

Abstract
Since its proposal in 1994, the amyloid cascade hypothesis has prevailed as the mainstream research subject on the molecular mechanisms leading to the Alzheimer's disease (AD). Most of the field had been historically based on the role of the different forms of aggregation of β-amyloid peptide (Aβ). However, a soluble intracellular fragment termed amyloid precursor protein (APP) intracellular domain (AICD) is produced in conjunction with Aβ fragments. This peptide had been shown to be highly toxic in both culture neurons and transgenic mice models. With the advent of this new toxic fragment, the centerpiece for the ethiology of the disease may be changed. This paper discusses the potential role of multiprotein complexes between the AICD and its adapter protein Fe65 and how this could be a potentially important new agent in the neurodegeneration observed in the AD.
1. Introduction
The APP is a type I transmembrane protein with characteristics of an orphan receptor, which shares with other members of its class a particular signaling mechanism termed regulated intramembrane proteolysis (RIP) [1].
RIP requires that the transmembrane protein undergoes two consecutive cleavage events. The first occurs outside the transmembrane domain, usually in response to ligand binding, inducing the release of the extracellular domain. This first cleavage event elicits a conformational change that triggers the second proteolytic cleavage which takes place on the transmembrane segment. The intracellular cytoplasmic fragment released translocates to the nucleus where it activates gene transcription [1]. This mechanism controls several cellular processes, such as the unfolded protein response [2], cholesterol synthesis [3], and cell fate instruction [4].
RIP of the APP is mediated by three different proteases. While α- and β-secretases catalyze extracellular cleavage, the γ-secretase complex cuts at the intramembrane domain and leads to the generation of two peptides: an APP active fragment, termed AICD and the Aβ [1]. The stoichiometry of both AICD and Aβ fragments has been a controversial issue. One study shows that the absence of the β-secretase does not affect AICD production [5]. In contrast, two independent groups indicate that AICD is produced mainly from the 695 aminoacids isoform of APP through the amyloidogenic pathway (dependent on β-secretase activity) [6, 7] and is therefore generated in equimolar quantities with Aβ [8]. The last one accumulation and the formation of various aggregates and deposits in the brain have been the main hypothesis to explain the neuropathological development of AD for almost 20 years [9]. Initially, the study of the functions associated with the AICD was limited by the hindrance in its detection [10]. However, recent studies showing that the levels of the AICD are increased in brains of AD patients and murine models reproducing the disease [11], open up the possibility that this fragment participates in the molecular mechanisms contributing to AD.
2. The AICD Interactome: Functions and Dysfunctions in the Route to AD
The AICD is the most evolutionarily-conserved region of the APP, accounting for its functional importance. Despite its relatively small size (59 aminoacids or less), it acts as a docking site for a particularly large group of intracellular proteins. Amongst this group of proteins are Pin1 [12], the X11 protein family [13], disabled (Dab)-1 [14], Shc [15], JNK-interacting protein (JIP)-1 [16], and the Fe65 protein family [17–19], which includes Fe65 itself and two closely related homologues, Fe65L1 and Fe65L2. Fe65 family members contain three protein-protein interaction domains: a WW domain at the N-terminal involved in interactions with proline-rich sequences and two phosphotyrosine binding domains (PTB1 and PTB2) located at the C-terminal. The second PTB domain (PTB2) is responsible for the interaction between Fe65 and the sequence 682YENPTY687 of the APP (following the numbering of the APP695 isoform). The interaction between these proteins occurs in a Tyr682 phosphorylation-independent manner [13]. The possibility of AICD to form multiprotein complexes through its association with Fe65 and its multiple ligands (Table 1) has unexpectedly expanded the potential roles of AICD.
Table 1.
Fe65 interactors and its functions.
| Protein | Domain involved | Putative functions of the interaction | References |
|---|---|---|---|
| Amyloid precursor protein (APP) | PTB2 | Regulation of Aβ secretion, nuclear signaling, and cytoskeleton regulation | [17] |
|
| |||
| Mammalian enabled (Mena) | WW | Actin polimerization | [20] |
|
| |||
| CP2/LSF/LBP1 | PTB1 | Transcriptional regulation, GSK-3β expression | [21] |
|
| |||
| Low-density lipoprotein receptor-related protein (LRP1) | PTB1 | APP trafficking, Aβ secretion | [22] |
|
| |||
| Abl tyrosine kinase | WW | Nuclear signaling | [23] |
|
| |||
| Tat-interacting protein 60 kDa (Tip60) | PTB1 | Nuclear signaling, DNA repair | [24] |
|
| |||
| Alcadein | ND | APP metabolism | [25] |
|
| |||
| Nucleosome assembly factor SET | WW | Transcriptional regulation | [26] |
|
| |||
| Tau | PTB1 | Cytoskeleton regulation | [27] |
|
| |||
| 14-3-3γ | Between WW and PTB1 | Nuclear signaling | [28] |
|
| |||
| P2X receptor | WW | Synaptic transmission | [29] |
|
| |||
| ApoER2 | PTB1 | APP trafficking, Aβ secretion | [30] |
|
| |||
| Estrogen receptor α | ND | Transcriptional regulation | [31] |
|
| |||
| NIMA-related kinase 6 | WW | Apoptosis | [32] |
|
| |||
| Glycogen synthase kinase-3β | WW | Kinase activation | [33] |
|
| |||
| Dexras1 | PTB2 | Nuclear signaling | [34] |
|
| |||
| Teashirt | PTB1 | Repression of caspase 4 expression | [35] |
|
| |||
| Neuronal precursor cell expressed developmentally down regulated 4-2 (Nedd 4-2) | WW | Fe65 ubiquitylation | [36] |
|
| |||
| Dab1 | ND | APP processing | [37] |
|
| |||
| Megalin | ND | Axonal branching, APP trafficking | [38] |
|
| |||
| Rac1 | ND | Fe65 expression | [39] |
ND: Not determined
2.1. Roles in APP Trafficking and Processing
AICD binds to Fe65 in a region that is essential for Aβ production, making Fe65 a good candidate for regulating APP processing. This could occur via two mutually-exclusive pathways: the amyloidogenic pathway, leading to Aβ production mediated by the β-secretase and the nonamyloidogenic pathway leading to the production of a large extracellular fragment (sAPPα), which is mediated by the α-secretase and prevents the generation of Aβ. Fe65 acts as a potent modulator by altering the balance between the two pathways. The overexpression of Fe65 in cell lines induces a dramatic increase in Aβ secretion [40], whereas Aβ secretion was decreased in Fe65 knockdown cells [41] and in hippocampal neurons of Fe65/Fe65L1 knockout (KO) mice [42]. The effect on the Aβ secretion appears to be dependent on the interaction between Fe65 and APP, because the knock-in mice carrying the Y682G mutation, that inhibits AICD binding to Fe65, show decreased levels of Aβ and a massive increase in sAPPα, as a consequence of the nonamyloidogenic pathway [43]. This is in agreement with a study showing that Fe65 is a potent suppressor of the nonamyloidogenic pathway in primate cells [44].
The mechanism by which Fe65 modulates Aβ secretion is related to its interaction with the apolipoprotein E (ApoE) receptors: the low density lipoprotein receptor-related protein (LRP) [22] and ApoE receptor 2 (ApoER2) [30].
