An official website of the United States government
Here's how you know
Official websites use .gov
A
.gov website belongs to an official
government organization in the United States.
Secure .gov websites use HTTPS
A lock (
) or https:// means you've safely
connected to the .gov website. Share sensitive
information only on official, secure websites.
As a library, NLM provides access to scientific literature. Inclusion in an NLM database does not imply endorsement of, or agreement with,
the contents by NLM or the National Institutes of Health.
Learn more:
PMC Disclaimer
|
PMC Copyright Notice
*Laboratory of Biophysics, University of Geneva, CH-1211 Geneva 4, Switzerland
†California Institute of Technology, Pasadena, California 91125
1
Minor corrections and editorial changes to the original typed manuscript were made by Costa Georgopoulos2 (University of Utah) and Dominique Belin2 (University of Geneva), who wrote the figure legends. For example, “phage” was systematically replaced by “bacteriophage.” Unfortunately, the three tables and one of the figures that are mentioned in the article were not recovered in R. H. Epstein's archives. The map of Figure 2 was drawn by pencil at an unknown date. Since the last-discovered version of the article must have been written after 1964, the total number of complementation groups was set at 56, according to Edgar and Epstein (1965). One of the mutants shown in Figure 2, amH17, maps to gene 52. Frank Stahl (University of Oregon) rectified punctuation and grammar and formatted the article for the GENETICS of the 21st century.
2
Corresponding authors: Department of Biochemistry, University of Utah, 15 N. Medical Dr. East, Room 4100 EEJ, Salt Lake City, UT 84112. E-mail: costa@biochem.utah.edu; and Department of Pathology and Immunology, University of Geneva, CH-1211 Geneva 4, Switzerland. E-mail: Dominique.Belin@unige.ch
3
Institute of Molecular Biology, University of Oregon, Eugene, Oregon 97403-1229. E-mail: fstahl@uoregon.edu
DICK Epstein and Bob Edgar, both with biology degrees in T4 genetics from the University of Rochester (Gus Doermann, advisor), pursued postdoctoral studies contemporaneously in Max Delbrück's lab at Caltech. T4 phage was (and is) such a beautiful object for molecular investigations that they continued working with it, attracting colleagues, including Charley Steinberg (Wu and Lindahl 2001), as they did so. Their work with T4 went stratospheric with the discovery that most of the essential genes of T4 could be identified by mutation to states that were lethal under some conditions but viable under others (“conditional lethal” mutants). [For particulars of this serendipitous realization, see Edgar and Epstein (1965), Edgar (1966), Stahl (1995), and Edgar (2004).] Armed with libraries of such mutants, and with the collaboration of visiting electron microscopist Edward Kellenberger and others, the Rochesterians famously demonstrated that phage development—at the molecular level—was now open for business (Epstein et al. 1964). That group effort was one of a number of papers that referred to work (“in press,” “unpublished,” or “in contemplation”) describing the birth particulars of the amber mutants of Doermann's T4 strain T4D. The unpublished paper, which should have been the official “birth certificate” of these versatile mutants, has remained unpublished for more than 50 years after its conception. We have submitted the paper (Epstein et al. 2012) now, not only for its historic interest but also because no one knows when a properly registered certificate of birth may become a matter of importance. The authors apparently anticipated that the article would appear as the first of a series on the ambers and their uses. We may speculate on why it was not.
Dick shared his thoughts (and his mutants) freely—he was a devotee of the Delbrückian view that science was a group effort. Writing papers? Not so much. In 1951, Dick explained to me that Don Charles, his first-year advisor at Rochester, was a very bright guy who rarely published because he lost interest in a project as soon as he knew the answer. Does that explain why the text of Epstein et al. (2012) stayed in Dick's filing cabinet until it was retrieved by his colleagues following Dick's death (March 6, 2011)? Probably so.
The T4 world had put Dick's amber mutants to work (Wiberg et al. 1962) before the birth certificate was ready for publication. When that reality was combined with the spectacular 1963 Cold Spring Harbor presentation (Epstein et al. 1964) and the comprehensive GENETICS paper (Edgar et al. 1964), Dick likely looked upon a birth certificate as an uninteresting formality. (The ambers have arrived, haven't they? What more could one want?) Those papers were soon followed by a cornucopia of classic “amber papers” from various labs, demonstrating, for example, circularity of the T4 linkage map, clustering of genes by function, programs of viral development, roles of chain-terminating codons, colinearity of gene and protein product, self-assembly of organelles, mechanisms of DNA replication and its coupling with genetic recombination, and control of gene expression at the translational level. In the face of such a tide of fundamental advances in molecular genetics, the confirming of references, editing, writing a cover letter, and submitting the intended birth certificate must have seemed quite unnecessary and all too boring.
