

Abstract
A leading hypothesis for the decimation of insulin-producing β-cells in type 2 diabetes attributes the cause to islet amyloid polypeptide (IAPP) for its deleterious effects on the cell membranes. This idea has produced extensive investigations on human IAPP (hIAPP) and its interactions with lipid bilayers. However, it is still difficult to correlate the peptide-lipid interactions with its effects on islet cells in culture. The hIAPP fibrils have been shown to interact with lipids and damage lipid bilayers, but appear to have no effect on islet cells in culture. Thus, a modified amyloid hypothesis assumes that the toxicity is caused by hIAPP oligomers, which are not preamyloid fibrils or protofibrils. However, so far such oligomers have not been isolated or identified. The hIAPP monomers also bind to lipid bilayers, but the mode of interaction is not clear. Here, we performed two types of experiments that, to our knowledge, have not been done before. We used x-ray diffraction, in conjunction with circular dichroism measurement, to reveal the location of the peptide bound to a lipid bilayer. We also investigated the effects of hIAPP on giant unilamellar vesicles at various peptide concentrations. We obtained the following qualitative results. Monomeric hIAPP binds within the headgroup region and expands the membrane area of a lipid bilayer. At low concentrations, such binding causes no leakage or damage to the lipid bilayer. At high concentrations, the bound peptides transform to β-aggregates. The aggregates exit the headgroup region and bind to the surface of lipid bilayers. The damage by the surface bound β-aggregates depends on the aggregation size. The initial aggregation extracts lipid molecules, which probably causes ion permeation, but no molecular leakage. However, the initial β-aggregates serve as the seed for larger fibrils, in the manner of the Jarrett-Lansbury seeded-polymerization model, that eventually disintegrate lipid bilayers by electrostatic and hydrophobic interactions.
Introduction
Diabetes affects nearly 8.3% of the American population and is the seventh leading cause of death in the United States. In adults, type 2 diabetes accounts for ∼90–95% of all diagnosed cases of diabetes (1). One possible cause of impaired insulin secretion in type 2 diabetes is attrition of insulin-producing β-cells (2). Prevailing hypotheses that explain the underlying cell attrition are the deleterious effects on the cell membranes by human islet amyloid polypeptide (hIAPP), a 37-residue peptide that is synthesized in pancreatic islet β-cells and cosecreted with insulin (3–7). A large body of research has described the phenomena of hIAPP interactions with lipid bilayers (see references below). However, it has been difficult to correlate the effects on lipid bilayers with the effects of hIAPP added to islet cells in culture, which indeed induce cell death (6,8). In this report, we studied the responses of giant unilamellar vesicles (GUVs) exposed to hIAPP in various conditions. To understand the physical process of hIAPP-GUV interactions, we also performed x-ray diffraction (XRD) in conjunction with circular dichroism (CD) measurements. The combined results provide what to our knowledge is new structural information about the hIAPP-membrane interactions, and reveal a low threshold peptide concentration for damaging the membranes. Monomeric hIAPP molecules at concentrations above the threshold convert to β-sheet aggregates via membrane binding, which subsequently damage the lipid bilayer. We use these results to speculate how islet amyloid polypeptide (IAPP) monomers may interact with cell membranes.
A great deal of IAPP studies made use of the contrast between hIAPP, which is amyloidogenic, and rodent IAPP (rIAPP), which is not (9,10); both are 37-residue peptides differing only in IAPP22-29 (10). Significantly, rodents do not develop diabetes-like symptoms even when rIAPP is overexpressed (11). Thus, the occurrence of diabetes in hIAPP-transgenic rodents has been regarded as the strongest evidence associating hIAPP with diabetes (12). However, the initial assumption that the formation of extracellular IAPP amyloid causes β-cell apoptosis has been disproved by the absence of correlation between cytotoxicity and extracellular amyloids (6,13). When amyloid fibrils were added to islet cells in culture, apoptosis was not induced. In contrast, if a freshly prepared aqueous solution of hIAPP was added to islet cells in culture, cell death was reproducibly induced and electron microscopy (EM) revealed the presence of small aggregates on cell membranes that were interpreted as hIAPP oligomers disrupting the cell membrane and penetrating the cell (3,8). Furthermore, the drug rifampicin inhibited hIAPP amyloid formation but failed to inhibit hIAPP cytotoxicity (13). Thus, in a current amyloid hypothesis, the toxicity is caused by hIAPP oligomers that are not preamyloid fibrils or protofibrils, but are an off-amyloid fibril pathway form of oligomer (6). However, so far the structures of these presumed toxic oligomers are unknown (such as their CD), nor have such oligomers been isolated (6). The possibility of hIAPP forming low-molecular-weight oligomers was recently examined by Vaiana et al. (14) using ultracentrifugation and by Soong et al. (7) using diffusion NMR spectroscopy and both obtained negative results.
The peptide hIAPP dissolves in aqueous solutions, initially in random coil configurations as indicated by CD, however the CD spectrum will gradually change to that of β-sheet indicating β-amyloid formation (4,8). EM showed the peptide turned into aggregates or fibrils (8,15,16). The x-ray fiber diffraction (16) showed a meridional reflection at 4.7 Å and an equatorial reflection at 9.5 Å, corresponding to the typical cross-β structure seen in the fibrils formed by other amyloid diseases (17,18). The speed of change from random coils to β-sheets in solution depended on the peptide concentration, temperature, and other solution conditions, such as pH, ions, and buffers (19). In general the higher the concentration, the faster was the change rate. It seemed possible that the peptide would remain a random coil at very low concentrations, for example at the physiological circulating concentrations of hIAPP (estimated to be 5–20 pM) (6,20). The transition from random coil to β-sheet in solution appeared not to involve α-helical configuration. This is in contrast to the conformation changes mediated by membrane binding (5, 21–23). Human IAPP spontaneously binds to lipid bilayers; particularly if the bilayers include anionic lipids (IAPP has three cationic amino acids). If IAPP was initially in random coils, the binding changed its CD to α-helical form initially but the spectrum invariably transformed to that of β-sheets in time (21,23). Again, the membrane bound aggregates and fibrils have been seen by EM (22) and by light microscope (24).
As summarized and reviewed recently by Gorbenko and Kinnunen (25) and by Jayasinghe and Langen (5), membranes have been implicated as the catalyst that facilitates fibril formation (21,22) and as the targets of IAPP toxicity (3,4). A theory for membranes as amyloid-catalyses has been advanced by Apostolidou et al. (26) who obtained the electron paramagnetic resonance-resolved α-helical core (residues 9–22) of hIAPP bound to lipid vesicles; they suggested that the lining up of the helical parts on the surface of membranes facilitates the aggregation and β-sheet formation of the amyloidogenic regions of hIAPP. The toxicity effect has been attributed to IAPP-induced membrane permeabilization, variably described as ion channels/pores (3,4,15,27–29) or membrane damage by the extraction of lipids from the bilayer (24,30–32). More recently Engel et al. (33) investigated the correlations between the growth of peptide fibril and their effects on lipid vesicles. They concluded that preformed or mature hIAPP fibrils did not interact or damage lipid bilayers, but during the growth of fibrils from membrane-bound monomers, the peptide-lipid interactions extracted lipids from the bilayers that caused membrane damages. On the other hand, Knight et al. (23) proposed that it is the aggregation of the IAPP α-helices on the membrane that induces toxicity. Somewhat similar to Soong et al. (7) who suggested that the toxicity was due to well-defined ion channels formed by aggregation of monomeric IAPPs bound to the membrane.