Related to the participation of the aforementioned receptors, the effect of Fe65 in the secretion of soluble APP fragments is lost in cells lacking LRP [45]. The functional relation with ApoER2 is more complex and depends on the presence of its extracellular ligand, reelin, and its intracellular adapter, Dab-1. Reelin reduces Aβ secretion by promoting the binding of Dab1 to the APP and displacing Fe65, because they share the same binding region [46]. A decrease in Reelin expression in the entorhinal cortex (the first region of the brain where Aβ deposits can be observed), displayed in PDAPP transgenic mice (which carry human APP with mutations Swedish (swe) and Indiana) and in AD patients [47], could seriously affect the balance of Dab1 and Fe65 in their binding to AICD, increasing Aβ secretion. This has been observed in transgenic mice which lack Reelin expression (reeler) and carry the mutations swedish and arctic in APP [48].
2.2. Roles in Transcription
A decade ago, a possible role for the RIP of APP was first suggested [24]. Since APP processing seems to be similar to Notch processing, it has been suggested that RIP of APP could be involved in transcriptional regulation. In fact, the fusion of the DNA binding domain of yeast Gal4 (Gal4DB) to the C-terminal of APP induced a strong transactivation of a luciferase reporter dependent on the formation of a trimeric complex with the adapter protein Fe65 and the histone acetyltransferase Tip60 [24]. A reciprocal experiment using Tip60 or Fe65 fused to the Gal4DB gave rise to some contradictory results [49, 50]. Nevertheless, a consensus model can be generated including the vast majority of observations derived from these studies (Figure 1).
Figure 1.
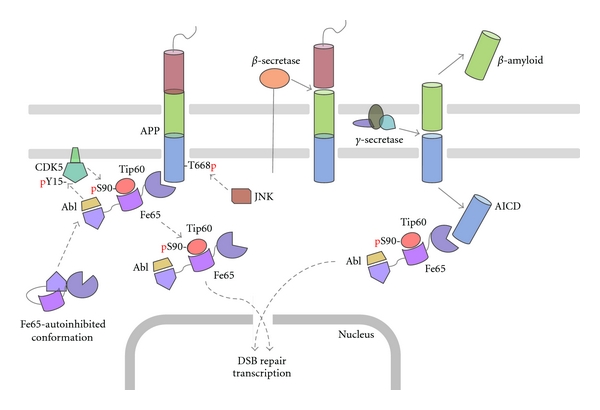
APP processing pathways involved in the activation and release of the AICD-associated complexes from the plasma membrane. Fe65 is in an autoinhibited conformation in the cytoplasm. The binding to the AICD triggers the exposure of Fe65 WW and PTB1 domains. These protein-protein domains elicit the recruitment to the subcortical domains of the plasma membrane of both c-Abl and Tip60. At the plasma membrane, c-Abl can phosphorylate and activate the protein kinase CDK-5 at Tyr15, and in turn, activated CDK-5 may phosphorylate Ser90 of Tip60. DNA damage or other unknown stimuli may then induce the release of the complex from the membrane through two complementary mechanisms: either by the activation of the γ-secretase or by JNK-dependent phosphorylation of Thr668 in the AICD. In spite of the preferred mechanisms involving the release of the Fe65-complex, it can be translocated to the nucleus where it activates transcription of target genes and is essential in the repair of the DNA double strand-breaks (DSB).
The APP acts as an anchor for Fe65 and Fe65-associated proteins that is,: Tip60, inducing its association with membrane compartments [51]. Membrane recruitment seems to be essential for the activation of the complex, since the overexpression of soluble AICD has no effect on transactivation [49].
The binding of APP to Fe65 induces a conformational change that “opens” the autoinhibited conformation of Fe65, produced by the association of the WW domain with a region flanked by the PTB1 and PTB2 domains [49].
The association with the plasma membrane allows the activation of the complex, induced by the phosphorylation of Tip60 by cyclin-dependent kinases (CDKs) [52]. An excellent prospective candidate is CDK-5, that can be found associated with plasma membranes through its activator p35 and displays high activity in the brain [53].
The release of the complex from the plasma membrane may be produced by the APP cleavage by γ-secretase [24] or additionally by the APP phosphorylation at Thr668 [54] which induces a conformational change in the region recognized by Fe65, decreasing the affinity for each other [55].
Although some groups have observed AICD in the nucleus [56], particularly in nuclear domains such as transcriptional factories [57], the splicing factor compartment [58] or directly at promoters of some genes [59–61], apparently in the artificial transactivation system, the nuclear translocation of AICD is not essential to enhance luciferase expression [49].
The N-terminal region of Fe65 that includes the WW domain is necessary for nuclear translocation [51] and therefore for its activity as a transactivating protein [24]. Although this region lacks a nuclear localization sequence (NLS), it could be directed to the nuclei by association with a protein carrying a functional NLS. A good candidate to perform this function would be the nucleosome assembly protein SET that binds the WW domain and is required for transactivation mediated by the Fe65Gal4DB fusion protein [26].
The phosphorylation of Tyr547 in the Fe65 PTB2 domain mediated by the Abl kinase stimulates its transactivational activity [62], possibly preventing the association of Fe65 with Dexras, a Ras family GTPase, that acts as an inhibitor of the complex [34].
The search for target genes regulated by the AICD has been complex and has yielded conflicting findings. It has been reported that the AICD/Fe65 complex regulates the APP expression itself [63], glycogen synthase kinase (GSK)-3β [63, 64], Tip60 [63], the β-secretase (BACE1) [63], the primate-specific caspase 4 [35], the Aβ degrading enzyme neprylisin [61, 65, 66], the tetraspanin KAI1 [26, 63], the lipoprotein receptor-related protein (LRP1) [60], the epidermal growth factor receptor (EGFR) [67], and the tumor suppressor p53 [68]. Nevertheless, many of these studies have been refuted by others, which using different strategies for modulating the AICD/Fe65 complex did not produce changes in the expression of the aforementioned genes [69–73].
The possible origin of the reported differences is unclear, but regarding the most intensively discussed target, neprilysin, recent data may shed light on the controversy. It was shown that the AICD-binding to neprilysin gene promoter is cell type-dependent [61, 74]. Furthermore, AICD-dependent gene regulation is influenced by the passage number and cell density [75], providing two likely experimental explanations for this disagreement.
