ten times the predicted KD were injected at a high flow rate (50 μl/min). Dimethylsulfoxide (DMSO) concentrations in all samples and running buffer were 2% (v/v). KDs were calculated using a 1:1 Langmuir model.
Cell culture and Western blots
Colorectal cancer cell lines SW480, SW620, SW837, HCT-116, HT-29, HCA-7, and A549 lung cancer cells were obtained from the American Tissue Type Culture Collection (ATCC). HT-29 and HCT-116 were maintained in McCoy's 5A from Cellgro (Herndon, VA) and cultured at 37°C and 5% CO2. HCA-7 and A549 were maintained in Dulbecco's Modified Eagle's Medium (DMEM) from Cellgro and cultured at 37°C and 5% CO2. SW837, SW480 and SW620 were maintained in Leibovitz's L-15 from ATCC (Manassas, VA) and cultured at 37°C without CO2 as instructed by the ATCC. All media were supplemented with 10% FBS from Gemini Bio-Products (Sacramento, CA) and 1x Penicillin-Streptomycin-Glutamine from Gibco (Grand Island, NY). Following treatments, cells were harvested and lysed in lysis buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1% NP-40, 20% SDS) supplemented with Protease Inhibitor Cocktail (Sigma-Aldrich), 0.4 mM PMSF (Sigma-Aldrich), 1 mM sodium orthovanadate (Sigma-Aldrich), 1 mM sodium fluoride (Sigma-Aldrich), and 1 mM sodium phosphate (Sigma-Aldrich). A 40 μg quantity of protein (quantified using Bradford Reagent from Bio-Rad, Hercules, CA) were loaded onto 10% NuPage gels from Invitrogen (Carlsbad, CA). The proteins were electrophoretically transferred onto PVDF membranes (PerkinElmer, Waltham, MA). The membranes were blocked and incubated with primary antibodies according to the product instructions sheet. Proteins were visualized by ECL reagents from Perkin-Elmer, and exposed to HyBlot CL films from Denville Scientific (Metuchen, NJ).
PGE2 production
Cells were seeded in 6-well plates and incubated overnight in DMEM/ 10% FBS. They were serum starved for the next 18 h. Cells were then treated with 10 ng/ml IL-1β and increasing concentration of compounds (dissolved in DMSO) in 1 ml serum-free medium. After 72 h incubation, the supernatants were collected for PGE2 level detection using the PGE2 EIA kit (R&D Systems).
PGE2 de novo synthesis assay
The assay was performed as described in reference [40] with some modification. Non-stimulated HCA-7 or A549 stimulated with IL-1β for 24 h were seeded in the 6-well plates and incubated overnight. Cells were then treated with serum-free DMEM containing vehicle (1% DMSO) or compounds dissolved in the same vehicle (i.e., 1% DMSO) for 2 h, and with 10 μM arachidonic acid (Cayman Chemical) for another 10 min, at 37°C and 5% CO2. The PGE2 and 6-keto PGF1α levels in the conditioned media were then determined using the respective EIA kits (from R&D Systems and Cayman Chemical).
mPGES-1, COX-2, and SPHK-1 cell free assays
In vitro mPGES-1 activity assay was performed following protocols below. Briefly, mPGES-1 recombinant protein (purchased from Cayman Chemical or purified as described above) or the membrane fraction of IL-1β-stimulated A549 [30] was diluted in a reaction buffer containing 0.1 M sodium phosphate buffer, pH 7.4, 0.3% TritonX-100, 1 mM EDTA, and 2.5 mM GSH. Compounds were then added to the solution to a final concentration of 20 μM. After 2 h incubation at room temperature, the reaction was started by adding cold PGH2 to a final concentration of 10 μM. The reaction was terminated immediately after 1.5 min by stop solution consisting of 20 mM FeCl2. Solution in each sample was diluted 30 times for measurement of PGE2 concentrations by an EIA kit (R&D Systems). Sphingosine kinase-1 (SPHK-1) activity assay was measured using a SPHK-1 inhibitor screening assay kit (Cayman Chemical) following manufacturer's instructions. COX-2 activity was measured by a COX Fluorescent inhibitor screen assay kit (Cayman Chemical) following the manufacturer's instructions. In both assays, 5 μM of PGE0001 was tested.
Anti-tumor activity
Approximately 1×106 SW837 rectal cancer cells or A549 lung cancer cells in log cell growth were resuspended in 0.1 ml phosphate buffered saline and injected subcutaneously (s.c.) into the flanks of female severe combined immunodeficient (SCID) mice. When the tumors reached volumes 100–150 mm3, the mice were stratified into groups of 8 animals having approximately equal mean tumor volumes and administration of compound PGE0001 suspended in 0.1% Tween-20 in water was started at a dose of 200 mg/kg (for SW837) or 100 mg/kg (for A549) i.p. daily for 5 days. The animals were weighed weekly and tumor diameters measured twice weekly at right angles (dshort and dlong) with electronic calipers and converted to volume by the formula volume = (dshort)2 × (dlong)/2 [41]. When the tumor volume reached ≥2,000 mm3 or became necrotic, the animals were euthanized. Anti-tumor effects are presented as %T/C (treatment-to-control ratio), where T and C represent the means of tumor volumes of the treatment and control mice, respectively [42].
Pharmacokinetic and pharmacodynamic studies
All studies involving animals were conducted in accordance with U.S. Public Health Service/U.S. Department of Agriculture guidelines and experimental protocols were approved by The University of Arizona Institutional Animal Care and Use Committee (IACUC). For pharmacokinetic studies, C57BL/6 mice received a single i.p. dose of compound PGE0001 at 200 mg/kg suspended in 0.1% Tween-20 in 0.9% NaCl. Mice were sacrificed after 30 min, 1, 2, 4, 6, 8, 14 and 24 h, blood was collected into heparinized tubes, and plasma was stored frozen at -80°C. Plasma levels of compound PGE0001 were measured by reverse-phase high-pressure liquid chromatography. For pharmacodynamic studies, SCID mice received a single i.p. dose of compound PGE0001 of 100 mg/kg. Mice were killed after 30 min, 1, 3, 5 or 24 h; blood was collected and indomethacin (Cayman Chemical) was added to the collection tubes immediately after drawn (final concentration 10 μg/ml). Serum was then collected and stored frozen at -80°C. Serum levels of PGE2 were measured by an enzyme immunoassay.
Statistical analyses
Data are presented as mean±S.D. Statistical analyses (Student's two-tailed t-tests) were performed using Stata software (Stata Corporation, College Station, TX).