In this work we hope to gain a better understanding of the physical process in which hIAPP induces membrane damage. We will gain this understanding by new experiments and also by comparing the effects of hIAPP with the known behaviors of other membrane-active peptides. In particular, the membrane-mediated amyloid formation of penetratin is a useful reference for the more complex reactions by hIAPP. Penetratin dissolves in solution as a random coil and has never been found in the β-amyloid form in solution. However, it binds to the lipid bilayer interface in the α-helical form as long as the peptide/lipid ratio (P/L) is below a lipid-dependent threshold value. As soon as P/L exceeds the threshold value, penetration progressively transforms to β-aggregates and exits from the lipid bilayer interface (34). The process of forming the β-aggregates extracts lipid molecules from the lipid bilayer. We will see that hIAPP exhibits a similar behavior.
Experiment
Materials
Human IAPP, KCNTATCATQRLANFLVHSSNNFGAILSSTNVGSNTY-NH2 (disulfide bridge: 2–7), was synthesized to HPLC purity > 95% by AnaSpec (Fremont, CA). 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1,2-di-(9Z-octadecenoyl)-sn-glycero-3-phospho-(1′-rac-glycerol) (DOPG), and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(Lissamine Rhodamine B Sulfonyl) (Rh-DOPE) were purchased from Avanti Polar Lipids (Alabaster, AL). All chemicals were purchased from Sigma-Aldrich (St. Louis, MO) and were used without further purification.
Sample preparation is separately specified for each experiment below. All experiments were performed at room temperature ∼25°C.
Preparation of monomeric hIAPP
We followed an established protocol for the preparation of monomeric hIAPP (4,7). The hIAPP was first dissolved in hexafluoroisopropanol at 0.5 mg/mL and sonicated for 5 min. The solution was then lyophilized. A stock solution of 100 μM hIAPP was prepared in 200 mM glucose solution. The peptide configuration in the solution was continuously monitored by CD. The result showed that hIAPP remained in random coil configurations for at least 1 week in pure glucose solution. On the other hand, if hIAPP was dissolved in 199 mM glucose and 1 mM Tris buffer (at pH 7.0), β-aggregate started to form in ∼4 h. For each run of the experiment, the stock solution was diluted with 199 mM glucose and 1mM Tris (at pH 7.0) to a desired hIAPP concentration. CD spectrum was measured to confirm that the peptide was in random coil form. The solution was used immediately after dilution and the experimental time for each run was ∼20 min.
Preparation of hIAPP fibrils
A stock solution of 200 μM hIAPP was made in 190 mM glucose and 10 mM Tris (at pH 7.0) solution. For the experiment, the stock solution was diluted with 199 mM glucose and 1 mM Tris (at pH 7.0) to a desired hIAPP concentration.
Fibrillization of peptide samples was monitored by a thioflavin T (ThT) dye binding assay (4,33). Diluted samples were mixed with ThT (2 μM) and its fluorescence was measured by a spectrophotometer (Ocean Optics, Dunedin, FL). We found that the ThT fluorescence saturated after ∼6 h (see the Supporting Material). CD spectrum was measured to check that the hIAPP was indeed in the β form. For experiments with mature hIAPP fibrils, the stock solution was kept at 4°C for 2 weeks.
Aspirated GUV experiments
The experiments were performed as described in Sun et al. (35). Briefly, GUVs of pure DOPC or 7:3 DOPC/DOPG, plus 0.5 mol % Rh-DOPE, were produced in 200 mM sucrose solution by electroformation (36), and were transferred to a control chamber containing 199 mM glucose and 1 mM Tris (pH 7.0). A GUV was aspirated by a micropipette with a small constant sucking pressure (∼100 Pa producing a membrane tension ∼0.4 mN/m) in the control chamber and then transferred, via a transfer pipette (35), to an observation chamber containing 199 mM glucose, 1mM Tris, and hIAPP at a specified concentration. The osmolality of every solution used in the GUV experiment was measured by a dew-point Wescor osmometer (model 5520) (Logan, UT). Solutions of the same osmolality were used inside and outside of the GUV in each experiment. The experiment was recorded by fluorescence imaging using a Nikon CoolSNAP HQ2 camera (Tokyo, Japan). The phase contrast between the sucrose solution inside the GUV and the glucose solution outside was inspected at randomly chosen times to determine if any change occurred.
The analysis of the GUV response is based on the change of the protrusion length Lp of the GUV in the micropipette (37): Under the condition of no volume change, the change of membrane area is , where Rp and Rv are radii of the micropipette and the GUV. All the values of Rp, Rv, and Lp were carefully measured and analyzed by using the Nikon NIS-Elements BR 2.30 software. To normalize the area changes in different GUVs, ΔLp was converted to the fractional change of the vesicle area ΔA/A.
HIAPP β-fibrils mixed with GUVs
10 μL of the GUV suspension was transferred to an observation chamber containing 199 mM glucose, 1 mM Tris at pH 7.0, and 2.5 μM hIAPP in β-fibril form. The GUVs settled to the bottom due to the density differential and were observed with a Nikon confocal microscope (Tokyo, Japan).
GUV leakage experiment
For the leakage experiment, the solution in the GUV production chamber also contained 10 μM calcein. Two observation chambers containing the same solution (199 mM glucose and 1mM Tris at pH 7.0) were set side by side. 10 μL of the GUV suspension was injected into the first observation chamber and were well mixed with the solution in the chamber by stirring with a micropipette. About 10 GUVs were transferred from the first to the second chamber via a transfer pipette. The purpose of this transferring procedure was to dilute the untrapped calcein. The GUVs settled at the bottom due to the density differential and were observed under an Olympus microscope (model IX81) (Tokyo, Japan) and recorded with a Hamamatsu Photonics digital charge-coupled device camera (model C10600-10B) (Hamamatsu City, Japan). 0.5 μL of the monomeric hIAPP stock solution was injected into the observation chamber (to a final concentration 0.1 μM) and GUVs were observed for 5 min. Another 0.5 μL was then injected and observed for the next 5 min. This process was repeated in the sequence of increasing hIAPP concentrations, until all the GUVs under observation ruptured. The highest final hIAPP concentration in the observation chamber was 0.5 μM. The leakage of GUV content was monitored by calcein fluorescence. During the experiment, the lipid dye and content dye were inspected alternately by switching between two dichroic/filter sets.