2.3. Roles in DNA Repair
The majority of the evidence pointing to a role of AICD in transcriptional responses derives from the use of artificial reporter systems that in fact measure the release of components from the membrane, without monitoring endogenous transcriptional activity. Besides the potential participation of Fe65 in promoting the expression of several genes described above, Fe65 has been also proposed to perform other nuclear functions such as the repair of DNA damage. Fe65 KO mice are more sensitive to DNA damage, and this can be overcome by increasing the availability of nuclear Fe65 [76]. Moreover, genotoxic damage produces a rapid translocation of Fe65 to the nuclear matrix [77] and stimulates APP processing by the γ-secretase complex [76] and APP phosphorylation in Thr668 [77], two mechanisms that allow translocation to the nucleus of the complexes associated with AICD. Fe65 is required for efficient repair of DNA double strand breaks (DSB), a function that depends on its interaction with Tip60 and AICD [78]. The Fe65-dependent recruitment of Tip60 to DSB sites is essential because the histone acetyltransferase activity leads to chromatin opening at the injury site, enabling the access of the complexes involved in repair [79]. On the other hand, Tip60 acetylates and activates the ataxia telangiectasia mutated (ATM) kinase [80] which in turn phosphorylates a histone H2A variant, called H2AX, which acts as a mark for the recruitment of the reparation machinery. Changes in H2AX phosphorylation could be also dependent on the stability of p53 in a mechanism that requires the accumulation of Fe65 in the nuclei [81, 82]. However, the fact that phosphorylated H2AX may be also increased in Fe65 KO cells under genotoxic damage [76] suggests that complementary mechanisms may regulate this behavior.
2.4. Roles in Brain Development
Fe65 is highly enriched in the brain where it is expressed as two isoforms produced by the alternative splicing of a 6 bp miniexon. The isoform that includes this exon (which encodes Arg-Glu inserted in the PTB1 domain) is expressed exclusively in neurons, whereas the isoform lacking these two aminoacids is expressed only in nonneuronal cells [83]. Fe65 protein expression may change during development [84] and also in pathological conditions such as AD [85], opening up the possibility that it participates in plastic processes in neurons, which is reflected in the phenotype of Fe65 and Fe65L1 double KO mouse. These mice exhibit defects in the positioning of cortical neurons characterized by the presence of ectopic neurons that break the pialmeningeal basement membrane and displace Cajal-Retzius neurons and also have serious defects in axonal projections [86]. Many of these phenotypical features are shared by mice lacking some of the Fe65-binding partners such as the APP family [87] and the mammalian homolog of Drosophila enabled (Mena) [88]. Mena belongs to a family of proteins that regulate actin dynamics and thereby modulate cell motility and morphology. Mena is located in areas of dynamic actin remodeling such as lamellipodia and growth cones and interacts with the actin-binding protein, profilin. Mena interacts with the Fe65 WW domain, assembling a macromolecular complex with APP [20] that regulates axonal branching [89], cell motility [90], and possibly the dynamics of actin at the growth cone and synapsis [91].
In a previous attempt to generate a Fe65 KO, it was expressed a truncated protein lacking the N-terminal domain and translated from Met261. This 60 kDa variant does not contain the WW domain and does not display the transactivation activity of the larger isoform [92]. In spite of the expression of this smaller protein, the animal shows defects in hippocampal-dependent learning and long-term potentiation (LTP) [93, 94]. However, it is difficult to assess whether these defects are due to the 97 kDa isoform loss or the appearance of this new 60 kDa isoform acting as a dominant negative protein. Behavioral studies in Fe65/Fe65L1 KO mice could help to clarify these points.
3. The AICD/Fe65 Transgenic Mice: New Perspectives in AD
Although the amyloid cascade hypothesis has become the mainstream in the study of AD neurobiological mechanisms, several groups have recently suggested that this should be at least reevaluated in the light of new findings [95–97]. Transgenic mice that overexpress the AICD and the adapter Fe65 in the forebrain (under the control of the CaMKIIα promoter) [98] display several neuropathological features observed in various transgenic models and in the AD patients brains, with the exception that they do not show Aβ accumulation in the brain [11]. The expression of AICD together with Fe65 seems to be essential to induce an AD-like phenotype in the transgenic model, since a single AICD transgenic mouse developed by an independent group does not present the characteristics of the double transgenic [99], indicating that the functional relationship between both proteins, discussed in the previous sections, is indeed essential.
3.1. Cell Signaling Alterations
As in the brain of patients with AD and several other transgenic models used to study AD, the AICD/Fe65 mice show an increase in GSK-3β activity. Interestingly, the double AICD/Fe65 transgenic does not affect the GSK-3β mRNA or protein levels, as would be expected from a previous study which suggests that the kinase should be transcriptionally regulated by the AICD/Fe65 complex [64]. Kinase activation in the double transgenic is indeed correlated with an increase in the Tyr216 activating phosphorylation and a decrease in the Ser9 inhibitory phosphorylation [98]. A molecular explanation for this may be related with the fact that Fe65, through its WW domain, interacts and promotes GSK-3β phosphorylation on Tyr216 [33]. Increased GSK-3β activity in the AICD/Fe65 mice produces hyperphosphorylation of two direct targets: the microtubule-binding proteins, collapsin-response mediator protein (CRMP)-2 and tau [11, 98]. Increased CRMP-2 phosphorylation is also found in transgenic mice expressing mutated forms of APP and presenilin (PS)-1 and also in the cerebral cortex of AD patients. Increased CRMP-2 phosphorylation is an early event that precedes the formation of amyloid plaques and neurofibrillary tangles. Interestingly, this posttranslational modification seems to be specific for AD, since it has not been reported in other neurodegenerative conditions like the frontotemporal dementia and Pick's Disease [100, 101].
Hyperphosphorylation of tau is the initial event in the pathway to tau self-aggregation, forming the paired helical filaments (PHFs). PHFs are found at the core of the highly insoluble intraneuronal neurofibrillary tangles, one of the two neuropathological lesions (another is the senile plaques) that characterize the AD patients brains. The AICD/Fe65 mouse shares with 3xTg mice [102] the capacity to promote the formation of tau insoluble aggregates, which are not observed in most mouse models for AD [11].
3.2. Neuronal Activity Impairments
The AICD/Fe65 double transgenic mouse has nonconvulsive seizures with aging, abnormal electroencephalogram (EEG) spiking, and a greater sensitivity to seizures induced by kainic acid (KA) in young animals [103]. It also presents several alterations in hippocampal neural circuits, characterized by abnormal sprouting of the mossy fiber terminals with increased neuropeptide Y (NPY) expression and loss of calbindin-positive neurons [104]. Alterations in the EEGs and seizures have been observed in AD patients and in mouse models for this pathology, such as mice R1.40 (with APPswe), APPPS1, and PDAPP [105, 106].
3.3. Memory Deficits and Neurodegeneration
Aged AICD/Fe65 animals (>18 months) show neurodegeneration in the CA3 hippocampal area, although the defects in working memory (evaluated by the Y maze paradigm) start at a young age (8 months). Interestingly, these changes occur in the absence of increased Aβ levels [11]. Since most of the mouse models for AD are based on the expression of mutant variants of the human APP or presenilin found in cases of familiar AD, the identity of neurotoxic APP fragments has not been clearly discerned yet. Several studies have shown that Aβ deposition in senile plaques does not correlate with neuronal death and cognitive deficits present in different transgenic models [107, 108]. For example, the overexpression of wild type hAPP in mice produces memory deficits, tau hyperphosphorylation, synaptic loss, and neurodegeneration without inducing an increase in Aβ levels [109]. Surprisingly, overexpression of hAPP together with β-secretase in mice induces a decrease in Aβ levels and plaque deposition, but the animals suffer severe neurodegenerative disorders and learning defects [110]. In both models, an accumulation of C-terminal fragments of APP including the AICD is observed [109, 110]. Is it therefore possible that this fragment generated along with the Aβ may be responsible for the alterations in transgenic models of AD? Interestingly, the AD model termed PDAPP, when combined with a mutated form of the AICD (D664A), shows a complete reversion of the neuropathological hallmarks of the disease, including synaptic loss, the dentate gyrus atrophy, the astrogliosis, the deficits in synaptic transmission and memory, and the behavioral abnormalities without affecting the Aβ levels or the plaque accumulation [111–114]. These results strongly suggest that the causal relationship between the Aβ accumulation and the neuropathological defects usually associated with AD may be challenged and position the AICD as a good candidate to explain the effects observed in various transgenic models based on mutations in APP and PS1.