Results
Discovery of an aminothiazole scaffold that binds to mPGES-1
We performed an in silico screen on the ~312,410 compound ChemBridge diversity library to identify novel mPGES-1 inhibitors. From the database searches and molecular docking, 13 compounds were identified as “potential hits” (Table 1). These hits were ranked according to their FlexiDock scores. These scores can be correlated to the binding of these compounds to the protein target (Table 1). Indeed, equilibrium dissociation constant KD of each compound was measured using expressed 6xHis-tagged mPGES-1 in E. coli and SPR spectroscopy. Among the 13 hits, 7 compounds presented the aminothiazole scaffold. They are 5807166 (#2 in Table 1), PGE0001 (#3), 5724933 (#4), 5935487 (#5), 7882458 (#6), 5662444 (#7), and 5933870 (#12). However, only PGE0001 and #7 were able to bind directly to mPGES-1 as revealed by their low KD.
Table 1.
Structures, calculated properties, docking scores and biological activities of initial hits
| Compound | ChemBridge ID | Structure | LogDa | FlexiDock scoreb (mPGES-1) | KDc (μM) | Relative PGE2 leveld (%) | FlexiDock scoreb (COX-2) | % COX-2 activitye |
|---|---|---|---|---|---|---|---|---|
| Triclosan | NA |
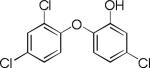
|
5.2 | –18.7 | 361.3±64.5 | 41.7±1.9 | ND†† | 97.0±19.3 |
| 1 | 5862295 |
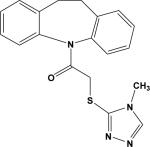
|
3.15 | –54.5 | NB† | 103.3±12.2 | ND | ND |
| 2 | 5807166 |
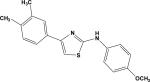
|
4.64 | –47.0 | NB | 22.4±5.2 | –49.9 | 30.7±5.4 |
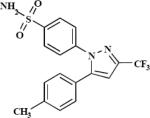
| 3 (PGE0001) | 5654455 |
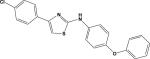
|
5.95 | –45.2 | 21.3±7.8 | 24.5±7.3 | –40.9 | 67.9±5.1 |
| 4 | 5724933 |
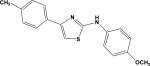
|
4.18 | –45.0 | NB | 41.2±30.5 | –41.4 | 37.6±11.7 |
| 5 | 5935487 |
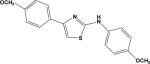
|
3.45 | –44.5 | NB | 19.3±10.1 | –41.1 | 33.2±3.2 |
| 6 | 7882458 |
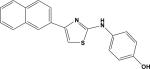
|
4.25 | –42.0 | 105.6±72.1 | 110.0±3.0 | –42.6 | 21.7±2.9 |
| 7 | 5662444 |
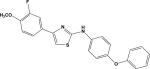
|
5.22 | –37.1 | 22.8±5.8 | 40.3±13.7 | –42.7 | 67.4±3.6 |
| 8 | 6239316 |
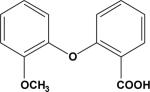
|
0.19 | –36.4 | NB | 92.6±3.5 | ND | ND |
| 9 | 6795274 |
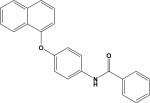
|
5.21 | –34.0 | NB | 40.4±16.6 | ND | ND |
| 10 | 7786927 |
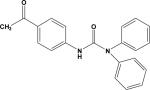
|
3.13 | –30.8 | NB | 103.4±1.8 | ND | ND |
| 11 | 7418129 |
|
3.82 | –27.8 | NB | 86.6±8.0 | ND | ND |
| 12 | 5933870 |
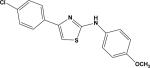
|
4.31 | –26.8 | NB | 104.4±21.1 | –34.7 | 67.9±14.8 |
| 13 | 7384071 |
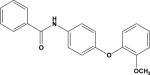
|
3.38 | –25.5 | NB | 94.3±11.0 | ND | ND |
| Celecoxib | NA |
|
ND | ND | ND | ND | –46.3 | 11.8±2.8 |
Calculated using ACDLabs 12.0.
Calculated using Sybyl 8.0 Tripos Inc. St. Louis. MS.
Equilibrium dissociation constants (KDs) were calculated using a 1:1 Langmuir model.
PGE2 production from IL-1β stimulated HCA-7 cells was measured using an EIA kit. 100% was reported for vehicle control. Compounds were tested at 1 μM.
COX-2 activity was measured as described in the Materials and methods. 100% of COX-2 was established in DMSO control.
No binding (NB), No response or KD>500 μM.
ND, not determined.
Representative dose response curves are shown in Fig. 1c for PGE0001 to recombinant mPGES-1 (KD =21.3±7.8 μM with an on rate of Ka×10–3=1.2±0.5 M–1s–1, and an off rate of Kd×10–3=25.8±4.8 s–1). These curves demonstrated a slow “on” and slow “off” rate binding pattern of the compound to mPGES-1. Molecular modeling of aminothiazole compounds with the protein showed extended interactions with the active site residues of mPGES-1. Thus, from the modeling studies, it is postulated that compound PGE0001 interacts in an extended conformation with the protein (Fig. 1a). The thiazole group appears well positioned for favorable interactions with the side chain of His72 of the protein, while the phenoxy ring extended into the active site near the Arg70 residue of the protein. More precisely, the oxygen atom of the phenoxy group (hydroxyl group) interacts with Arg70 and the amino group interacts with Glu77 via hydrogen bonding interactions (Fig. 1b).
Fig. 1.

Interaction of PGE0001 with mPGES-1. (a) Interaction model for PGE0001 interacting with the active site of human mPGES-1. (b) Schematics of the key interactions of PGE0001 with selected active site residues in the protein. The mPGES-1 protein is displayed as MOLCAD surface (colored by potential) and red-colored ribbon. PGE0001 is shown as atom-colored capped sticks. (c) SPR sensograms of PGE0001. Representative dose response curves obtained using SPR (Biacore 2000) with increasing concentrations (0, 1, 5, 10, 15, and 20 μM) of compound PGE0001 (lowest at the bottom and highest at the top). The recombinant 6xHis- human mPGES-1 was loaded on a CM5 chip and the compound was flowed through at a rate 50 μg/min.