Preparation of multilayer samples for CD and XRD
The procedure for preparing peptide/lipid mixtures into aligned multiple layers has been fully described in Ludtke et al. (38). IAPP was first dissolved in tetrafluoroethylene. Appropriate amounts of IAPP and DOPC for a chosen peptide/lipid molar ratio, P/L, were mixed in 1:1 (v/v) chloroform and tetrafluoroethylene. The mixture was uniformly spread on a thoroughly cleaned quartz substrate (0.05 mg peptide on a 1 cm2 quartz plate for P/L > 1/30; 0.3 mg of lipid on a 1 cm2 plate for P/L ≤ 1/30). After the solvent evaporated in vacuum, the sample was hydrated with 50% relative humidity (RH) water vapor at room temperature overnight. The results were well-aligned parallel bilayers, as proven by XRD. During the experiment, the samples were housed in a temperature humidity chamber in which the hydration level of the sample was controlled by the RH of water vapor (39). All measurements were performed at 25°C and 60–98% RH.
CD experiment
CD spectra were measured in a Jasco (Tokyo, Japan) J-810 spectropolarimeter. The substrates of multilayer samples were oriented normal to the incident light, as for the measurement of oriented circular dichroism (40). The background spectrum for each sample was the spectrum for the same amount of lipid on the same substrate. After the background correction, the spectra of different P/Ls were normalized to the same amount of hIAPP.
XRD experiment
The x-ray samples were prepared and measured in a temperature-humidity chamber at the same conditions as for the CD experiment. ω-2θ diffraction was collected on a four-circle Huber goniometer (Huber Diffraktionstechnik, Rimsting, Germany), with a vertical line-focused Cu Kα source (λ = 1.542 Å) operating at 35 kV and 15–30 mA. The incident beam was collimated by a horizontal soller slit and two vertical slits on the front and the back sides of the soller slit. The horizontal and vertical divergences of the incident beam were 0.23° and 0.3°, respectively. The diffracted beam first passed through a vertical slit and then was discriminated by a bent graphite monochromator before entering a scintillation detector, which was biased to discriminate against x rays other than 1.542 Å, including Compton scattering and fluorescence, and against x rays from air scattering. This diffractometer was designed to minimize the background signal, as a result allowing the measurement of high diffraction orders.
Before the measurement, the aligned multilayer sample was carefully positioned at the center of the x-ray beam and was oriented so that ω = 0° and θ = 0° coincided. We have established an elaborate routine for positioning and orienting the sample as described in Wu et al. (41). A two-dimensional ω-θ around the second or the third Bragg order was used to check the alignment of the ω angle and the mosaic of the multilayers alignment. Typically the full width at half-maximum of the peak on the ω axis (of the ω-θ scan) was 0.2–0.3° (example in (42)). Once the sample was properly positioned and aligned on the diffractometer, each ω-2θ scan was performed from ω = 0.5° to ω = 7.5° with a step size of Δω = 0.01°. An attenuator was used to prevent the first-order Bragg peak from saturating the detector. The scan was repeated 2–3 times for each hydration level and then averaged for data analysis.
The procedure of data reduction was described in many of our previous works (43,44). Briefly, the measured diffraction intensity was first corrected for the attenuator absorption and for the detector's deadtime factor. After removing the background, data were corrected for sample absorption and diffraction volume. The integrated peak intensities were then corrected for the polarization and the Lorentz factors. The relative magnitude of the diffraction amplitude was the square-root of the integrated intensity. The phases were determined by the swelling method. With their phases determined, the diffraction amplitudes were used to reconstruct the electron density profile of the bilayer. Across the bilayer profile, the phosphate peak-to-phosphate peak distance (PtP) was measured for the bilayer thickness. Measurements by the previously described procedure have been done for various peptide-lipid mixtures for more than a decade, e.g. (41,42,44). We found in each case the measured PtP reproducible within ±0.2 Å.
Results
Aspirated GUV exposed to monomeric hIAPP
Besides the apparently different binding affinities to neutral and anionic lipid bilayers, the reaction of GUVs of phosphocholine and of phosphocholine/phosphoglycerol to hIAPP binding is entirely similar. The reaction of an aspirated GUV when exposed to monomeric hIAPP depended on the peptide concentration. At low concentrations (e.g., 0.05 μM), the binding of hIAPP simply lengthened the GUV protrusion length (i.e., expanded the membrane area). At high concentrations (e.g., 1 μM), the binding of hIAPP caused the protrusion to increase to a maximum and then decreased. As the protrusion length decreased, small aggregates containing lipid dye appeared on the surface of the GUV; some of these aggregates would come off the GUV surface (Fig. 1, also Supporting Material). Of importance, we found the phase contrast between the GUV interior (sucrose) and the solution background (glucose) did not change during the whole process, indicating that no leakage of the GUV content occurred.
Figure 1.
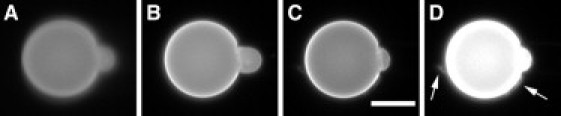
Time sequence of a GUV (7:3 DOPC/DOPG, plus 0.5 mol % Rh-DOPE) containing 200 mM sucrose exposed to 0.25 μM monomeric hIAPP in 199 mM glucose and 1 mM Tris buffer (pH 7.0). (A) t = 0; (B) the protrusion length first increased; (C) the protrusion length then decreased; (D) the high-contrast fluorescence image of (C) shows aggregates (indicated by arrows)—some appeared to be coming off the surface of GUV (see Movie S1 in the Supporting Material). Scale bar = 20 μm.
The protrusion length decrease could be due to either a GUV volume increase at constant membrane area or a membrane area decrease at constant volume (37,45,46). One possibility for volume increase is due to the formation of pores in GUV, as in the case of melittin binding (45,47) or magainin binding (46). The finite-sized pores would allow the permeation of the smaller glucose into the vesicle more than the permeation of the larger sucrose out of the vesicle; and the resulted osmolality imbalance would induce a net water influx to cause a volume increase at constant membrane area—hence the protrusion length decrease (45,47). This did not happen with hIAPP, because the phase contrast of the GUV did not change (47). To further prove that the hIAPP-induced protrusion decrease was not by pore formation, we repeated the experiment both inside and outside of the GUV in sucrose solutions. In this case, the protrusion length would not decrease (because there would be no net influx or efflux) if the effect of hIAPP was inducing pores (as shown in (46)); but the protrusion length did decrease (see the Supporting Material). This proved that the observed decrease of protrusion length was due to a membrane area decrease. This phenomenon is similar to the binding of penetratin to GUV reported earlier (34). In both cases, we observed small aggregates of lipids coming off the surface of GUV while the protrusion length was decreasing.