4. Conclusions
The two hallmarks of AD, the amyloid plaques, and neurofibrillary tangles, which are elegantly related through the amyloid cascade hypothesis, are the main components in the current research on the molecular mechanisms leading to this pathology. Since its origin, the amyloid cascade hypothesis has accumulated substantial evidence in its support, which has virtually overshadowed the fact that clinical trials based on this hypothesis have been shown to be unsuccessful [115]. One of many possibilities to explain the failure of clinical trials could be related with the fact that several mouse models express the human-mutated APP found in familial AD, so it is unclear which abnormalities detected in these models are product of specific Aβ species (like oligomers) or another toxic metabolites of APP (like AICD) or simply due to effects of overexpression of hAPP. However, the evidence collected from the transgenic models here reviewed could help to discern whether the Aβ species or the AICD are the key elements triggering neurodegeneration. Three independent transgenic mice lines (a single transgenic of hAPP, a double AICD/Fe65 transgenic, and the double hAPP/β-secretase transgenic) recapitulate the neuropathological alterations of the disease without any increase in Aβ secretion. All of these models have an accumulation of the APP C-terminal fragments. Moreover, the introduction of a point mutation in the AICD in transgenic mice expressing the hAPP with the swe and Indiana mutations, the AD-like phenotype is reversed, in spite of increased Aβ production. All of these evidences suggest that the AICD could be acting as the bona fide toxic intermediate in the AD progression and could become a target for future therapeutic interventions against this devastating disease.
Acknowledgments
This work was supported by CONICYT Fellowship D-21070173 and 24090187 (to DB) and Grants FONDECYT 1095089 and ICM P05-001-F (to CG-B).
References
- 1.Ebinu JO, Yankner BA. A RIP tide in neuronal signal transduction. Neuron. 2002;34(4):499–502. doi: 10.1016/s0896-6273(02)00704-3. [DOI] [PubMed] [Google Scholar]
- 2.Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Molecular Biology of the Cell. 1999;10(11):3787–3799. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang X, Sato R, Brown MS, Hua X, Goldstein JL. SREBP-1, a membrane-bound transcription factor released by sterol-regulated proteolysis. Cell. 1994;77(1):53–62. doi: 10.1016/0092-8674(94)90234-8. [DOI] [PubMed] [Google Scholar]
- 4.Schroeter EH, Kisslinger JA, Kopan R. Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature. 1998;393(6683):382–386. doi: 10.1038/30756. [DOI] [PubMed] [Google Scholar]
- 5.Frigerio CS, Fadeeva JV, Minogue AM, et al. β-Secretase cleavage is not required for generation of the intracellular C-terminal domain of the amyloid precursor family of proteins. FEBS Journal. 2010;277(6):1503–1518. doi: 10.1111/j.1742-4658.2010.07579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goodger ZV, Rajendran L, Trutzel A, Kohli BM, Nitsch RM, Konietzko U. Nuclear signaling by the APP intracellular domain occurs predominantly through the amyloidogenic processing pathway. Journal of Cell Science. 2009;122(20):3703–3714. doi: 10.1242/jcs.048090. [DOI] [PubMed] [Google Scholar]
- 7.Belyaev ND, Kellett KA, Beckett C, et al. The transcriptionally active amyloid precursor protein (APP) intracellular domain is preferentially produced from the 695 isoform of APP in a β-secretase-dependent pathway. Journal of Biological Chemistry. 2010;285(53):41443–41454. doi: 10.1074/jbc.M110.141390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kakuda N, Funamoto S, Yagishita S, et al. Equimolar production of amyloid β-protein and amyloid precursor protein intracellular domain from β-carboxyl-terminal fragment by γ-secretase. Journal of Biological Chemistry. 2006;281(21):14776–14786. doi: 10.1074/jbc.M513453200. [DOI] [PubMed] [Google Scholar]
- 9.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 10.Pimplikar SW, Suryanarayana A. Detection of APP intracellular domain in brain tissue. Methods in Molecular Biology. 2011;670:85–91. doi: 10.1007/978-1-60761-744-0_7. [DOI] [PubMed] [Google Scholar]
- 11.Ghosal K, Vogt DL, Liang M, Shen Y, Lamb BT, Pimplikar SW. Alzheimer’s disease-like pathological features in transgenic mice expressing the APP intracellular domain. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(43):18367–18372. doi: 10.1073/pnas.0907652106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pastorino L, Sun A, Lu PJ, et al. The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-β production. Nature. 2006;440(7083):528–534. doi: 10.1038/nature04543. [DOI] [PubMed] [Google Scholar]
- 13.Borg JP, Ooi J, Levy E, Margolis B. The phosphotyrosine interaction domains of X11 and FE65 bind to distinct sites on the YENPTY motif of amyloid precursor protein. Molecular and Cellular Biology. 1996;16(11):6229–6241. doi: 10.1128/mcb.16.11.6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Howell BW, Lanier LM, Frank R, Gertler FB, Cooper JA. The disabled 1 phosphotyrosine-binding domain binds to the internalization signals of transmembrane glycoproteins and to phospholipids. Molecular and Cellular Biology. 1999;19(7):5179–5188. doi: 10.1128/mcb.19.7.5179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tarr PE, Roncarati R, Pelicci G, Pelicci PG, D’Adamio L. Tyrosine phosphorylation of the β-amyloid precursor protein cytoplasmic tail promotes interaction with Shc. Journal of Biological Chemistry. 2002;277(19):16798–16804. doi: 10.1074/jbc.M110286200. [DOI] [PubMed] [Google Scholar]
- 16.Matsuda S, Yasukawa T, Homma Y, et al. c-Jun N-terminal kinase (JNK)-interacting protein-1b/islet-brain-1 scaffolds Alzheimer’s amyloid precursor protein with JNK. Journal of Neuroscience. 2001;21(17):6597–6607. doi: 10.1523/JNEUROSCI.21-17-06597.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fiore F, Zambrano N, Minopoli G, Donini V, Duilio A, Russo T. The regions of the Fe65 protein homologous to the phosphotyrosine interaction/phosphotyrosine binding domain of Shc bind the intracellular domain of the Alzheimer’s amyloid precursor protein. Journal of Biological Chemistry. 1995;270(52):30853–30856. doi: 10.1074/jbc.270.52.30853. [DOI] [PubMed] [Google Scholar]