Biological activities of the compounds in cancer cells
In order to determine the effect of the 13 compounds on PGE2 production, HCA-7 colon cancer cells which express both COX-2 and mPGES-1 [18] were treated with the compounds at 1 μM and stimulated with IL-1β. The relative PGE2 levels in the culture media in comparison with vehicle (DMSO) control are listed in Table 1. Out of 13 compounds identified, 6 compounds (PGE0001, #2, #4, #5, #7, and #9) inhibited IL-1β induced PGE2 production by more than 50%. However, the PGE2 reduction activity of #2, #4, and #5 was attributed to COX-2 inhibition. Indeed, these compounds were tested for COX-2 inhibition in vitro (Table 1). Compound # 6 inhibited COX-2 enzyme activity by ~78% at 5 μM. However, other aminothiazoles showed only 30–60% inhibition of COX-2 at 5 μM.
Both PGE0001 and #7, showed low KD value, reduced IL-1β induced PGE2 production by ~80% and ~60%, respectively and only inhibited COX-2 activity by ~30%, which may represent an insignificant effect on COX-2. Hence, PGE0001 and #7 were identified as our selective lead molecules. In order to further characterize PGE0001 in colon and lung cancer cells, we measured the effects of increasing concentrations of the compound on cytokine-induced PGE2 production. In colon cancer cells lines (HCA-7 and HT-29) and A549 lung cancer cells, mPGES-1 expression is induced by IL-1β (Fig. 2a and [43]). On the contrary, SW837 cells were shown to express high levels of mPGES-1 constitutively (Fig. 3a). The treatment of the cells with increasing concentrations of PGE0001 decreased IL-1β-induced PGE2 production in HCA-7 and A549 cells in a dose dependent manner with an EC50=0.29±0.08 μM and EC50=0.32±0.09 μM, respectively (Fig. 2b). PGE0001 also exhibited similar activity in two other colorectal cancer cell lines SW837 (EC50=0.76±0.14 μM) and in HT-29 (EC50=0.87±0.39 μM) (data not shown). Celecoxib (COX-2 inhibitor) and MK-886 (dual inhibitor of mPGES-1 and 5-lipoxygenase activating protein) were tested at 1 μM and used as comparable controls. The overall levels of COX-2 and mPGES-1 expression did not change in the presence of the compounds (data not shown), hence it was concluded that the changes in PGE2 production were due to the overall inhibition of the pathway.
Fig. 2.

Effects of PGE0001 on PGE2 production in cancer cells. (a) HCA-7 colon cancer cells (left) and A549 lung cancer cells (right) were stimulated with IL-1β (+) or non-stimulated (– for control). COX-2 and mPGES-1 induction were detected by Western blotting using specific antibodies. β-actin was used as loading control. (b) Left panel: HCA-7 cells were treated as described above and incubated for 72 hrs with increasing concentrations of PGE0001 (0.03, 0.1, 0.3, 1, and 3 μM), Celecoxib (1 μM), or MK-886 (1 μM). The release of PGE2 in the culture media was measured using an enzyme immunoassay kit for PGE2 detection. Right panel: A549 lung cancer cells were stimulated with IL-1β, incubated for 48 h with increasing concentrations of PGE0001 (0.1, 0.5, 1, and 10 μM), Celecoxib (1 μM) or MK-886 (1 μM). PGE2 was measured as described. Values are relative and are the means of at least 3 determinations. * p<0.05; ** p<0.01 (Student's t-tests) compared to control stimulated with IL-1β. (c) HCA-7 cells (left panel) or IL-1β-stimulated A549 cells (right panel) were pre-treated with PGE0001 (10 μM), Celecoxib (10 μM), MK-886 (10 μM) or DMSO as vehicle control for 2 h. The cells were then treated with AA and the media were collected for PGE2 and 6-keto PGF1α measurements as described in the Materials and Methods section. Values are relative and are the means of at least 3 determinations ± SD. Statistical analysis (Student's t-tests) for PGE2 levels: * p<0.05; ** p<0.01 compared to the vehicle control. Statistical analysis (Student's t-tests) for 6-keto PGF1α levels: # p<0.05; ## p<0.01 compared to the vehicle control.
Fig. 3.

In vivo effects of PGE0001. (a) Colorectal cancer cell lines and A549 lung cancer cells were analyzed for the expression of COX-2, mPGES-1, mPGES-2 and cPGES by Western blotting using specific antibodies. β-actin was used as loading control. Note that only SW837 exhibited high constitutively expressed mPGES-1 levels as compared to all other cell lines tested. (b) SCID mice were s.c. inoculated in the right flank with 1 × 106 SW837 rectal cancer cells. When the average tumor volume reached ~150 mm3, mice were pair matched (8 mice/ group) and injected i.p. with PGE0001 at a dose of 200 mg/kg for 5 days. The arrow at day 22 represents the first day of injection. (c) SCID mice were inoculated s.c. with 1 × 106 A549 cells and were injected i.p. with PGE0001 (100 mg/kg) for 5 days. The arrows at day 21 and day 41 indicate the first day of injection for each treatment cycle. Values are means and error bars are SE. * p<0.1, ** p<0.05 (Student's t-tests). (d) Pharmacokinetics of compound PGE0001 in mice. C57BL/6 mice were i.p. administered compound PGE0001 at 200 mg/kg. Values are means of 3 mice per time point and error bars are SE. (e) Pharmacodynamics of compound PGE0001 in mice. SCID mice were i.p. administered compound PGE0001 at 100 mg/kg. Values are means of 2 mice per time point and error bars are SD. Serum PGE2 levels were measured by an enzyme immunoassay.
In order to distinguish COX-2 inhibition versus PGE2 synthase inhibition, we performed a de novo PGE2 synthesis assay as described recently by Mbalaviele et al. [40]. Cells were pre-treated with compounds, and then induced with arachidonic acid for 10 min. PGE2 and 6-keto PGF1α (metabolite of PGI2) levels were measured in the media using separate EIA kits (Fig. 2c). In resting HCA-7 cells (left panel, Fig. 2c) or IL-1β-stimulated A549 cells (right panel, Fig. 2c) cells, Celecoxib inhibited both PGE2 and 6-keto PGF1α de novo synthesis, whereas MK-886 reduced PGE2 specifically. A mixed effect of PGE0001 was observed and neither PGE2 nor 6-keto PGF1α was greatly decreased. Finally, PGE0001 slightly inhibited cellular proliferation of colon cancer cell lines as measured by a MTT assay and caused ~20% of apoptosis with 20 μM of PGE0001 as measured by an acridine-orange stain. PGE0001 induced PARP cleavage as well in these cells (data not shown).