Fig. 2 A shows representative curves of ΔA/A, the fractional change of vesicle area, as a function of time, for three different concentrations of hIAPP. At 1 μM, the increase-decrease of protrusion length occurred after ∼5 s of the GUVs exposure to hIAPP and then the GUV ruptured. At 0.5 μM of hIAPP, the increase-decrease occurred ∼10 s after the hIAPP exposure. At 0.05 μM hIAPP, the protrusion length steadily increased for most of the time.
Figure 2.
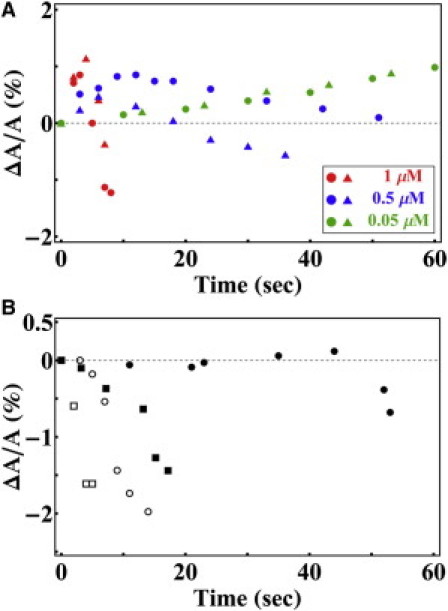
(A) GUV (7:3 DOPC/DOPG, plus 0.5 mol % Rh-DOPE) containing 200 mM sucrose exposed to three different concentrations of monomeric hIAPP in 199 mM glucose and 1 mM Tris buffer (pH 7.0). The change of protrusion length was converted to ▵A/A as a function of time (only two curves are shown for each concentration): 1 μM (red), 0.5 μM (blue), 0.05 μM (green) of monomeric hIAPP. (B) Same as (A) except that the monomeric hIAPP was replaced by hIAPP β-fibrils at 0.25 μM. In both A and B, different symbols represent different runs. With the exception of the case of 0.05 μM (green) in A, the GUV ruptured after the last data point.
Leakage experiment with monomeric hIAPP
The purpose of the leakage experiment is to investigate if there is hIAPP-induced leakage from the GUV before its rupture. Free (not aspirated) GUVs containing solution dye were first exposed to monomeric hIAPP at 0.1 μM for 5 min. No dye leakage was observed, but some small aggregates came off the surface from a few GUVs (same as Fig. 1 D). When the peptide concentration was increased to 0.2 μM for the next 5 min and then to 0.3 μM for another 5 min, more aggregates were seen coming off the surface of GUVs and a few GUVs ruptured. As the peptide concentration was increased to 0.4 μM and then 0.5 μM, the rupture became more frequent. Upon rupture, the dye content of GUV leaked out instantly and the GUV disintegrated (Fig. 3). We have never observed gradual leakage by hIAPP. There was no leakage from the GUVs before rupture. The reaction of GUVs to monomeric hIAPP was distinctly different from the reaction of GUVs to antimicrobial peptides, such as magainin (48,49) and melittin (47), which are known to induce pores in the membranes. First, antimicrobial peptides at similar concentrations did not cause GUV disintegration. Second, the leakage by antimicrobial peptides was gradual and its rate changed with the peptide concentration; the smaller the concentration the slower the rate. For example, the complete leak-out time by magainin was ∼1 min at concentration ∼1 μM, but lengthened to ∼10 min as concentrations decreased to sub-μM (48).
Figure 3.
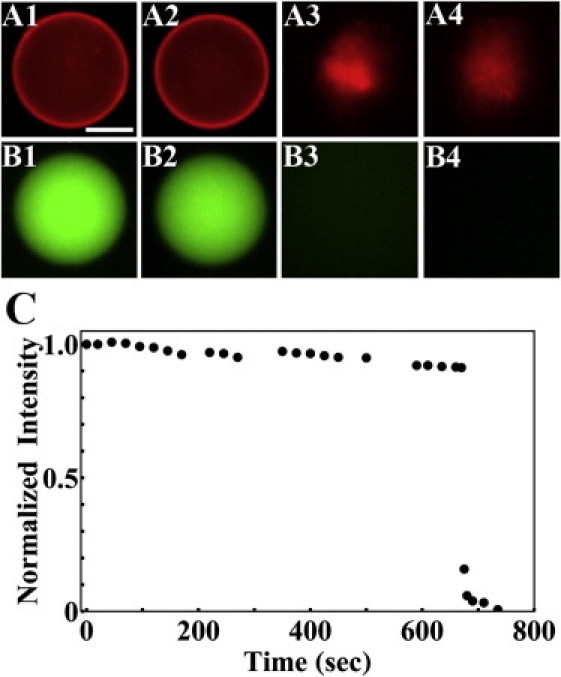
Leakage experiment. GUVs (7:3 DOPC/DOPG, plus 0.5 mol % Rh-DOPE) containing 10 μM calcein were exposed to increasing concentrations of monomeric hIAPP at 0.1, 0.2, …, 0.5 μM in 5 min intervals. No gradual leakage was detected. The figure shows an example of rupture occurring at concentration 0.3 μM: the lipid dye fluorescence images A1 at 0 s; A2 at 660 s; A3 at 670 s; A4 at 680 s. The content dye fluorescence images (Bs) were recorded ∼1 s after the corresponding lipid dye images. (C) Shows the content dye fluorescence intensity in time. The gradual decrease before rupture was due to photobleaching. The slight variations were due to varying fluorescence background. Scale bar = 20 μm.
GUV interaction with hIAPP fibrils
When an aspirated GUV was exposed to hIAPP fibrils, the fibrils were seen attached to the surface of GUV (it was visible because the fibrils extracted lipid molecules including dye from the bilayer). However, unlike monomeric hIAPP, fibrils did not cause an increase of the protrusion length; they only caused its decrease (Fig. 2 B) and caused GUV rupture.
When a suspension of GUVs was injected into a suspension of hIAPP fibrils, the vesicles were disintegrated and the lipids became part of the fibril network (Fig. 4).
Figure 4.
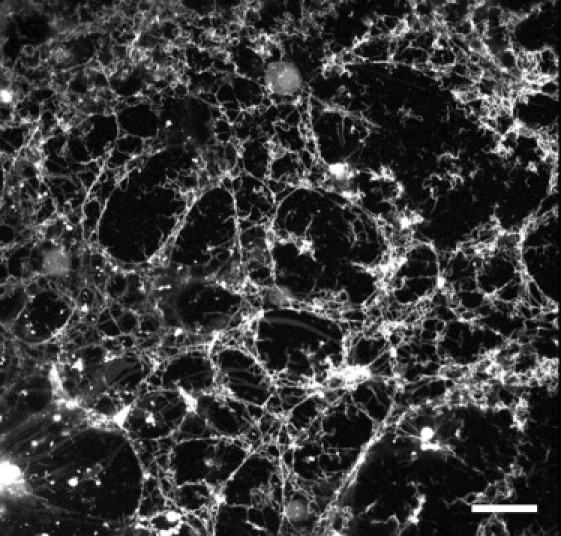
Confocal lipid-dye-fluorescence image of GUVs (7:3 DOPC/DOPG, plus 0.5 mol % Rh-DOPE) exposed to hIAPP β-fibrils (2.5 μM) in 199 mM glucose and 1 mM Tris (at pH 7.0) solution. The GUVs disintegrated and lipids became part of the fibril network. Scale bar =100 μm.