The belated publication by GENETICS of a famously overdue article confirms, in a formal way, the legitimacy of the ambers. In clerical detail, it tells how the mutants were induced, isolated, mapped, and sorted into complementation groups. With a few exceptions, the language and style is that of the era in which it was written. However, there has been some copy editing, presumably like that that would have occurred in the normal course of publication. Its proper place in the literature would have been as a companion paper with Edgar and Lielausis (1964) and Edgar et al. (1964), all three of which would likely have been dated a year or two earlier if progress had been orderly. Read it, and appreciate a time and a man.
Literature Cited
Edgar R. S., 1966. Conditional lethals, pp. 166–170
Phage and the Origins of Molecular Biology, edited by
Cairns J., Stent G. S., Watson J. D.
Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
Edgar R. S., Epstein R. H., 1965. The genetics of a bacterial virus. Sci. Am.
212: 70–78. [DOI] [PubMed] [Google Scholar]
Edgar R. S., Lielausis I., 1964. Temperature-sensitive mutants of bacteriophage T4D: their isolation and genetic characterization. Genetics
49: 649–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
Edgar R. S., Denhardt G. H., Epstein R. H., 1964. A comparative genetic study of conditional lethal mutations of bacteriophage T4D. Genetics
49: 635–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
Epstein R. H., Bolle A., Steinberg C. M., Kellenberger E., Boy de la Tour E., et al. , 1964. Physiological studies on conditional lethal mutants of bacteriophage T4D. Cold Spring Harb. Symp. Quant. Biol.
28: 375–394. [Google Scholar]
Epstein R. H., Bolle A., Steinberg C. M., 2012. Amber mutants of bacteriophage T4D: their isolation and genetic characterization. Genetics
190: 831–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wiberg J. F., Dirksen M. L., Epstein R. H., Luria S. E., Buchanan J. M., 1962. Early enzyme synthesis and its control in E. coli infected with some amber mutants of bacteriophage T4. Proc. Natl. Acad. Sci. USA
48: 293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
We have isolated a large number of mutants of bacteriophage T4D that are unable to form plaques on strain B of Escherichia coli, but are able to grow (nearly) normally on some other strains of E. coli, in particular strain CR63. These mutants, designated amber (am), have been characterized by complementation tests, by genetic crosses, and by their response to chemical mutagens. It is concluded that a particular subclass of base substitution mutations may give rise to amber mutants and that such mutants occur in many genes, which are widely distributed over the T4 genome.
ALTHOUGH information exists concerning the fine structure of a number of genes in various organisms, knowledge of the detailed organization of the genome of any one organism is lacking. Genetic studies with bacteria have indicated a clustered arrangement of genes having related functions, but, due to the genome size in these organisms, it is unlikely that the organization of the total structure will soon be available. It is, however, of interest to know if the clustered arrangement of related genes is a more general feature of a genome, and, further, if additional relations exist between clusters. The study of such problems necessarily involves, as a preliminary step, the isolation and mapping of mutants in most of the genes of a given organism. Such an isolation could in principle be carried out by the selection of mutants with specific requirements, but in practice any such procedure is limited both by absence of prior knowledge of possible defects and, more importantly, by the fact that some defects are not dispensable. Thus, mutations that occur in genes essential for the propagation of the organism are useful for genetic studies only when conditions permitting the growth of the mutants can be found. Ideally, then, a system of conditional lethal mutations is needed, which is not restricted to a minority of the genes of the organism.
In an interesting study of bacteriophage λ (Jacob et al. 1957), mutants were selected using the criterion of inability to produce infective particles following induction of lysogenic bacteria. Although the mutants could be propagated in the prophage state, the difficulty of obtaining infective mutant particles greatly hampered genetic analysis. Campbell (1961) has reported some studies with conditional lethal mutants of bacteriophage λ with which this difficulty is circumvented. These mutants were sensitive to the presence or absence of a suppressor mutation in the bacterial hosts employed, the expression of the mutant being lethal in the absence of the suppressor and nonlethal in the host carrying the suppressor. Similar mutations in the rII region of bacteriophage T4B have been studied by Benzer and Champe (1961, 1962).