Effects of PGE0001 on tumor growth
The effects of PGE0001 were evaluated on xenografts mouse tumor growth. SW837 rectal cancer cell line has a constitutive over-expression of mPGES-1 reflecting at best clinical observations where mPGES-1 is constitutively over-expressed in more than 80% of human colorectal cancers [44] (Fig. 3a–c). Mice were inoculated with 1×106 SW837 cells subcutaneously in the right flank. When the average tumor volume reached ~150 mm3, mice were randomly pair-matched into 8 mice per group: a control group and a group where the mice received PGE0001 (200 mg/kg i.p. for 5 days). Preliminary studies showed no toxicity of single doses up to 200 mg/kg, which was the maximum dose for compound PGE0001 that could be conveniently administered i.p. The results of this experiment are summarized in Table 2 and Fig. 3b. Compound PGE0001 exhibited a significant anti-tumor activity in SW837 xenografts with T/C 39.9 % (p<0.05). We also tested the anti-tumor effect of PGE0001 in the A549 xenograft mouse model where mice were treated with 2 cycles of PGE0001 (100 mg/kg i.p. for 5 days). As shown in Table 2 and Fig. 3c, the tumor burden of the PGE0001-treated group was lower. The T/C was 37.9 % (p<0.05). Significance was achieved for the compound after the second cycle of drug treatment until the end of the experiment as compared to controls. Finally, the compound also exhibited anti-tumor activity in HCA-7 colon cancer xenografts at 200 mg/kg for 5 days (Table 2). However, the significance was only achieved right after the treatment period ended and tumor growth resumed at its original rate when the drug was removed.
Table 2.
Antitumor activity of PGE0001
| Tumora | Doseb (mg/kg) | Volume at start (mm3) | Number of days of growth | Growth rate (mm3/7 days1, 10 days2, or 15 days3) | T/C (%) | p valuec |
|---|---|---|---|---|---|---|
| HCA-7 colon | Controld | 132 | 7 | 104±421 | ||
| 200 | 7 | 63±191 | 60.8 | 0.039 | ||
| SW837 rectum | Controld | 144 | 15 | 118±353 | ||
| 200 | 15 | 47±423 | 39.9 | 0.043 | ||
| A549 lung | Controld | 144 | 15 | 453±2823 | ||
| 100 | 15 | 385±1993 | 85.1 | 0.707 | ||
| Controld,e | 10 | 850±3862 | ||||
| 100 | 10 | 322±1442 | 37.9 | 0.043 |
8 mice per group.
SID for 5 days.
Compared to vehicle control.
Control received vehicle only (0.1 ml).
Animals were given a second regimen of 5 days.
Early pharmacokinetic studies showed that plasma levels of PGE0001 following i.p. administration to mice at a dose of 200 mg/kg was best described by a two compartment open model (Fig. 3d). Absorption was rapid, without a lag phase and Cmax was 5.6 μg/ml was reached within 1 h following dosing. PGE0001 terminal half-life was 7.7 h and plasma clearance was 6.6 l/h/kg with a terminal concentration of 0.1 μg/ml 24 h after dosing. The concentration was calculated to be higher than the EC50 for PGE2 reduction in cells. In order to determine the effect of the compound PGE0001 on blood PGE2 level, mice were injected i.p. with a single dose of PGE0001 (100 mg/kg) and serum samples were collected at different time points for PGE2 measurements. This dose produced up to 70% inhibition at 1 and 5 h with almost a return to untreated levels by 24 h (Fig. 3e). These results correlated well with the plasma concentrations of PGE0001 after the single dose. Taken together, in vivo studies demonstrated that PGE0001 reduced serum PGE2 and exhibited good anti-tumor activity.
Mechanism of action for PGE0001
In order to fully define the mechanism of action for PGE0001, we tested the effects of the compound on mPGES-1, COX-2 and sphingosine kinase-1 (SPHK-1) using cell-free assays (Fig. 4). Surprisingly, we demonstrated that 20 μM of PGE0001 did not inhibit mPGES-1 (regardless of the protein source) and that 5 μM of PGE0001 did not affect the activity of COX-2 or SPHK-1 in vitro (Fig. 4). Fig. 4a represents the activity of recombinant human mPGES-1 from Cayman Chemical. Similar results were obtained using our recombinant human mPGES-1 (expressed by E. coli, data not shown) as well as membrane preparation from A549 cells (data not shown) according to the published protocol [30]. PGE0001 reduced COX-2 activity by ~30% at 5 μM which appears much less significant when compared to Celecoxib, which produced a ~90% inhibition of the activity at the same concentration (Fig. 4b). PGE0001 did not affect SPHK-1 activity (Fig. 4c).
Fig. 4.

In vitro effects of PGE0001. (a) mPGES-1 activity was measured as described in the Materials and methods section. PGE0001 and MK-886 (20 μM) were pre-incubated with the enzyme and the remaining activity of mPGES-1 as ng/ml of PGE2 produced was measured using an EIA kit. (b) COX-2 activity was measured as described. PGE0001, Celecoxib and MK-886 (5 μM) were pre-incubated with the enzyme and the remaining relative COX-2 enzymatic activity was measured according to the manufacturer's instructions. Control activity (in the presence of DMSO) was reported at 100%. (c) SPHK-1 activity was measured as described. SPHK-1 inhibitor and PGE0001 (5 μM) were pre-incubated with the enzyme and the remaining relative enzymatic activity was measured. * p<0.05; ** p<0.01 (Student's t-tests).
Discussion
Although there is accumulated evidence supporting the role of mPGES-1 in carcinogenesis [25,28], the effect of mPGES-1 deletion on tumorigenesis, at least in gastrointestinal cancer, is still controversial [45,25]. Pre-clinical tests for mPGES-1 inhibitors have been limited to models of inflammation and pain [46,47]. To the best of our knowledge, no information about an in vivo anti-tumor activity of mPGES-1 inhibitor has been published. In this study, we used docking models to evaluate the compounds for their interaction with mPGES-1 active site. The FlexiDock scores showed good correlation with KD values (for binding to mPGES-1) measured using SPR technology. Compounds #8, #10, #11, #12, and #13 were hypothesized to bind poorly to mPGES-1 active site were subsequently determined not to bind the protein. These compounds did not inhibit PGE2 production in the cells. Interestingly, #2, #4, #5 and #6 were predicted to bind better to mPGES-1 but did not bind the target. These compounds reduced PGE2 production and were subsequently shown to inhibit between ~60 to 80% the activity of COX-2 in a cell free assay. Compound #6 inhibited strongly COX-2 but was not able to reduce PGE2 production probably due to a poor bioavailability or stability in the cells. Thus, molecular modeling combined with SPR allowed us to focus on PGE0001 and #7, which exhibited a good KD value and the ability to decrease 60 to 80% of PGE2 production without affecting COX-2 activity as measured in the cell free assay and the de novo PGE2 synthesis assay. Indeed, no reduction of 6-keto PGF1α was observed in the presence of PGE0001.