Combined results of CD and XRD
Note that the samples were prepared in a dry condition of 50% RH (the reason will become clear). We started CD measurement from 60% RH and upward to 98% RH. The sample was in each RH condition for at least 15 min. Hydration above 98% would make the sample too fluid to be held vertically (necessary for both oriented CD and XRD). DOPC/hIAPP mixtures from P/L = 1/100 to 1/20 were measured. The CD results were independent of the peptide concentration. Fig. 5 shows the representative result. Below ∼92% RH, hIAPPs bound in lipid bilayers were in the α-helical form. According to the oriented CD spectra (40), the helical axes were parallel to the plane of the bilayers. As long as the sample was kept below 92% RH, the α-helical CD was unchanged. However, when the hydration changed to ≥96% RH, the CD spectrum began to change to that of β-sheet (the CD measurement time was 3.6 min). This change of spectrum was irreversible. If a hydrated sample was dehydrated to below 96% RH, the spectrum remained β-sheet like, independent of the hydration condition.
Figure 5.
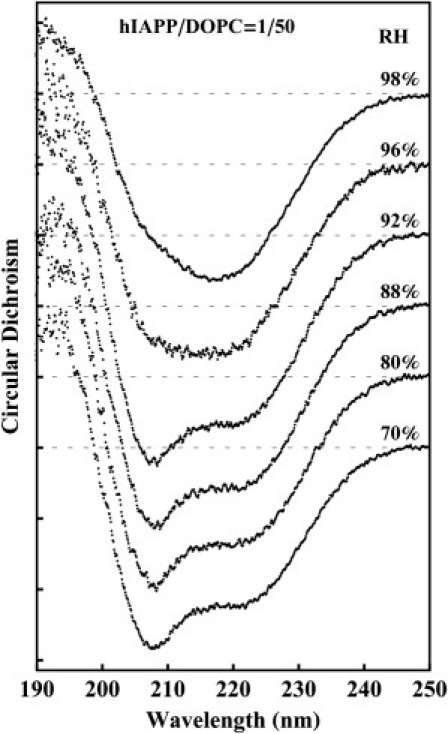
CD spectra of hIAPP in DOPC multiple bilayers at P/L = 1/50, prepared by first codissolving in organic solvent and, after the solvent evaporated, equilibrating at 50% RH. The CD was measured in the sequence of increasing hydrations. At and below 92% RH, hIAPP was in α-helical form. At hydrations above 96% RH, hIAPP transformed to β-configurations. The configuration change is irreversible. The result is independent of the P/L ratio from 1/100 to 1/20.
The CD samples were also measured by XRD in the hydration sequence from 70% to 98% RH and then reversed from 98% to 70% RH. The diffraction patterns showed well-ordered bilayers in each sample of P/L ranging from 1/80 to 1/30. From the reconstructed electron density profiles for the peptide-lipid bilayers, the changes of the phosphate-to-phosphate distance across the bilayer (PtP) are shown in Fig. 6. From the CD results, we know that the peptides were in α-helical configuration during the hydration sequence from 70% to 92% RH. However, once the sample was hydrated to 98% RH, the peptides turned into β-sheet configurations and remained in that configuration when the sample was once again dehydrated. Fig. 6 A shows that the thickness of pure DOPC was reversible by the hydration-dehydration cycle, but not for the lipid bilayers containing hIAPP. Once hIAPP turned into β-configurations, the bilayer thickness at full hydration became the same as pure lipid. In fact the electron density profiles obtained from the multilayers containing hIAPP in the β form are identical to that of pure lipid bilayers (Fig. 6 B). Clearly hIAPP molecules in the β form were not within the lipid bilayers, otherwise the bilayer profiles would have been altered from the free bilayer form. The values of PtP at 92% RH are plotted as a function of P/L in Fig. 6 C, showing that when the bound hIAPP was in the α-helical form, the bilayer thickness was reduced in proportion to P/L. Membrane thinning in proportion to P/L implies that the peptide in the α-helical form was bound within the headgroup region (34,38,41,50).
Figure 6.
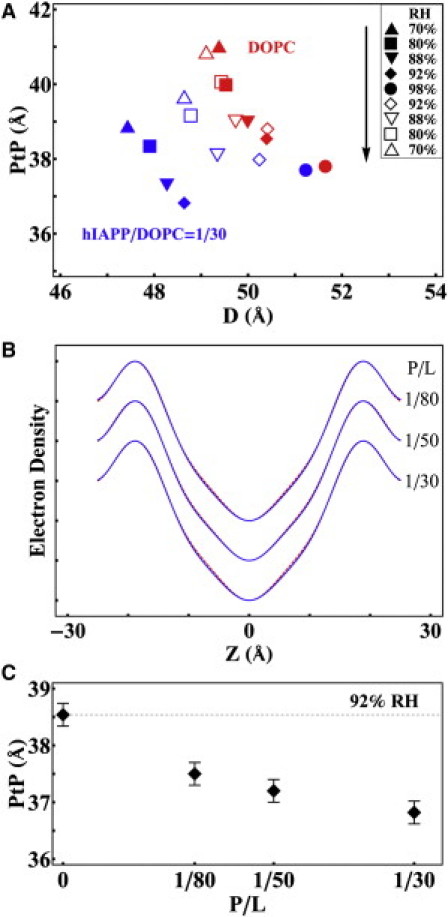
CD samples (Fig. 5) were measured by XRD. (A) The phosphate-to-phosphate distance (PtP) of the bilayer electron density profiles obtained from DOPC multilayers containing hIAPP (blue symbols) compared with that of pure DOPC (red symbols) in the sequence of hydration-dehydration cycle indicated in the inset. (B) The electron density profiles of DOPC multilayers containing hIAPP (blue line) compared with that of pure DOPC (red line; mostly overlaying the blue line) at 98% RH. The profiles are the same within experimental resolution. (C) PtP of DOPC/hIAPP changes with P/L at 92% RH.
Many independent experiments (5,21,23) have shown that monomeric hIAPPs bind to lipid vesicles initially in the α-helical form, and then convert to β-configurations. Our oriented circular dichroism and x-ray experiments with multilamellar samples are too slow to measure this transitional α-helical state in full hydration. We trapped α-helical hIAPP in dry lipid bilayers. However, by the time the samples were hydrated and measured (taking ∼5 min), the peptide had already converted to β-configurations and exited from the headgroup region.