In this article we will report on suppressor-sensitive mutants isolated in bacteriophage T4D. These mutations, designated amber (am), are conditional lethals in the sense that they form plaques on Escherichia coli strain CR63, but not on strain B. Wild-type bacteriophage form plaques on both strains. We will show that these mutations are widely distributed in the genome of T4. Edgar and Lielausis (1964) describe similar experiments with a second type of conditional lethal system, mutations resulting in a temperature-sensitive (Ts) phenotype.
Materials and Methods
Media
A description of the media used in the preparation of plates (EHA-bottom agar, EHA-top agar) and for the growth of bacteria and bacteriophage (H-broth) can be found in a recent publication (Steinberg and Edgar 1962). A synthetic medium, M9S (Champe and Benzer 1962), was also used as a liquid nutrient medium in the preparation of some bacteriophage stocks and in some isolations of am mutants employing mutagens.
Bacterial strains
E. coli B, S/6 (a smooth colony variant of strain B made resistant to bacteriophage T6), E. coli C600 (Appleyard 1954), and CR63 (Appleyard et al. 1956) were used as indicator strains and as host cells in experiments. Strains C600 and CR63 are derivatives of K12S and were kindly supplied by Jean Weigle. Plating bacteria were prepared by diluting an inoculum from a saturated overnight culture 100-fold in prewarmed (37°) H-broth. This culture was then grown with vigorous aeration for 2 hr, centrifuged, and then resuspended in 1/10 the volume of fresh H-broth. Bacteria to be used as host cells in one-step growth experiments were prepared in an identical manner except that the dilution from the overnight culture was 1000-fold and the concentration of bacteria was adjusted during resuspension to 4 × 108 cells/ml.
Bacteriophage
All of the bacteriophage used in this study originated from the Doermann strain of T4 (T4D). The rI mutant, r48, has been described by Doermann and Hill (1953). The ts endolysin mutant eC103 was obtained from George Streisinger. The am mutants employed in this study were isolated as described below. Bacteriophage stocks were prepared on strain CR63. The lysates were sterilized with chloroform and centrifuged to remove bacterial debris. In some cases, the stocks were filtered through Mandler candles.
Isolation of mutants
Random plaque technique:
Samples from bacteriophage suspensions were plated at a density of ∼200 plaques per plate on strain CR63. After 12–16 hr of incubation at 37°, plaques from these plates were stabbed with a sterile wire and transferred to a plate containing a layer of S/6 and then to a plate containing a layer of CR63. The bacterial layers were prepared ∼15 min before the plaque transfers. Putative mutants, which failed to grow on S/6, were recovered from the CR63 plates and grown as bacteriophage stocks.
Double-layer technique:
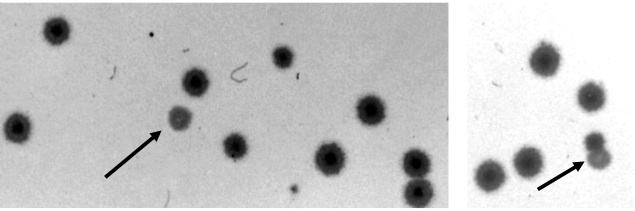
In general, the plaques formed by am mutants and by wild-type bacteriophage on strain CR63 are not morphologically distinguishable. However, wild-type and mutant plaques are distinguishable on a double layer of S/6 and CR63 (Figure 1). A layer of 1.5 ml EHA top agar (modified to contain 0.5% agar) to which S/6 plating bacteria have been added is poured over the surface of a dry plate (4–5 days storage at room temperature) containing ∼50 ml of EHA-bottom agar. After 15–20 min at room temperature, a second layer (1.5 ml of the 0.5% EHA-top agar) containing CR63 bacteria and a sample of bacteriophage is poured over the first. The second layer is allowed to harden and the plates are then incubated at 37° for 12–16 hr. Since only the wild-type bacteriophage can grow on both bacterial strains, the mutant plaques are readily distinguished from wild type by their turbid appearance. The genotypes of plaques exhibiting the turbid phenotype were verified by stab transfers as described in Random plaque technique. About 50–75% of turbid plaques were found to be am mutants in subsequent testing; the remainders were mostly turbid mutants or host-range mutants.