PGE0001 exhibited promising anti-tumor activity. PGE2 production was also inhibited in vivo after a single dose of 100 mg/kg of PGE0001. Maximum inhibition was observed between 30 min to 5 h after administration of PGE0001, with the timing corresponding to its peak plasma concentration. There was also anti-tumor activity with complete cessation of tumor growth and even some regression following the administration of PGE0001 in SW837 colorectal xenografts. Absorption, metabolism, distribution and elimination properties of PGE0001 were predicted that low cardiotoxicity may be expected as evaluated by hERG channel activity (data not shown). Additionally, physicochemical properties such as polar surface area (PSA) and logD were calculated as indicators of cellular permeability and solubility respectively. The values were: tPSA: 33.62 and logD: 5.95, suggesting a high likelihood of the compound passively diffusing into cells and permeating the small intestine cell wall. The compound also has high logD which may indicate poor water solubility. However observations solubility was sufficient in water for accurate biological evaluation. Noteworthy, the compound was well tolerated in animals up to the dose of 200 mg/kg, where a significant anti-tumor activity was observed.
Surprisingly, PGE0001 did not inhibit COX-2 nor mPGES-1 activity in vitro sufficiently enough to explain the reduction of cellular PGE2 production in cancer cells and the in vivo anti-tumor effect observed in mouse xenografts. Interestingly, the chemical structure of PGE0001 is strangely similar to a compound known reported as a SPHK-1 inhibitor [48]. Sphingosine-1 phosphate (S1P), the product of SPHK-1, has been shown to induce COX-2 expression [49-51]. Therefore, the measured PGE2 reduction effect of PGE0001 in cancer cells could also result from the inhibition of SPHK-1. However, PGE0001 did not inhibit SPHK-1 activity in vitro when tested in a cell free assay. Among other possible off-targets that would affect PGE2 biosynthesis, we have identified two kinases as potential targets. These alternative mechanisms are currently under investigation.
In conclusion, PGE0001 showed binding to the expressed mPGES-1 protein and exhibited in vitro PGE2 reduction in colorectal and lung cancer cell lines. Although the mechanism of action of such compound may remain to be clarified, its promising anti-tumor activity made it a worthwhile compound to study. Derivatization of one of the aminothiazole compounds may lead to the identification of a novel generation of active small molecules that may represent a good starting point and chemical probe for future anti-cancer studies.
Acknowledgments
This work was supported by the following grants to EJM: American Cancer Society Institutional Research Grant (ACS-IRG); Arizona Cancer Center Specialized Program of Research Excellence (SPORE) pilot grant; and a National Institutes of Health National Cancer Institute R01 grant [CA138702] to EJM. The authors would like to thank the Experimental Mouse Shared Service (EMSS) at the Arizona Cancer Center as well as at MD Anderson Cancer Center for help provided with the animal experiments.
Abbreviations
- AA
arachidonic acid
- COX-2
cyclooxygenase-2
- DMSO
dimethylsulfoxide
- FLAP
5-lipoxygenase-activating protein
- GSH
glutathione
- IL-1β
interleukin-1 beta
- PARP
poly (ADP-ribose) polymerase
- PGES
prostaglandin E2 synthase
- PGE2
prostaglandin E2
- PGH2
prostaglandin H2
- PLA2
phospholipase A2
- SPR
surface plasmon resonance
- SCID
severe combined immunodeficiency disease
- TNF-α
tumor necrosis factor-alpha
Footnotes
Conflict of Interest
The authors declare that they have no conflict of interest.
Contributor Information
Hui-Hua Chang, Arizona Cancer Center The University of Arizona 1515 N. Campbell Ave. Tucson, AZ 85724, USA; Department of Nutritional Sciences The University of Arizona Tucson, AZ 85721, USA.
Zuohe Song, Arizona Cancer Center The University of Arizona 1515 N. Campbell Ave. Tucson, AZ 85724, USA; Department of Nutritional Sciences The University of Arizona Tucson, AZ 85721, USA.
Lee Wisner, Arizona Cancer Center The University of Arizona 1515 N. Campbell Ave. Tucson, AZ 85724, USA.
Tina Tripp, Department of Nutritional Sciences The University of Arizona Tucson, AZ 85721, USA.
Vijay Gokhale, Department of Pharmacology and Toxicology The University of Arizona Tucson, AZ 85721.
Emmanuelle J. Meuillet, Arizona Cancer Center The University of Arizona 1515 N. Campbell Ave. Tucson, AZ 85724, USA Department of Nutritional Sciences The University of Arizona Tucson, AZ 85721, USA; Department of Molecular and Cellular Biology The University of Arizona Tucson, AZ 85721.