- 18.Guénette SY, Chen J, Jondro PD, Tanzi RE. Association of a novel human FE65-like protein with the cytoplasmic domain of the β-amyloid precursor protein. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(20):10832–10837. doi: 10.1073/pnas.93.20.10832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duilio A, Faraonio R, Minopoli G, Zambrano N, Russo T. Fe65L2: a new member of the Fe65 protein family interacting with the intracellular domain of the Alzheimer’s β-amyloid precursor protein. Biochemical Journal. 1998;330(1):513–519. doi: 10.1042/bj3300513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ermekova KS, Zambrano N, Linn H, et al. The WW domain of neural protein FE65 interacts with proline-rich motifs in Mena, the mammalian homolog of Drosophila enabled. Journal of Biological Chemistry. 1997;272(52):32869–32877. doi: 10.1074/jbc.272.52.32869. [DOI] [PubMed] [Google Scholar]
- 21.Zambrano N, Minopoli G, de Candia P, Russo T. The Fe65 adaptor protein interacts through its PID1 domain with the transcription factor CP2/LSF/LBP1. Journal of Biological Chemistry. 1998;273(32):20128–20133. doi: 10.1074/jbc.273.32.20128. [DOI] [PubMed] [Google Scholar]
- 22.Trommsdorff M, Borg JP, Margolis B, Herz J. Interaction of cytosolic adaptor proteins with neuronal apolipoprotein E receptors and the amyloid precursor protein. Journal of Biological Chemistry. 1998;273(50):33556–33560. doi: 10.1074/jbc.273.50.33556. [DOI] [PubMed] [Google Scholar]
- 23.Zambrano N, Bruni P, Minopoli G, et al. The β-amyloid precursor protein APP is tyrosine-phosphorylated in cells expressing a Constitutively active form of the Abl protoncogene. Journal of Biological Chemistry. 2001;276(23):19787–19792. doi: 10.1074/jbc.M100792200. [DOI] [PubMed] [Google Scholar]
- 24.Cao X, Sudhof TC. A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science. 2001;293(5527):115–120. doi: 10.1126/science.1058783. [DOI] [PubMed] [Google Scholar]
- 25.Araki Y, Miyagi N, Kato N, et al. Coordinated metabolism of Alcadein and amyloid β-protein precursor regulates FE65-dependent gene transactivation. Journal of Biological Chemistry. 2004;279(23):24343–24354. doi: 10.1074/jbc.M401925200. [DOI] [PubMed] [Google Scholar]
- 26.Telese F, Bruni P, Donizetti A, et al. Transcription regulation by the adaptor protein Fe65 and the nucleosome assembly factor SET. EMBO Reports. 2005;6(1):77–82. doi: 10.1038/sj.embor.7400309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barbato C, Canu N, Zambrano N, et al. Interaction of Tau with Fe65 links tau to APP. Neurobiology of Disease. 2005;18(2):399–408. doi: 10.1016/j.nbd.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 28.Sumioka A, Nagaishi S, Yoshida T, Lin A, Miura M, Suzuki T. Role of 14-3-3γ in FE65-dependent gene transactivation mediated by the amyloid β-protein precursor cytoplasmic fragment. Journal of Biological Chemistry. 2005;280(51):42364–42374. doi: 10.1074/jbc.M504278200. [DOI] [PubMed] [Google Scholar]
- 29.Masin M, Kerschensteiner D, Dümke K, Rubio ME, Soto F. Fe65 interacts with P2X2 subunits at excitatory synapses and modulates receptor function. Journal of Biological Chemistry. 2006;281(7):4100–4108. doi: 10.1074/jbc.M507735200. [DOI] [PubMed] [Google Scholar]
- 30.Hoe HS, Magill LA, Guenette S, Fu Z, Vicini S, Rebeck GW. FE65 interaction with the ApoE receptor ApoEr2. Journal of Biological Chemistry. 2006;281(34):24521–24530. doi: 10.1074/jbc.M600728200. [DOI] [PubMed] [Google Scholar]
- 31.Bao J, Cao C, Zhang X, Jiang F, Nicosia SV, Bai W. Suppression of β-amyloid precursor protein signaling into the nucleus by estrogens mediated through complex formation between the estrogen receptor and Fe65. Molecular and Cellular Biology. 2007;27(4):1321–1333. doi: 10.1128/MCB.01280-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee EJ, Hyun SH, Chun J, Kang SS. Human NIMA-related kinase 6 is one of the Fe65 WW domain binding proteins. Biochemical and Biophysical Research Communications. 2007;358(3):783–788. doi: 10.1016/j.bbrc.2007.04.203. [DOI] [PubMed] [Google Scholar]
- 33.Lee EJ, Hyun S, Chun J, et al. The PPLA motif of glycogen synthase kinase 3β is required for interaction with Fe65. Molecules and Cells. 2008;26(1):100–105. [PubMed] [Google Scholar]
- 34.Lau KF, Chan WM, Perkinton MS, et al. Dexras1 interacts with FE65 to regulate FE65-amyloid precursor protein-dependent transcription. Journal of Biological Chemistry. 2008;283(50):34728–34737. doi: 10.1074/jbc.M801874200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kajiwara Y, Akram A, Katsel P, et al. FE65 binds teashirt, inhibiting expression of the primate-specific caspase-4. PLoS One. 2009;4(4) doi: 10.1371/journal.pone.0005071. Article ID e5071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eun JL, Hyun S, Chun J, Sung HS, Kang SS. Ubiquitylation of Fe65 adaptor protein by neuronal precursor cell expressed developmentally down regulated 4-2 (Nedd4-2) via the WW domain interaction with Fe65. Experimental and Molecular Medicine. 2009;41(8):555–568. doi: 10.3858/emm.2009.41.8.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kwon OY, Hwang K, Kim JA, et al. Dab1 binds to Fe65 and diminishes the effect of Fe65 or LRP1 on APP processing. Journal of Cellular Biochemistry. 2010;111(2):508–519. doi: 10.1002/jcb.22738. [DOI] [PubMed] [Google Scholar]
- 38.Alvira-Botero X, Pérez-Gonzalez R, Spuch C, et al. Megalin interacts with APP and the intracellular adapter protein FE65 in neurons. Molecular and Cellular Neuroscience. 2010;45(3):306–315. doi: 10.1016/j.mcn.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 39.Wang P-L, Niidome T, Kume T, Akaike A, Kihara T, Sugimoto H. Functional and molecular interactions between Rac1 and FE65. NeuroReport. 2011;22(14):716–720. doi: 10.1097/WNR.0b013e32834aca9d. [DOI] [PubMed] [Google Scholar]
- 40.Sabo SL, Lanier LM, Ikin AF, et al. Regulation of β-amyloid secretion by FE65, an amyloid protein precursor-binding protein. Journal of Biological Chemistry. 1999;274(12):7952–7957. doi: 10.1074/jbc.274.12.7952. [DOI] [PubMed] [Google Scholar]
- 41.Xie Z, Dong Y, Maeda U, Xia W, Tanzi RE. RNA interference silencing of the adaptor molecules ShcC and Fe65 differentially affect amyloid precursor protein processing and Aβ generation. Journal of Biological Chemistry. 2007;282(7):4318–4325. doi: 10.1074/jbc.M609293200. [DOI] [PubMed] [Google Scholar]
- 42.Suh J, Lyckman A, Wang L, Eckman EA, Guénette SY. FE65 proteins regulate NMDA receptor activation-induced amyloid precursor protein processing. Journal of Neurochemistry. 2011;119(2):377–388. doi: 10.1111/j.1471-4159.2011.07419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barbagallo AP, Weldon R, Tamayev R, et al. Tyr682 in the intracellular domain of APP regulates amyloidogenic APP processing in vivo. PLoS One. 2010;5(11) doi: 10.1371/journal.pone.0015503. Article ID e15503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu Q, Wang L, Yang Z, Cool BH, Zitnik G, Martin GM. Endoproteolytic cleavage of FE65 converts the adaptor protein to a potent suppressor of the sAPPα pathway in primates. Journal of Biological Chemistry. 2005;280(13):12548–12558. doi: 10.1074/jbc.M411855200. [DOI] [PubMed] [Google Scholar]