Double-layer technique. Under these plating conditions, am bacteriophage form turbid plaques (indicated by arrows) while am+ bacteriophage form clear plaques.
Late-lysis isolation technique:
Some of our mutants were isolated by selecting for late-lysing mutants. Mutagen-treated bacteriophage were added to log-phase E. coli B (2 × 108/ml) at a multiplicity of 0.1. After 10 min for adsorption of bacteriophage to bacteria, anti-T4 serum (association constant K ∼60) was added to the adsorption tube. Sixty minutes after the addition of serum, the contents of the adsorption tubes were centrifuged three times (in the cold), and the pellet fraction from the final centrifugation was resuspended in fresh medium. (In some cases, unlysed bacteria were separated from the other contents of the adsorption tube by filtration through Millipore filters.) Bacteriophage were liberated from the unlysed bacteria in the pellet fraction by treatment with CHCl3 or lysozyme plus Versene chelating agent. Mutants were then identified by the double-layer technique.
Sources of am mutants
HNO2-induced mutants:
Wild-type T4D bacteriophage at a titer of 109 particles per milliliter in 0.5 M potassium acid phthalate buffer (pH 5) were exposed to HNO2 at a final concentration of 0.5 M. The suspension was incubated at 21°; at intervals, samples were removed from the suspension and diluted 100-fold into M/15 phosphate buffer (pH 7) to terminate the reaction. Within a few hours after sampling, the treated bacteriophage were cycled on strain CR63 to permit the segregation of mutational heterozygotes (Vielmetter and Wieder 1959). For cycling, CR63 (log-phase culture concentrated to 4 × 108 cells/ml) was infected at a multiplicity of 10−2 with treated bacteriophage; the infected cultures were lysed with chloroform 50 min after the addition of bacteriophage.
The increase in the proportion of mutants, as judged from assays of r mutant frequencies in the samples of treated bacteriophage, was linear with time of treatment (0.3% r mutants per minute). Samples containing 0.25–0.5% r mutants were used as source material for the isolation of am mutants. Induced am mutants were recovered either by random plaque isolations or by use of the double-layer technique.
5-bromodeoxyuridine induced mutants:
An overnight culture of strain CR63 was diluted 1000-fold into H-broth and grown with aeration at 30° for 1.5 hr. At this time, 5-bromodeoxyuridine (5-BdU; to a final concentration of 5 μg/ml) was added to the culture, and growth was continued with the same conditions for an additional hour. The bacterial culture was then infected (multiplicity of 10−2) with wild-type bacteriophage and then incubated until the culture cleared. The yield of r mutants was ∼0.5%; am mutants were isolated by the double-layer technique.
EMS-induced mutants:
The procedure of Loveless and Howarth was followed (Loveless and Howarth 1959) except that the temperature chosen for treatment was 45°. As in the case of the nitrous acid mutants, bacteriophage sample that had been treated with ethyl methanesulfonate (EMS) (Shell) was cycled within 2 hr after treatment. Mutants were isolated from samples containing 0.5% r mutants by means of the double-layer technique.
Proflavine-induced mutants:
Strain CR63 was infected with wild-type T4D at a multiplicity of 4. At the time of infection, proflavine (to give a final concentration of 4 μg/ml) was added to the culture. After 25 min of incubation at 30°, samples from the infected culture were diluted 100-fold into H-broth. The diluted samples were grown with aeration for an additional 50 min and then lysed with chloroform. The frequency of r mutants was 0.6%. The double-layer technique was used to search for am mutants.
2-amino-purine-induced mutants:
A culture of strain CR63, grown in M9S at 37° to 107 bacteria per milliliter, was infected at a multiplicity of 10−2 with wild-type T4D bacteriophage. At the time of infection, 2-amino purine (2-AP; to give a final concentration of 1 mg/ml) was added to the culture, and growth was continued until the culture cleared. am mutants were identified by the double-layer technique.
Spontaneous mutants:
A wild-type stock of T4D was cycled on strain CR63. Mutants were isolated by the double-layer technique.