Discussion
The new (to our knowledge) structural information obtained from our experiments is that the β-aggregates of hIAPP bind on the surface of lipid bilayers; they do not penetrate into the bilayer structure. This is true even when the β-aggregates originated from α-helical hIAPPs bound inside the headgroup region of the lipid bilayer. This behavior of hIAPP is identical to the previous finding with the amyloidogenic peptide penetratin (34). Thus, it could be a common pattern of interaction between amyloid-forming peptides and lipid bilayers, i.e., the peptides bind as helices inside the headgroup region of the bilayer, but once the peptides convert to β-aggregates, they bind on the aqueous surface of the bilayer.
The GUV experiments with monomeric hIAPP imply that there is a threshold bound-peptide/lipid ratio, below which the bound peptide does not convert to β-aggregates. This behavior of hIAPP is also similar to penetratin. Such a threshold is important because the binding of hIAPP does not damage the lipid bilayer until the peptide converts into β-aggregates.
Interaction of hIAPP β-fibrils with lipid bilayers
Binding of hIAPP β-fibrils to lipid vesicles was observed in two ways. When an aspirated GUV was exposed to hIAPP β-fibrils, the peptide binding did not cause a membrane expansion—no protrusion length increase, consistent with surface binding. Instead, the GUV protrusion length quickly decreased and the GUV ruptured (Fig. 2 B). On the other hand, if lipid vesicles were dispersed into a β-fibrils suspension, vesicles and lipids were seen attached to the fibrils and became part of the fibril aggregates. We stress that hIAPP β-fibrils bind to both neutral (DOPC) and anionic lipids (7:3 DOPC/DOPG) and that the same binding phenomena were observed during the growth of β-fibrils as well as with preformed β-fibrils. This is contrary to the observation by Engel et al. (33) who reported interactions only during the growth of fibrils.
Eisenberg and colleagues (17,18) have resolved the crystalline structures of β-amyloid formed by a small peptide GNNQQNY. The universal cross-β structure for β-amyloids has three levels of organization: The first is the alignment of the peptide molecules to form a β-sheet; the second is the self-complementation of two sheets, forming the pair-of-sheets structure with a dry interface; in the third level, pair-of-sheets interact to form a fibril. For an extended peptide like hIAPP, the organization at each level is bound to be imperfect (unlike the small peptide GNNQQNY), namely, there will be unpaired (positive) charges and uncovered hydrophobic regions exposed on the surface of the fibrils. It is well known that multivalent cations adsorb to the lipid bilayers of both zwitterionic and acidic headgroups (51). Therefore, it is not surprising that the positive charges on the surface of hIAPP β-fibrils bind to the surface of lipid bilayers. It is also not surprising that lipid molecules bind to the hydrophobic regions of the fibril surface.
Given such deleterious effects on lipid bilayers, why were there no interactions between hIAPP β-fibrils and cell membranes in culture (3,8)? We speculate that perhaps the β-fibrils formed in solution cannot penetrate the carbohydrate layer (glycocalyx) (52) that covers the cell surface, so the fibrils do not come into contact with plasma membranes.
Interaction of hIAPP monomers with lipid bilayers
The hIAPP monomers in solution bind to both the neutral (zwitterionic) and the anionic lipid bilayers. As expected, the binding affinity to the latter is stronger. In our experiments, pure lipid GUVs were introduced into a peptide solution at t = 0. Thus, the bound peptide/lipid ratio P/L increased with time. In all concentrations, the initial binding expanded the membrane area without causing molecular leakage. At the peptide concentration ≲ 0.05 μM, the GUV remained in this condition within our experimental time. No aggregates were observed on the GUV surface and no damage to lipid bilayers occurred. This clearly implies that before P/L reaches a threshold value, the bound peptides do not transform to β-aggregates. However, the exact value of the threshold P/L is too small (<1/100) to be measured by the multilayer experiments. In contrast, the threshold P/L for penetratin has been measured accurately by the multilayer experiments: four different thresholds, between 1/65 and 1/20, in four different lipids (34).
When an aspirated GUV was exposed to monomeric hIAPP at concentrations ≳0.5 μM, the initial membrane expansion reached a limit and then the expansion was reversed. During the decrease of protrusion length, we observed small aggregates, which included lipid dye that appeared on the membrane surface, but there was no molecular leakage (the vesicle phase contrast remained constant). Eventually the GUV ruptured.
Thus, we can summarize the physical process of interaction between monomeric hIAPP with lipid bilayers as follows. The hIAPP spontaneously binds to lipid bilayers. If the bound P/L is below a certain threshold value, the binding causes a membrane expansion but otherwise causes no leakage or damage. However, if the P/L exceeds the threshold value, the bound peptides convert to β-aggregates, which exit from the bilayer and bind on the bilayer surface. The process of conversion to the β-aggregates probably extracts lipid molecules from the bilayer. As long as the β-aggregates are sufficiently small, the extraction of lipid does not cause molecular leakage. However, small β-aggregates seed the formation of fibrils, as demonstrated by the Jarrett-Lansbury model (53). The large size fibrils eventually rupture the lipid bilayer via electrostatic and hydrophobic interactions.
Our observation at high concentrations of monomeric hIAPP essentially agrees with Engel et al. (33). Many previous investigations have reported ion conduction caused by hIAPP monomers while detecting no molecular leakage (3,4,15,27–29). This phenomenon may be correlated to the extraction of lipid molecules from the bilayer during the hIAPPs conversion to β-aggregates. It has been reported that defects of a lipid bilayer allow ion conduction, for example near the fluid-gel phase transition point, but no molecular leakage (54).
Finally, as stated previously, we speculate that β-fibrils cannot pass through the glycocalyx to reach the plasma membranes of cells, but we assume that hIAPP monomers can pass through the glycocalyx. The latter assumption is based on the observation that antimicrobial peptides (which are monomeric in solution and many of them are of similar size of hIAPP) can readily reach the plasma membranes (55). Based on our knowledge of how hIAPP interacts with lipid bilayers, the soluble monomers is the most likely form of hIAPP that could affect cell membranes in culture. The possible damage strongly depends on the peptide concentration. The damage caused by high concentrations of hIAPP may not occur in low concentrations.
Acknowledgments
This work was supported by National Institutes of Health grant GM55203 and by Robert A. Welch Foundation grant C-0991.
Supporting Material
References
- 1.CDC (Centers for Disease Control and Prevention). http://www.cdc.gov/diabetes/pubs/pdf/ndfs_2011.pdf.