- 45.Pietrzik CU, Yoon IS, Jaeger S, Busse T, Weggen S, Koo EH. FE65 constitutes the functional link between the low-density lipoprotein receptor-related protein and the amyloid precursor protein. Journal of Neuroscience. 2004;24(17):4259–4265. doi: 10.1523/JNEUROSCI.5451-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hoe HS, Tran TS, Matsuoka Y, Howell BW, Rebeck GW. DAB1 and reelin effects on amyloid precursor protein and ApoE receptor 2 trafficking and processing. Journal of Biological Chemistry. 2006;281(46):35176–35185. doi: 10.1074/jbc.M602162200. [DOI] [PubMed] [Google Scholar]
- 47.Chin J, Massaro CM, Palop JJ, et al. Reelin depletion in the entorhinal cortex of human amyloid precursor protein transgenic mice and humans with Alzheimer’s disease. Journal of Neuroscience. 2007;27(11):2727–2733. doi: 10.1523/JNEUROSCI.3758-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kocherhans S, Madhusudan A, Doehner J, et al. Reduced Reelin expression accelerates amyloid-β plaque formation and Tau pathology in transgenic Alzheimer’s disease mice. Journal of Neuroscience. 2010;30(27):9228–9240. doi: 10.1523/JNEUROSCI.0418-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cao X, Sudhof TC. Dissection of amyloid-β precursor protein-dependent transcriptional transactivation. Journal of Biological Chemistry. 2004;279(23):24601–24611. doi: 10.1074/jbc.M402248200. [DOI] [PubMed] [Google Scholar]
- 50.Yang Z, Cool BH, Martin GM, Hu Q. A dominant role for FE65 (APBB1) in nuclear signaling. Journal of Biological Chemistry. 2006;281(7):4207–4214. doi: 10.1074/jbc.M508445200. [DOI] [PubMed] [Google Scholar]
- 51.Minopoli G, De Candia P, Bonetti A, Faraonio R, Zambrano N, Russo T. The β-amyloid precursor protein functions as a cytosolic anchoring site that prevents Fe65 nuclear translocation. Journal of Biological Chemistry. 2001;276(9):6545–6550. doi: 10.1074/jbc.M007340200. [DOI] [PubMed] [Google Scholar]
- 52.Hass MR, Yankner BA. A γ-secretase-independent mechanism of signal transduction by the amyloid precursor protein. Journal of Biological Chemistry. 2005;280(44):36895–36904. doi: 10.1074/jbc.M502861200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Patrick GN, Zukerberg L, Nikolic M, De La Monte S, Dikkes P, Tsai LH. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402(6762):615–622. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- 54.Nakaya T, Suzuki T. Role of APP phosphorylation in FE65-dependent gene transactivation mediated by AICD. Genes to Cells. 2006;11(6):633–645. doi: 10.1111/j.1365-2443.2006.00968.x. [DOI] [PubMed] [Google Scholar]
- 55.Radzimanowski J, Simon B, Sattler M, Beyreuther K, Sinning I, Wild K. Structure of the intracellular domain of the amyloid precursor protein in complex with Fe65-PTB2. EMBO Reports. 2008;9(11):1134–1140. doi: 10.1038/embor.2008.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kimberly WT, Zheng JB, Guénette SY, Selkoe DJ. The intracellular domain of the β-amyloid precursor protein is stabilized by Fe65 and translocates to the nucleus in a notch-like manner. Journal of Biological Chemistry. 2001;276(43):40288–40292. doi: 10.1074/jbc.C100447200. [DOI] [PubMed] [Google Scholar]
- 57.Konietzko U, Goodger ZV, Meyer M, et al. Co-localization of the amyloid precursor protein and Notch intracellular domains in nuclear transcription factories. Neurobiology of Aging. 2010;31(1):58–73. doi: 10.1016/j.neurobiolaging.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Muresan Z, Muresan V. A phosphorylated, carboxy-terminal fragment of β-amyloid precursor protein localizes to the splicing factor compartment. Human Molecular Genetics. 2004;13(5):475–488. doi: 10.1093/hmg/ddh054. [DOI] [PubMed] [Google Scholar]
- 59.Baek SH, Ohgi KA, Rose DW, Koo EH, Glass CK, Rosenfeld MG. Exchange of N-CoR corepressor and Tip60 coactivator complexes links gene expression by NF-κB and β-amyloid precursor protein. Cell. 2002;110(1):55–67. doi: 10.1016/s0092-8674(02)00809-7. [DOI] [PubMed] [Google Scholar]
- 60.Liu Q, Zerbinatti CV, Zhang J, et al. Amyloid precursor protein regulates brain apolipoprotein E and cholesterol metabolism through lipoprotein receptor LRP1. Neuron. 2007;56(1):66–78. doi: 10.1016/j.neuron.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Belyaev ND, Nalivaeva NN, Makova NZ, Turner AJ. Neprilysin gene expression requires binding of the amyloid precursor protein intracellular domain to its promoter: implications for Alzheimer disease. EMBO Reports. 2009;10(1):94–100. doi: 10.1038/embor.2008.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Perkinton MS, Standen CL, Lau KF, et al. The c-Abl tyrosine kinase phosphorylates the Fe65 adaptor protein to stimulate Fe65/amyloid precursor protein nuclear signaling. Journal of Biological Chemistry. 2004;279(21):22084–22091. doi: 10.1074/jbc.M311479200. [DOI] [PubMed] [Google Scholar]
- 63.von Rotz RC, Kohli BM, Bosset J, et al. The APP intracellular domain forms nuclear multiprotein complexes and regulates the transcription of its own precursor. Journal of Cell Science. 2004;117(19):4435–4448. doi: 10.1242/jcs.01323. [DOI] [PubMed] [Google Scholar]
- 64.Kim HS, Kim EM, Lee JP, et al. C-terminal fragments of amyloid precursor protein exert neurotoxicity by inducing glycogen synthase kinase-3β expression. FASEB Journal. 2003;17(13):1951–1953. doi: 10.1096/fj.03-0106fje. [DOI] [PubMed] [Google Scholar]
- 65.Pardossi-Piquard R, Petit A, Kawarai T, et al. Presenilin-dependent transcriptional control of the Aβ-degrading enzyme neprilysin by intracellular domains of βAPP and APLP. Neuron. 2005;46(4):541–554. doi: 10.1016/j.neuron.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 66.Pardossi-Piquard R, Dunys J, Yu G, George-Hyslop PS, Alves Da Costa C, Checler F. Neprilysin activity and expression are controlled by nicastrin. Journal of Neurochemistry. 2006;97(4):1052–1056. doi: 10.1111/j.1471-4159.2006.03822.x. [DOI] [PubMed] [Google Scholar]