References
- 1.Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294(5548):1871–1875. doi: 10.1126/science.294.5548.1871. doi:10.1126/science.294.5548.1871 294/5548/1871 [pii] [DOI] [PubMed] [Google Scholar]
- 2.Legler DF, Bruckner M, Uetz-von Allmen E, Krause P. Prostaglandin E2 at new glance: novel insights in functional diversity offer therapeutic chances. Int J Biochem Cell Biol. 2010;42(2):198–201. doi: 10.1016/j.biocel.2009.09.015. doi:S1357-2725(09)00271-4 [pii] 10.1016/j.biocel.2009.09.015. [DOI] [PubMed] [Google Scholar]
- 3.Bingham CO, 3rd, Austen KF. Phospholipase A2 enzymes in eicosanoid generation. Proc Assoc Am Physicians. 1999;111(6):516–524. doi: 10.1046/j.1525-1381.1999.99321.x. [DOI] [PubMed] [Google Scholar]
- 4.Pugh S, Thomas GA. Patients with adenomatous polyps and carcinomas have increased colonic mucosal prostaglandin E2. Gut. 1994;35(5):675–678. doi: 10.1136/gut.35.5.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Williams CS, Smalley W, DuBois RN. Aspirin use and potential mechanisms for colorectal cancer prevention. J Clin Invest. 1997;100(6):1325–1329. doi: 10.1172/JCI119651. doi:10.1172/JCI119651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldman AP, Williams CS, Sheng H, Lamps LW, Williams VP, Pairet M, Morrow JD, DuBois RN. Meloxicam inhibits the growth of colorectal cancer cells. Carcinogenesis. 1998;19(12):2195–2199. doi: 10.1093/carcin/19.12.2195. [DOI] [PubMed] [Google Scholar]
- 7.Rahme E, Barkun AN, Toubouti Y, Bardou M. The cyclooxygenase-2-selective inhibitors rofecoxib and celecoxib prevent colorectal neoplasia occurrence and recurrence. Gastroenterology. 2003;125(2):404–412. doi: 10.1016/s0016-5085(03)00880-1. doi:S0016508503008801 [pii] [DOI] [PubMed] [Google Scholar]
- 8.Bresalier RS, Sandler RS, Quan H, Bolognese JA, Oxenius B, Horgan K, Lines C, Riddell R, Morton D, Lanas A, Konstam MA, Baron JA. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med. 2005;352(11):1092–1102. doi: 10.1056/NEJMoa050493. doi:NEJMoa050493 [pii] 10.1056/NEJMoa050493. [DOI] [PubMed] [Google Scholar]
- 9.Solomon SD, McMurray JJ, Pfeffer MA, Wittes J, Fowler R, Finn P, Anderson WF, Zauber A, Hawk E, Bertagnolli M. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N Engl J Med. 2005;352(11):1071–1080. doi: 10.1056/NEJMoa050405. doi:NEJMoa050405 [pii] 10.1056/NEJMoa050405. [DOI] [PubMed] [Google Scholar]
- 10.Samuelsson B, Morgenstern R, Jakobsson PJ. Membrane prostaglandin E synthase-1: a novel therapeutic target. Pharmacol Rev. 2007;59(3):207–224. doi: 10.1124/pr.59.3.1. doi:59/3/207 [pii] 10.1124/pr.59.3.1. [DOI] [PubMed] [Google Scholar]
- 11.Friesen RW, Mancini JA. Microsomal prostaglandin E2 synthase-1 (mPGES-1): a novel anti-inflammatory therapeutic target. J Med Chem. 2008;51(14):4059–4067. doi: 10.1021/jm800197b. doi:10.1021/jm800197b. [DOI] [PubMed] [Google Scholar]
- 12.Jakobsson PJ, Thoren S, Morgenstern R, Samuelsson B. Identification of human prostaglandin E synthase: a microsomal, glutathione-dependent, inducible enzyme, constituting a potential novel drug target. Proc Natl Acad Sci U S A. 1999;96(13):7220–7225. doi: 10.1073/pnas.96.13.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Watanabe K, Kurihara K, Suzuki T. Purification and characterization of membrane-bound prostaglandin E synthase from bovine heart. Biochim Biophys Acta. 1999;1439(3):406–414. doi: 10.1016/s1388-1981(99)00084-0. doi:S1388-1981(99)00084-0 [pii] [DOI] [PubMed] [Google Scholar]
- 14.Tanioka T, Nakatani Y, Semmyo N, Murakami M, Kudo I. Molecular identification of cytosolic prostaglandin E2 synthase that is functionally coupled with cyclooxygenase-1 in immediate prostaglandin E2 biosynthesis. J Biol Chem. 2000;275(42):32775–32782. doi: 10.1074/jbc.M003504200. doi:10.1074/jbc.M003504200 M003504200 [pii] [DOI] [PubMed] [Google Scholar]
- 15.Jakobsson PJ, Thoren S, Morgenstern R, Samuelsson B. Characterization of microsomal, glutathione dependent prostaglandin E synthase. Adv Exp Med Biol. 2002;507:287–291. doi: 10.1007/978-1-4615-0193-0_44. [DOI] [PubMed] [Google Scholar]
- 16.Yoshimatsu K, Altorki NK, Golijanin D, Zhang F, Jakobsson PJ, Dannenberg AJ, Subbaramaiah K. Inducible prostaglandin E synthase is overexpressed in non-small cell lung cancer. Clin Cancer Res. 2001;7(9):2669–2674. [PubMed] [Google Scholar]
- 17.Yoshimatsu K, Golijanin D, Paty PB, Soslow RA, Jakobsson PJ, DeLellis RA, Subbaramaiah K, Dannenberg AJ. Inducible microsomal prostaglandin E synthase is overexpressed in colorectal adenomas and cancer. Clin Cancer Res. 2001;7(12):3971–3976. [PubMed] [Google Scholar]
- 18.Kamei D, Murakami M, Nakatani Y, Ishikawa Y, Ishii T, Kudo I. Potential role of microsomal prostaglandin E synthase-1 in tumorigenesis. J Biol Chem. 2003;278(21):19396–19405. doi: 10.1074/jbc.M213290200. doi:10.1074/jbc.M213290200 M213290200 [pii] [DOI] [PubMed] [Google Scholar]
- 19.van Rees BP, Sivula A, Thoren S, Yokozaki H, Jakobsson PJ, Offerhaus GJ, Ristimaki A. Expression of microsomal prostaglandin E synthase-1 in intestinal type gastric adenocarcinoma and in gastric cancer cell lines. Int J Cancer. 2003;107(4):551–556. doi: 10.1002/ijc.11422. doi:10.1002/ijc.11422. [DOI] [PubMed] [Google Scholar]
- 20.Mehrotra S, Morimiya A, Agarwal B, Konger R, Badve S. Microsomal prostaglandin E2 synthase-1 in breast cancer: a potential target for therapy. J Pathol. 2006;208(3):356–363. doi: 10.1002/path.1907. doi:10.1002/path.1907. [DOI] [PubMed] [Google Scholar]