- 2.Gepts W., Lecompte P.M. The pancreatic islets in diabetes. Am. J. Med. 1981;70:105–115. doi: 10.1016/0002-9343(81)90417-4. [DOI] [PubMed] [Google Scholar]
- 3.Janson J., Ashley R.H., Butler P.C. The mechanism of islet amyloid polypeptide toxicity is membrane disruption by intermediate-sized toxic amyloid particles. Diabetes. 1999;48:491–498. doi: 10.2337/diabetes.48.3.491. [DOI] [PubMed] [Google Scholar]
- 4.Anguiano M., Nowak R.J., Lansbury P.T., Jr. Protofibrillar islet amyloid polypeptide permeabilizes synthetic vesicles by a pore-like mechanism that may be relevant to type II diabetes. Biochemistry. 2002;41:11338–11343. doi: 10.1021/bi020314u. [DOI] [PubMed] [Google Scholar]
- 5.Jayasinghe S.A., Langen R. Membrane interaction of islet amyloid polypeptide. Biochim. Biophys. Acta. 2007;1768:2002–2009. doi: 10.1016/j.bbamem.2007.01.022. [DOI] [PubMed] [Google Scholar]
- 6.Haataja L., Gurlo T., Butler P.C. Islet amyloid in type 2 diabetes, and the toxic oligomer hypothesis. Endocr. Rev. 2008;29:303–316. doi: 10.1210/er.2007-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soong R., Brender J.R., Ramamoorthy A. Association of highly compact type II diabetes related islet amyloid polypeptide intermediate species at physiological temperature revealed by diffusion NMR spectroscopy. J. Am. Chem. Soc. 2009;131:7079–7085. doi: 10.1021/ja900285z. [DOI] [PubMed] [Google Scholar]
- 8.Konarkowska B., Aitken J.F., Cooper G.J. The aggregation potential of human amylin determines its cytotoxicity towards islet beta-cells. FEBS J. 2006;273:3614–3624. doi: 10.1111/j.1742-4658.2006.05367.x. [DOI] [PubMed] [Google Scholar]
- 9.Sanke T., Bell G.I., Steiner D.F. An islet amyloid peptide is derived from an 89-amino acid precursor by proteolytic processing. J. Biol. Chem. 1988;263:17243–17246. [PubMed] [Google Scholar]
- 10.Westermark P., Engström U., Betsholtz C. Islet amyloid polypeptide: pinpointing amino acid residues linked to amyloid fibril formation. Proc. Natl. Acad. Sci. USA. 1990;87:5036–5040. doi: 10.1073/pnas.87.13.5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang C.J., Lin C.Y., Butler P.C. High expression rates of human islet amyloid polypeptide induce endoplasmic reticulum stress mediated beta-cell apoptosis, a characteristic of humans with type 2 but not type 1 diabetes. Diabetes. 2007;56:2016–2027. doi: 10.2337/db07-0197. [DOI] [PubMed] [Google Scholar]
- 12.Matveyenko A.V., Butler P.C. Islet amyloid polypeptide (IAPP) transgenic rodents as models for type 2 diabetes. ILAR J. 2006;47:225–233. doi: 10.1093/ilar.47.3.225. [DOI] [PubMed] [Google Scholar]
- 13.Meier J.J., Kayed R., Butler P.C. Inhibition of human IAPP fibril formation does not prevent beta-cell death: evidence for distinct actions of oligomers and fibrils of human IAPP. Am. J. Physiol. Endocrinol. Metab. 2006;291:E1317–E1324. doi: 10.1152/ajpendo.00082.2006. [DOI] [PubMed] [Google Scholar]
- 14.Vaiana S.M., Ghirlando R., Hofrichter J. Sedimentation studies on human amylin fail to detect low-molecular-weight oligomers. Biophys. J. 2008;94:L45–L47. doi: 10.1529/biophysj.107.125146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kayed R., Sokolov Y., Glabe C.G. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J. Biol. Chem. 2004;279:46363–46366. doi: 10.1074/jbc.C400260200. [DOI] [PubMed] [Google Scholar]
- 16.Sumner Makin O., Serpell L.C. Structural characterisation of islet amyloid polypeptide fibrils. J. Mol. Biol. 2004;335:1279–1288. doi: 10.1016/j.jmb.2003.11.048. [DOI] [PubMed] [Google Scholar]
- 17.Balbirnie M., Grothe R., Eisenberg D.S. An amyloid-forming peptide from the yeast prion Sup35 reveals a dehydrated beta-sheet structure for amyloid. Proc. Natl. Acad. Sci. USA. 2001;98:2375–2380. doi: 10.1073/pnas.041617698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nelson R., Sawaya M.R., Eisenberg D. Structure of the cross-beta spine of amyloid-like fibrils. Nature. 2005;435:773–778. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nielsen L., Khurana R., Fink A.L. Effect of environmental factors on the kinetics of insulin fibril formation: elucidation of the molecular mechanism. Biochemistry. 2001;40:6036–6046. doi: 10.1021/bi002555c. [DOI] [PubMed] [Google Scholar]
- 20.Butler P.C., Chou J., Rizza R.A. Effects of meal ingestion on plasma amylin concentration in NIDDM and nondiabetic humans. Diabetes. 1990;39:752–756. doi: 10.2337/diab.39.6.752. [DOI] [PubMed] [Google Scholar]
- 21.Jayasinghe S.A., Langen R. Lipid membranes modulate the structure of islet amyloid polypeptide. Biochemistry. 2005;44:12113–12119. doi: 10.1021/bi050840w. [DOI] [PubMed] [Google Scholar]
- 22.Knight J.D., Miranker A.D. Phospholipid catalysis of diabetic amyloid assembly. J. Mol. Biol. 2004;341:1175–1187. doi: 10.1016/j.jmb.2004.06.086. [DOI] [PubMed] [Google Scholar]
- 23.Knight J.D., Hebda J.A., Miranker A.D. Conserved and cooperative assembly of membrane-bound alpha-helical states of islet amyloid polypeptide. Biochemistry. 2006;45:9496–9508. doi: 10.1021/bi060579z. [DOI] [PubMed] [Google Scholar]
- 24.Sparr E., Engel M.F., Killian J.A. Islet amyloid polypeptide-induced membrane leakage involves uptake of lipids by forming amyloid fibers. FEBS Lett. 2004;577:117–120. doi: 10.1016/j.febslet.2004.09.075. [DOI] [PubMed] [Google Scholar]
- 25.Gorbenko G.P., Kinnunen P.K. The role of lipid-protein interactions in amyloid-type protein fibril formation. Chem. Phys. Lipids. 2006;141:72–82. doi: 10.1016/j.chemphyslip.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 26.Apostolidou M., Jayasinghe S.A., Langen R. Structure of alpha-helical membrane-bound human islet amyloid polypeptide and its implications for membrane-mediated misfolding. J. Biol. Chem. 2008;283:17205–17210. doi: 10.1074/jbc.M801383200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Quist A., Doudevski I., Lal R. Amyloid ion channels: a common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. USA. 2005;102:10427–10432. doi: 10.1073/pnas.0502066102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kawahara M., Kuroda Y., Rojas E. Alzheimer's beta-amyloid, human islet amylin, and prion protein fragment evoke intracellular free calcium elevations by a common mechanism in a hypothalamic GnRH neuronal cell line. J. Biol. Chem. 2000;275:14077–14083. doi: 10.1074/jbc.275.19.14077. [DOI] [PubMed] [Google Scholar]