Reversion studies
In general, the procedures of Champe and Benzer (1962) were followed. The procedure for mutagenesis with 5-BdU was modified as follows: E. coli CR63 bacteria at 2 × 108/ml were infected with wild-type T4D at a multiplicity of ∼4. After 5 min for adsorption of bacteriophage to bacteria, the culture was diluted by a factor of 10 and divided into two aliquots. To one aliquot, sufficient 5-BdU was added to give a final concentration of 10 μg/ml; the other aliquot was kept as a control. These tubes were transferred to a shaker and, after 50 min at 30°, the cultures were lysed with chloroform and assayed for total bacteriophage and for am+ revertants.
Complementation tests
Spot test:
A suspension of S/6 in soft agar was poured over the surface of a 3-day-old plate. After 15 min to permit hardening of the layer, a drop from a test suspension of one mutant (at a concentration of 108 bacteriophage per milliliter) was applied to the surface of the plate with a capillary pipette or with a wire loop. After a few minutes, this was overlaid with a drop from a second mutant suspension. All pairwise combinations of mutants and, as controls, each mutant with itself were represented in the tests. Plates were scored after overnight incubation at 37°. Further discussion of this test will be found in Results.
One-step growth test:
Strain B of E. coli was infected with two different am mutants at a multiplicity of infection of ∼5 of each type. The bacteriophage yield 90 min after infection was measured and compared to that obtained in similar infections with wild-type bacteriophage. The yield in the mixed infection was also compared to the yields obtained in control infections with either mutant type alone.
Spot-test identification of am mutants
This test has been used for the determination of output allele ratios in am × am crosses and for the isolation of am double mutants when the mutants belong to different complementation groups. For this purpose, two test plates, one for each of the parents in the cross, are prepared by plating 107–108 bacteriophage with two drops of a strain S/6 plating culture. Plaques obtained from platings of the cross lysate on strain CR63 are transferred by means of a sterile wire to each of the test plates and, in addition, to a plate containing only the nonselective indicator CR63 so that desired genotypes can be recovered after the test results have been obtained. The genotype of any bacteriophage tested can be determined from a comparison of the results of the separate tests against the two parents—wild-type bacteriophage produce a clear spot on both test plates, parental genotypes a clear spot on one of the plates and a turbid spot on the other, and double-am genotypes a turbid spot on both plates.
Crosses
Host bacteria were infected in the presence of M/500 KCN at multiplicities of ∼15 with a bacteriophage mixture containing equal numbers of the two mutant types. The adsorption tubes were agitated by shaking during the 10-min adsorption period. Unadsorbed bacteriophage were determined in a sample taken from the adsorption tube at the end of the adsorption period. Infected bacteria present in these samples (which do not yet contain active progeny virus) were destroyed with chloroform. In all crosses, the unadsorbed bacteriophage were <10% of the input and made a negligible contribution to the final yield of bacteriophage. Following the adsorption period, a 5 × 104-fold dilution of the adsorption mixture was made. The last dilution, containing ∼2 × 103 infected bacteria per milliliter, was kept as a growth tube and incubated at 30°. After a 90-min incubation, the growth tube was shaken with chloroform to ensure complete lysis of the infected bacteria. Determinations of recombinant frequencies (as described below) and total bacteriophage yields were made using appropriate dilutions of the growth tube.
Mutant nomenclature
Mutants are designated by one or two letters indicating the origin of the mutant followed by a number determined by isolation order. The letter designations have the following meanings: S—spontaneous; B—5-bromodeoxyuridine induced; A—2-aminopurine induced; N—nitrous acid induced; E—ethyl methanesulfonate induced; and L—isolated by late-lysis technique. Thus, amN112 is one of a series of nitrous acid-induced amber mutants.
Results
More than 250 mutants of bacteriophage T4D have been isolated that have in common the property that they do not plate on certain strains of E. coli, although they grow normally, or nearly so, on other closely related strains of E. coli. The details of the growth properties of these am mutants on various strains and under various environmental conditions will be the subject of another article; for the present purpose it is sufficient to mention only that the amber mutants form plaques on strains C600 and CR63, but not on strains B or S/6.