- 21.Rask K, Zhu Y, Wang W, Hedin L, Sundfeldt K. Ovarian epithelial cancer: a role for PGE2-synthesis and signalling in malignant transformation and progression. Mol Cancer. 2006;5:62. doi: 10.1186/1476-4598-5-62. doi:1476-4598-5-62 [pii] 10.1186/1476-4598-5-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hasan S, Satake M, Dawson DW, Funahashi H, Angst E, Go VL, Reber HA, Hines OJ, Eibl G. Expression analysis of the prostaglandin E2 production pathway in human pancreatic cancers. Pancreas. 2008;37(2):121–127. doi: 10.1097/MPA.0b013e31816618ba. doi:10.1097/MPA.0b013e31816618ba 00006676-200808000-00001 [pii] [DOI] [PubMed] [Google Scholar]
- 23.Hanaka H, Pawelzik SC, Johnsen JI, Rakonjac M, Terawaki K, Rasmuson A, Sveinbjornsson B, Schumacher MC, Hamberg M, Samuelsson B, Jakobsson PJ, Kogner P, Radmark O. Microsomal prostaglandin E synthase 1 determines tumor growth in vivo of prostate and lung cancer cells. Proc Natl Acad Sci U S A. 2009;106(44):18757–18762. doi: 10.1073/pnas.0910218106. doi:0910218106 [pii] 10.1073/pnas.0910218106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kamei D, Murakami M, Sasaki Y, Nakatani Y, Majima M, Ishikawa Y, Ishii T, Uematsu S, Akira S, Hara S, Kudo I. Microsomal prostaglandin E synthase-1 in both cancer cells and hosts contributes to tumour growth, invasion and metastasis. Biochem J. 2010;425(2):361–371. doi: 10.1042/BJ20090045. doi:BJ20090045 [pii] 10.1042/BJ20090045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakanishi M, Montrose DC, Clark P, Nambiar PR, Belinsky GS, Claffey KP, Xu D, Rosenberg DW. Genetic deletion of mPGES-1 suppresses intestinal tumorigenesis. Cancer Res. 2008;68(9):3251–3259. doi: 10.1158/0008-5472.CAN-07-6100. doi:68/9/3251 [pii] 10.1158/0008-5472.CAN-07-6100. [DOI] [PubMed] [Google Scholar]
- 26.Baron JA, Sandler RS, Bresalier RS, Lanas A, Morton DG, Riddell R, Iverson ER, Demets DL. Cardiovascular events associated with rofecoxib: final analysis of the APPROVe trial. Lancet. 2008;372(9651):1756–1764. doi: 10.1016/S0140-6736(08)61490-7. doi:S0140-6736(08)61490-7 [pii] 10.1016/S0140-6736(08)61490-7. [DOI] [PubMed] [Google Scholar]
- 27.Cheng Y, Wang M, Yu Y, Lawson J, Funk CD, Fitzgerald GA. Cyclooxygenases, microsomal prostaglandin E synthase-1, and cardiovascular function. J Clin Invest. 2006;116(5):1391–1399. doi: 10.1172/JCI27540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakanishi M, Gokhale V, Meuillet EJ, Rosenberg DW. mPGES-1 as a target for cancer suppression: A comprehensive invited review “Phospholipase A2 and lipid mediators”. Biochimie. 2010;92(6):660–664. doi: 10.1016/j.biochi.2010.02.006. doi:S0300-9084(10)00050-7 [pii] 10.1016/j.biochi.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Swingle KF, Moore GG, Grant TJ. 4-nitro-2-phenoxymethanesulfonanilide (R-805): a chemically novel anti-inflammatory agent. Arch Int Pharmacodyn Ther. 1976;221(1):132–139. [PubMed] [Google Scholar]
- 30.Thoren S, Jakobsson PJ. Coordinate up- and down-regulation of glutathione-dependent prostaglandin E synthase and cyclooxygenase-2 in A549 cells. Inhibition by NS-398 and leukotriene C4. Eur J Biochem. 2000;267(21):6428–6434. doi: 10.1046/j.1432-1327.2000.01735.x. doi:ejb1735 [pii] [DOI] [PubMed] [Google Scholar]
- 31.Arai I, Hamasaka Y, Futaki N, Takahashi S, Yoshikawa K, Higuchi S, Otomo S. Effect of NS-398, a new nonsteroidal anti-inflammatory agent, on gastric ulceration and acid secretion in rats. Res Commun Chem Pathol Pharmacol. 1993;81(3):259–270. [PubMed] [Google Scholar]
- 32.Futaki N, Yoshikawa K, Hamasaka Y, Arai I, Higuchi S, Iizuka H, Otomo S. NS-398, a novel non-steroidal anti-inflammatory drug with potent analgesic and antipyretic effects, which causes minimal stomach lesions. Gen Pharmacol. 1993;24(1):105–110. doi: 10.1016/0306-3623(93)90018-s. [DOI] [PubMed] [Google Scholar]
- 33.Riendeau D, Aspiotis R, Ethier D, Gareau Y, Grimm EL, Guay J, Guiral S, Juteau H, Mancini JA, Methot N, Rubin J, Friesen RW. Inhibitors of the inducible microsomal prostaglandin E2 synthase (mPGES-1) derived from MK-886. Bioorg Med Chem Lett. 2005;15(14):3352–3355. doi: 10.1016/j.bmcl.2005.05.027. [DOI] [PubMed] [Google Scholar]
- 34.Koeberle A, Siemoneit U, Buhring U, Northoff H, Laufer S, Albrecht W, Werz O. Licofelone suppresses prostaglandin E2 formation by interference with the inducible microsomal prostaglandin E2 synthase-1. J Pharmacol Exp Ther. 2008;326(3):975–982. doi: 10.1124/jpet.108.139444. [DOI] [PubMed] [Google Scholar]
- 35.Koeberle A, Zettl H, Greiner C, Wurglics M, Schubert-Zsilavecz M, Werz O. Pirinixic acid derivatives as novel dual inhibitors of microsomal prostaglandin E2 synthase-1 and 5-lipoxygenase. J Med Chem. 2008;51(24):8068–8076. doi: 10.1021/jm801085s. [DOI] [PubMed] [Google Scholar]
- 36.Koeberle A, Bauer J, Verhoff M, Hoffmann M, Northoff H, Werz O. Green tea epigallocatechin-3-gallate inhibits microsomal prostaglandin E(2) synthase-1. Biochem Biophys Res Commun. 2009;388(2):350–354. doi: 10.1016/j.bbrc.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 37.Koeberle A, Pollastro F, Northoff H, Werz O. Myrtucommulone, a natural acylphloroglucinol, inhibits microsomal prostaglandin E(2) synthase-1. Br J Pharmacol. 2009;156(6):952–961. doi: 10.1111/j.1476-5381.2009.00070.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thoren S, Weinander R, Saha S, Jegerschold C, Pettersson PL, Samuelsson B, Hebert H, Hamberg M, Morgenstern R, Jakobsson PJ. Human microsomal prostaglandin E synthase-1: purification, functional characterization, and projection structure determination. J Biol Chem. 2003;278(25):22199–22209. doi: 10.1074/jbc.M303227200. doi:10.1074/jbc.M303227200 M303227200 [pii] [DOI] [PubMed] [Google Scholar]