- 29.Mirzabekov T.A., Lin M.C., Kagan B.L. Pore formation by the cytotoxic islet amyloid peptide amylin. J. Biol. Chem. 1996;271:1988–1992. doi: 10.1074/jbc.271.4.1988. [DOI] [PubMed] [Google Scholar]
- 30.Green J.D., Kreplak L., Aebi U. Atomic force microscopy reveals defects within mica supported lipid bilayers induced by the amyloidogenic human amylin peptide. J. Mol. Biol. 2004;342:877–887. doi: 10.1016/j.jmb.2004.07.052. [DOI] [PubMed] [Google Scholar]
- 31.Harroun T.A., Bradshaw J.P., Ashley R.H. Inhibitors can arrest the membrane activity of human islet amyloid polypeptide independently of amyloid formation. FEBS Lett. 2001;507:200–204. doi: 10.1016/s0014-5793(01)02972-6. [DOI] [PubMed] [Google Scholar]
- 32.Zhao H., Tuominen E.K., Kinnunen P.K. Formation of amyloid fibers triggered by phosphatidylserine-containing membranes. Biochemistry. 2004;43:10302–10307. doi: 10.1021/bi049002c. [DOI] [PubMed] [Google Scholar]
- 33.Engel M.F., Khemtémourian L., Höppener J.W. Membrane damage by human islet amyloid polypeptide through fibril growth at the membrane. Proc. Natl. Acad. Sci. USA. 2008;105:6033–6038. doi: 10.1073/pnas.0708354105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee C.C., Sun Y., Huang H.W. Membrane-mediated peptide conformation change from alpha-monomers to beta-aggregates. Biophys. J. 2010;98:2236–2245. doi: 10.1016/j.bpj.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun Y., Lee C.C., Huang H.W. The bound states of amphipathic drugs in lipid bilayers: study of curcumin. Biophys. J. 2008;95:2318–2324. doi: 10.1529/biophysj.108.133736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Angelova M.I. Liposome electroformation. In: Luisi P.L., Walde P., editors. Giant Vesicles. John Wiley & Sons; Chichester: 2000. pp. 27–36. [Google Scholar]
- 37.Kwok R., Evans E. Thermoelasticity of large lecithin bilayer vesicles. Biophys. J. 1981;35:637–652. doi: 10.1016/S0006-3495(81)84817-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ludtke S., He K., Huang H. Membrane thinning caused by magainin 2. Biochemistry. 1995;34:16764–16769. doi: 10.1021/bi00051a026. [DOI] [PubMed] [Google Scholar]
- 39.Yang L., Huang H.W. A rhombohedral phase of lipid containing a membrane fusion intermediate structure. Biophys. J. 2003;84:1808–1817. doi: 10.1016/S0006-3495(03)74988-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu Y., Huang H.W., Olah G.A. Method of oriented circular dichroism. Biophys. J. 1990;57:797–806. doi: 10.1016/S0006-3495(90)82599-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu Y., He K., Huang H.W. X-ray diffraction study of lipid bilayer membranes interacting with amphiphilic helical peptides: diphytanoyl phosphatidylcholine with alamethicin at low concentrations. Biophys. J. 1995;68:2361–2369. doi: 10.1016/S0006-3495(95)80418-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weiss T.M., van der Wel P.C., Huang H.W. Hydrophobic mismatch between helices and lipid bilayers. Biophys. J. 2003;84:379–385. doi: 10.1016/S0006-3495(03)74858-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Olah G.A., Huang H.W., Wu Y.L. Location of ion-binding sites in the gramicidin channel by X-ray diffraction. J. Mol. Biol. 1991;218:847–858. doi: 10.1016/0022-2836(91)90272-8. [DOI] [PubMed] [Google Scholar]
- 44.Harroun T.A., Heller W.T., Huang H.W. Experimental evidence for hydrophobic matching and membrane-mediated interactions in lipid bilayers containing gramicidin. Biophys. J. 1999;76:937–945. doi: 10.1016/S0006-3495(99)77257-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Longo M.L., Waring A.J., Hammer D.A. Area expansion and permeation of phospholipid membrane bilayer by influenza fusion peptides and melittin. Langmuir. 1998;14:2385–2395. [Google Scholar]
- 46.Sun Y., Hung W.C., Huang H.W. Interaction of tea catechin (-)-epigallocatechin gallate with lipid bilayers. Biophys. J. 2009;96:1026–1035. doi: 10.1016/j.bpj.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee M.T., Hung W.C., Huang H.W. Mechanism and kinetics of pore formation in membranes by water-soluble amphipathic peptides. Proc. Natl. Acad. Sci. USA. 2008;105:5087–5092. doi: 10.1073/pnas.0710625105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tamba Y., Yamazaki M. Single giant unilamellar vesicle method reveals effect of antimicrobial peptide magainin 2 on membrane permeability. Biochemistry. 2005;44:15823–15833. doi: 10.1021/bi051684w. [DOI] [PubMed] [Google Scholar]
- 49.Tamba Y., Ariyama H., Yamazaki M. Kinetic pathway of antimicrobial peptide magainin 2-induced pore formation in lipid membranes. J. Phys. Chem. B. 2010;114:12018–12026. doi: 10.1021/jp104527y. [DOI] [PubMed] [Google Scholar]
- 50.Huang H.W. Free energies of molecular bound states in lipid bilayers: lethal concentrations of antimicrobial peptides. Biophys. J. 2009;96:3263–3272. doi: 10.1016/j.bpj.2009.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lis L.J., Lis W.T., Rand R.P. Adsorption of divalent cations to a variety of phosphatidylcholine bilayers. Biochemistry. 1981;20:1771–1777. doi: 10.1021/bi00510a010. [DOI] [PubMed] [Google Scholar]
- 52.Alberts B., Johnson A., Walter P. Garland Science; New York: 2002. Molecular Biology of the Cell. [Google Scholar]
- 53.Jarrett J.T., Lansbury P.T., Jr. Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie? Cell. 1993;73:1055–1058. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- 54.Heimburg T. Lipid ion channels. Biophys. Chem. 2010;150:2–22. doi: 10.1016/j.bpc.2010.02.018. [DOI] [PubMed] [Google Scholar]
- 55.Boman H.G., Marsh J., Goode J.A., editors. Antimicrobial peptides. Ciba Foundation Symposium 186. John Wiley and Sons; Chichester: 1994. pp. 1–272. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.