- 67.Zhang YW, Wang R, Liu Q, Zhang H, Liao FF, Xu H. Presenilin/γ-secretase-dependent processing of β-amyloid precursor protein regulates EGF receptor expression. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(25):10613–10618. doi: 10.1073/pnas.0703903104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Alves da Costa C, Sunyach C, Pardossi-Piquard R, et al. Presenilin-dependent gamma-secretase-mediated control of p53-associated cell death in Alzheimer’s disease. The Journal of Neuroscience. 2006;26(23):6377–6385. doi: 10.1523/JNEUROSCI.0651-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen AC, Selkoe DJ. Response to: Pardossi-Piquard etal., "Presenilin-dependent transcriptional control of the Aβ-degrading enzyme neprilysin by intracellular domains of βAPP and APLP." Neuron 46, 541–554. Neuron. 2007;53(4):479–483. doi: 10.1016/j.neuron.2007.01.023. [DOI] [PubMed] [Google Scholar]
- 70.Waldron E, Isbert S, Kern A, et al. Increased AICD generation does not result in increased nuclear translocation or activation of target gene transcription. Experimental Cell Research. 2008;314(13):2419–2433. doi: 10.1016/j.yexcr.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 71.Hébert SS, Serneels L, Tolia A, et al. Regulated intramembrane proteolysis of amyloid precursor protein and regulation of expression of putative target genes. EMBO Reports. 2006;7(7):739–745. doi: 10.1038/sj.embor.7400704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pardossi-Piquard R, Dunys J, Kawarai T, et al. Response to correspondence: Pardossi-Piquard etal., "Presenilin-dependent transcriptional control of the Aβ-degrading enzyme neprilysin by Intracellular Domains of βAPP and APLP." Neuron 46, 541–554. Neuron. 2007;53(4):483–486. doi: 10.1016/j.neuron.2007.01.024. [DOI] [PubMed] [Google Scholar]
- 73.Aydin D, Filippov MA, Tschäpe J, et al. Comparative transcriptome profiling of amyloid precursor protein family members in the adult cortex. BMC Genomics. 2011;12:p. 160. doi: 10.1186/1471-2164-12-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hong Y, Beckett C, Belyaev ND, Turner AJ. The impact of amyloid precursor protein signalling and histone deacetylase inhibition on neprilysin expression in human prostate cells. International Journal of Cancer. 2012;130(4):775–786. doi: 10.1002/ijc.26028. [DOI] [PubMed] [Google Scholar]
- 75.Bauer C, Pardossi-Piquard R, Dunys J, et al. γ-secretase-mediated regulation of neprilysin: influence of cell density and aging and modulation by Imatinib. Journal of Alzheimer’s Disease. 2011;27(3):511–520. doi: 10.3233/JAD-2011-110746. [DOI] [PubMed] [Google Scholar]
- 76.Minopoli G, Stante M, Napolitano F, et al. Essential roles for Fe65, Alzheimer amyloid precursor-binding protein, in the cellular response to DNA damage. Journal of Biological Chemistry. 2007;282(2):831–835. doi: 10.1074/jbc.C600276200. [DOI] [PubMed] [Google Scholar]
- 77.Nakaya T, Kawai T, Suzuki T. Regulation of FE65 nuclear translocation and function by amyloid β-protein precursor in osmotically stressed cells. Journal of Biological Chemistry. 2008;283(27):19119–19131. doi: 10.1074/jbc.M801827200. [DOI] [PubMed] [Google Scholar]
- 78.Stante M, Minopoli G, Passaro F, Raia M, del Vecchio L, Russo T. Fe65 is required for Tip60-directed histone H4 acetylation at DNA strand breaks. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(13):5093–5098. doi: 10.1073/pnas.0810869106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Murr R, Loizou JI, Yang YG, et al. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nature Cell Biology. 2006;8(1):91–99. doi: 10.1038/ncb1343. [DOI] [PubMed] [Google Scholar]
- 80.Sun Y, Jiang X, Chen S, Fernandes N, Price BD. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(37):13182–13187. doi: 10.1073/pnas.0504211102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kawai T, Nakaya T, Suzuki T. Roles of the intramolecular regions of FE65 in its trans-accumulation and in p53 stabilization in the nuclear matrix of osmotically stressed cells. FEBS Letters. 2010;584(4):765–769. doi: 10.1016/j.febslet.2009.12.040. [DOI] [PubMed] [Google Scholar]
- 82.Nakaya T, Kawai T, Suzuki T. Metabolic stabilization of p53 by FE65 in the nuclear matrix of osmotically stressed cells. FEBS Journal. 2009;276(21):6364–6374. doi: 10.1111/j.1742-4658.2009.07349.x. [DOI] [PubMed] [Google Scholar]
- 83.Hu QA, Hearn MG, Jin LW, Bressler SL, Martin GM. Alternatively spliced isoforms of FE65 serve as neuron-specific and non- neuronal markers. Journal of Neuroscience Research. 1999;58(5):632–640. doi: 10.1002/(sici)1097-4547(19991201)58:5<632::aid-jnr4>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 84.Kesavapany S, Banner SJ, Lau KF, et al. Expression of the Fe65 adapter protein in adult and developing mouse brain. Neuroscience. 2002;115(3):951–960. doi: 10.1016/s0306-4522(02)00422-0. [DOI] [PubMed] [Google Scholar]
- 85.Hu Q, Jin LW, Starbuck MY, Martin GM. Broadly altered expression of the mRNA isoforms of FE65, a facilitator of beta amyloidogenesis, in Alzheimer cerebellum and other brain regions. Journal of Neuroscience Research. 2000;60(1):73–86. doi: 10.1002/(SICI)1097-4547(20000401)60:1<73::AID-JNR8>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 86.Guénette S, Chang Y, Hiesberger T, et al. Essential roles for the FE65 amyloid precursor protein-interacting proteins in brain development. EMBO Journal. 2006;25(2):420–431. doi: 10.1038/sj.emboj.7600926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Herms J, Anliker B, Heber S, et al. Cortical dysplasia resembling human type 2 lissencephaly in mice lacking all three APP family members. EMBO Journal. 2004;23(20):4106–4115. doi: 10.1038/sj.emboj.7600390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lanier LM, Gates MA, Witke W, et al. Mena is required for neurulation and commissure formation. Neuron. 1999;22(2):313–325. doi: 10.1016/s0896-6273(00)81092-2. [DOI] [PubMed] [Google Scholar]
- 89.Ikin AF, Sabo SL, Lanier LM, Buxbaum JD. A macromolecular complex involving the amyloid precursor protein (APP) and the cytosolic adapter FE65 is a negative regulator of axon branching. Molecular and Cellular Neuroscience. 2007;35(1):57–63. doi: 10.1016/j.mcn.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sabo SL, Ikin AF, Buxbaum JD, Greengard P. The Alzheimer amyloid precursor protein (APP) and FE65, an APP-binding protein, regulate cell movement. Journal of Cell Biology. 2001;153(7):1403–1414. doi: 10.1083/jcb.153.7.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sabo SL, Ikin AF, Buxbaum JD, Greengard P. The amyloid precursor protein and its regulatory protein, FE65, in growth cones and synapses in vitro and in vivo. Journal of Neuroscience. 2003;23(13):5407–5415. doi: 10.1523/JNEUROSCI.23-13-05407.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cool BH, Zitnik G, Martin GM, Hu Q. Structural and functional characterization of a novel FE65 protein product up-regulated in cognitively impaired FE65 knockout mice. Journal of Neurochemistry. 2010;112(2):410–419. doi: 10.1111/j.1471-4159.2009.06456.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang B, Hu Q, Hearn MG, et al. Isoform-Specific Knockout of FE65 Leads to Impaired Learning and Memory. Journal of Neuroscience Research. 2004;75(1):12–24. doi: 10.1002/jnr.10834. [DOI] [PubMed] [Google Scholar]