- 39.Mahadevan D, Powis G, Mash EA, George B, Gokhale VM, Zhang S, Shakalya K, Du-Cuny L, Berggren M, Ali MA, Jana U, Ihle N, Moses S, Franklin C, Narayan S, Shirahatti N, Meuillet EJ. Discovery of a novel class of AKT pleckstrin homology domain inhibitors. Mol Cancer Ther. 2008;7(9):2621–2632. doi: 10.1158/1535-7163.MCT-07-2276. doi:7/9/2621 [pii] 10.1158/1535-7163.MCT-07-2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mbalaviele G, Pauley AM, Shaffer AF, Zweifel BS, Mathialagan S, Mnich SJ, Nemirovskiy OV, Carter J, Gierse JK, Wang JL, Vazquez ML, Moore WM, Masferrer JL. Distinction of microsomal prostaglandin E synthase-1 (mPGES-1) inhibition from cyclooxygenase-2 inhibition in cells using a novel, selective mPGES-1 inhibitor. Biochem Pharmacol. 2010;79(10):1445–1454. doi: 10.1016/j.bcp.2010.01.003. doi:S0006-2952(10)00010-9 [pii] 10.1016/j.bcp.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 41.Paine-Murrieta GD, Taylor CW, Curtis RA, Lopez MH, Dorr RT, Johnson CS, Funk CY, Thompson F, Hersh EM. Human tumor models in the severe combined immune deficient (scid) mouse. Cancer Chemother Pharmacol. 1997;40(3):209–214. doi: 10.1007/s002800050648. [DOI] [PubMed] [Google Scholar]
- 42.Meuillet EJ, Zuohe S, Lemos R, Ihle N, Kingston J, Watkins R, Moses SA, Zhang S, Du-Cuny L, Herbst R, Jacoby JJ, Zhou LL, Ahad AM, Mash EA, Kirkpatrick DL, Powis G. Molecular pharmacology and antitumor activity of PHT-427, a novel Akt/phosphatidylinositide-dependent protein kinase 1 pleckstrin homology domain inhibitor. Mol Cancer Ther. 2010;9(3):706–717. doi: 10.1158/1535-7163.MCT-09-0985. doi:1535-7163.MCT-09-0985 [pii] 10.1158/1535-7163.MCT-09-0985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Terzuoli E, Donnini S, Giachetti A, Iniguez MA, Fresno M, Melillo G, Ziche M. Inhibition of hypoxia inducible factor-1alpha by dihydroxyphenylethanol, a product from olive oil, blocks microsomal prostaglandin-E synthase-1/vascular endothelial growth factor expression and reduces tumor angiogenesis. Clin Cancer Res. 2010;16(16):4207–4216. doi: 10.1158/1078-0432.CCR-10-0156. doi:1078-0432.CCR-10-0156 [pii] 10.1158/1078-0432.CCR-10-0156. [DOI] [PubMed] [Google Scholar]
- 44.Lim SC, Cho H, Lee TB, Choi CH, Min YD, Kim SS, Kim KJ. Impacts of cytosolic phospholipase A2, 15-prostaglandin dehydrogenase, and cyclooxygenase-2 expressions on tumor progression in colorectal cancer. Yonsei Med J. 2010;51(5):692–699. doi: 10.3349/ymj.2010.51.5.692. doi:201009692 [pii] 10.3349/ymj.2010.51.5.692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Elander N, Ungerback J, Olsson H, Uematsu S, Akira S, Soderkvist P. Genetic deletion of mPGES-1 accelerates intestinal tumorigenesis in APC(Min/+) mice. Biochem Biophys Res Commun. 2008;372(1):249–253. doi: 10.1016/j.bbrc.2008.05.026. doi:S0006-291X(08)00933-9 [pii] 10.1016/j.bbrc.2008.05.026. [DOI] [PubMed] [Google Scholar]
- 46.Cote B, Boulet L, Brideau C, Claveau D, Ethier D, Frenette R, Gagnon M, Giroux A, Guay J, Guiral S, Mancini J, Martins E, Masse F, Methot N, Riendeau D, Rubin J, Xu D, Yu H, Ducharme Y, Friesen RW. Substituted phenanthrene imidazoles as potent, selective, and orally active mPGES-1 inhibitors. Bioorg Med Chem Lett. 2007;17(24):6816–6820. doi: 10.1016/j.bmcl.2007.10.033. doi:S0960-894X(07)01202-4 [pii] 10.1016/j.bmcl.2007.10.033. [DOI] [PubMed] [Google Scholar]
- 47.Xu D, Rowland SE, Clark P, Giroux A, Cote B, Guiral S, Salem M, Ducharme Y, Friesen RW, Methot N, Mancini J, Audoly L, Riendeau D. MF63 [2-(6-chloro-1H-phenanthro[9,10-d]imidazol-2-yl)-isophthalonitrile], a selective microsomal prostaglandin E synthase-1 inhibitor, relieves pyresis and pain in preclinical models of inflammation. J Pharmacol Exp Ther. 2008;326(3):754–763. doi: 10.1124/jpet.108.138776. doi:jpet.108.138776 [pii] 10.1124/jpet.108.138776. [DOI] [PubMed] [Google Scholar]
- 48.French KJ, Schrecengost RS, Lee BD, Zhuang Y, Smith SN, Eberly JL, Yun JK, Smith CD. Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer Res. 2003;63(18):5962–5969. [PubMed] [Google Scholar]
- 49.Pettus BJ, Bielawski J, Porcelli AM, Reames DL, Johnson KR, Morrow J, Chalfant CE, Obeid LM, Hannun YA. The sphingosine kinase 1/sphingosine-1-phosphate pathway mediates COX-2 induction and PGE2 production in response to TNF-alpha. FASEB J. 2003;17(11):1411–1421. doi: 10.1096/fj.02-1038com. doi:10.1096/fj.02-1038com 17/11/1411 [pii] [DOI] [PubMed] [Google Scholar]
- 50.Ohama T, Okada M, Murata T, Brautigan DL, Hori M, Ozaki H. Sphingosine-1-phosphate enhances IL-1{beta}-induced COX-2 expression in mouse intestinal subepithelial myofibroblasts. Am J Physiol Gastrointest Liver Physiol. 2008;295(4):G766–775. doi: 10.1152/ajpgi.90423.2008. doi:ajpgi.90423.2008 [pii] 10.1152/ajpgi.90423.2008. [DOI] [PubMed] [Google Scholar]
- 51.Snider AJ, Kawamori T, Bradshaw SG, Orr KA, Gilkeson GS, Hannun YA, Obeid LM. A role for sphingosine kinase 1 in dextran sulfate sodium-induced colitis. FASEB J. 2009;23(1):143–152. doi: 10.1096/fj.08-118109. doi:fj.08-118109 [pii] 10.1096/fj.08-118109. [DOI] [PMC free article] [PubMed] [Google Scholar]