- 94.Wang Y, Zhang M, Moon C, et al. The APP-interacting protein FE65 is required for hippocampus-dependent learning and long-term potentiation. Learning and Memory. 2009;16(9):537–544. doi: 10.1101/lm.1499309. [DOI] [PubMed] [Google Scholar]
- 95.Pimplikar SW, Nixon RA, Robakis NK, Shen J, Tsai LH. Amyloid-independent mechanisms in Alzheimer’s disease pathogenesis. Journal of Neuroscience. 2010;30(45):14946–14954. doi: 10.1523/JNEUROSCI.4305-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pimplikar SW. Reassessing the amyloid cascade hypothesis of Alzheimer’s disease. International Journal of Biochemistry and Cell Biology. 2009;41(6):1261–1268. doi: 10.1016/j.biocel.2008.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Castellani RJ, Smith MA. Compounding artefacts with uncertainty, and an amyloid cascade hypothesis that is “too big to fail”. Journal of Pathology. 2011;224(2):147–152. doi: 10.1002/path.2885. [DOI] [PubMed] [Google Scholar]
- 98.Ryan KA, Pimplikar SW. Activation of GSK-3 and phosphorylation of CRMP2 in transgenic mice expressing APP intracellular domain. Journal of Cell Biology. 2005;171(2):327–335. doi: 10.1083/jcb.200505078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Giliberto L, d’Abramo C, Acker CM, Davies P, D’Adamio L. Transgenic expression of the amyloid-β precursor protein-intracellular domain does not induce Alzheimer’s disease-like traits in vivo. PLoS One. 2010;5(7) doi: 10.1371/journal.pone.0011609. Article ID e11609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cole AR, Noble W, Aalten LV, et al. Collapsin response mediator protein-2 hyperphosphorylation is an early event in Alzheimer’s disease progression. Journal of Neurochemistry. 2007;103(3):1132–1144. doi: 10.1111/j.1471-4159.2007.04829.x. [DOI] [PubMed] [Google Scholar]
- 101.Williamson R, van Aalten L, Mann DM, et al. CRMP2 hyperphosphorylation is characteristic of Alzheimer’s Disease and not a feature vommon to other neurodegenerative diseases. Journal of Alzheimer’s Disease. 2011;27(3):615–625. doi: 10.3233/JAD-2011-110617. [DOI] [PubMed] [Google Scholar]
- 102.Oddo S, Caccamo A, Shepherd JD, et al. Triple-transgenic model of Alzheimer’s Disease with plaques and tangles: intracellular Aβ and synaptic dysfunction. Neuron. 2003;39(3):409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 103.Vogt DL, Thomas D, Galvan V, Bredesen DE, Lamb BT, Pimplikar SW. Abnormal neuronal networks and seizure susceptibility in mice overexpressing the APP intracellular domain. Neurobiology of Aging. 2009;32(9):1725–1729. doi: 10.1016/j.neurobiolaging.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ghosal K, Pimplikar SW. Aging and excitotoxic stress exacerbate neural circuit reorganization in amyloid precursor protein intracellular domain transgenic mice. Neurobiology of Aging. 2010;32(12):E1–E9. doi: 10.1016/j.neurobiolaging.2010.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Palop JJ, Chin J, Roberson ED, et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron. 2007;55(5):697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wang J, Ikonen S, Gurevicius K, Van Groen T, Tanila H. Alteration of cortical EEG in mice carrying mutated human APP transgene. Brain Research. 2002;943(2):181–190. doi: 10.1016/s0006-8993(02)02617-3. [DOI] [PubMed] [Google Scholar]
- 107.Gruart A, López-Ramos JC, Muñoz MD, Delgado-García JM. Aged wild-type and APP, PS1, and APP + PS1 mice present similar deficits in associative learning and synaptic plasticity independent of amyloid load. Neurobiology of Disease. 2008;30(3):439–450. doi: 10.1016/j.nbd.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 108.Koistinaho M, Ort M, Cimadevilla JM, et al. Specific spatial learning deficits become severe with age in β-amyloid precursor protein transgenic mice that harbor diffuse β-amyloid deposits but do not form plaques. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(25):14675–14680. doi: 10.1073/pnas.261562998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Simon AM, Schiapparelli L, Salazar-Colocho P, et al. Overexpression of wild-type human APP in mice causes cognitive deficits and pathological features unrelated to Aβ levels. Neurobiology of Disease. 2009;33(3):369–378. doi: 10.1016/j.nbd.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 110.Rockenstein E, Mante M, Alford M, et al. High β-secretase activity elicits neurodegeneration in transgenic mice despite reductions in amyloid-β levels: implications for the treatment of Alzheimer disease. Journal of Biological Chemistry. 2005;280(38):32957–32967. doi: 10.1074/jbc.M507016200. [DOI] [PubMed] [Google Scholar]
- 111.Zhang J, Gorostiza OF, Tang H, Bredesen DE, Galvan V. Reversal of learning deficits in hAPP transgenic mice carrying a mutation at Asp664: a role for early experience. Behavioural Brain Research. 2010;206(2):202–207. doi: 10.1016/j.bbr.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Galvan V, Zhang J, Gorostiza OF, et al. Long-term prevention of Alzheimer’s disease-like behavioral deficits in PDAPP mice carrying a mutation in Asp664. Behavioural Brain Research. 2008;191(2):246–255. doi: 10.1016/j.bbr.2008.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Galvan V, Gorostiza OF, Banwait S, et al. Reversal of Alzheimer’s-like pathology and behavior in human APP transgenic mice by mutation of Asp664. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(18):7130–7135. doi: 10.1073/pnas.0509695103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Saganich MJ, Schroeder BE, Galvan V, Bredesen DE, Koo EH, Heinemann SF. Deficits in synaptic transmission and learning in amyloid precursor protein (APP) transgenic mice require C-terminal cleavage of APP. Journal of Neuroscience. 2006;26(52):13428–13436. doi: 10.1523/JNEUROSCI.4180-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hardy J. The amyloid hypothesis for Alzheimer’s disease: a critical reappraisal. Journal of Neurochemistry. 2009;110(4):1129–1134. doi: 10.1111/j.1471-4159.2009.06181.x. [DOI] [PubMed] [Google Scholar]