Mutation studies
A number of mutagens have been tested for their effectiveness in the production of am mutants. The frequencies of the induced am mutants were compared with the frequencies of r mutants induced in the same exposed population. Spontaneous mutants of the am type are rare. The occurrence of induced am mutants is also rare when proflavine is used as a mutagen although proflavine induces r mutants. In contrast, the mutagens 2-AP, EMS, 5-BdU, and nitrous acid are quite effective in the induction of mutants of both types.
Of 185 mutants examined in some detail, 28 were induced with 5-BdU, 43 with EMS, 100 with HNO2, and 12 with 2-AP; the remaining two mutants were of spontaneous origin. In all isolations with mutagens, the frequency of am mutants was about one-half the r mutant frequency. With the exception of one 5-BdU isolation, the r mutant frequencies were <0.6%.
With one exception, all of the mutants that can be tested revert spontaneously to wild type, and the reversion indices (Benzer 1955) range from 10−3 to 10−8. For three mutants investigated, the reversion appears to be a change at the site of the am mutation and not elsewhere (i.e., in crosses of the revertants to wild type in each case no am segregants were found among ∼104 progeny examined). We have found that ∼20% of our mutants exhibit a temperature-sensitive phenotype on the permissive host CR63. In most cases, revertants of these mutants are no longer temperature sensitive, again suggesting that the reversion is a true return to wild type.
A sample of some 30 mutants of different origins has been studied with respect to their revertibility by NH2OH and by the base analog 2-AP and 5-BdU. Quite independently of the origin of the mutant, these mutants are highly revertible only with 2-AP. Some mutants have been tested for revertibility by proflavine; no increase in revertants was observed.
Complementation studies
While amber mutants do not form plaques on strain B, the restriction of growth is not complete, and some bacteria produce bacteriophage. The fraction of infected B bacteria capable of yielding at least one infective particle (active on strain CR63) depends upon the mutant tested and varies from 1.0 for “leaky” mutants to ∼10−4 for “non-leaky” mutants. There is no obvious correlation between the fraction of yielders and the frequency of back mutation to wild type. In almost all cases that have been examined, the yield per infected bacterium is low, and it is clear that the few particles issuing from the bacteria are predominantly of the am genotype.
While infections of strain B or S/6 with a single am mutant are usually nonproductive, mixed infections with an am mutant and wild type are productive, typically with a normal yield of progeny bacteriophage. Both the am and am+ genotypes appear in the progeny, and the input and output allele frequencies are roughly equal. Thus, the wild-type bacteriophage can supply information that permits both genotypes to grow in a strain in which the mutant alone cannot grow. This result leads to the following question: Are the am mutation alterations in a single essential gene required in the development of T4D, or are they affecting many different genetic functions? This question can be answered by studying mixed infection with two am mutants.
Mixed infection of strain B with two am mutants may be either productive, as in the case of mixed infection with wild-type and am bacteriophage, or nonproductive, as in the case of infection with a single am mutant. Following the usual terminology, mutant pairs that lead to productive infection are said to be complementing, and mutant pairs that do not give rise to active progeny particles are said to be noncomplementing. Extensive complementation tests have been carried out with 197 am mutants by the spot-test method (for details, see Materials and Methods). In this test, drops from suspensions of two mutants are mixed on an agar surface spread with strain B or S/6 bacteria. Drops from suspensions of each mutant are placed separately upon the surface as controls for the mixtures of mutants. The plates are then incubated to permit growth of bacteriophage in the bacterial layer. Since a mutant, by itself, cannot reproduce in strain S/6, the points of application of the control spots are turbid; a mixture of two mutants is also turbid when no complementation occurs. For mutant pairs that complement, repeated cycles of infection and lysis give rise to a clear spot. It should be noted that in some pairwise comparisons distinct wild-type plaques appear on the turbid background of the spot, although the control spots are turbid. These wild-type plaques arise by recombination, and the mutants are classified as non-allelic although noncomplementing; appropriate crosses have confirmed this classification.
Eighty-one of the 5-BdU- and HNO2-induced mutants were tested in all pairwise combinations and were found to fall into 28 discrete groups. All mutants in a group are mutually noncomplementing; all intergroup tests give complementation. From these 81 mutants, one mutant from each group was selected as a tester for use in additional complementation tests with other mutants. Sixty-six other mutants (HNO2-, EMS-, and 5-BdU-induced) were tested with each other in all pairwise combinations and with each of the 28 testers mentioned above. Twelve groups that were not identical to those from the first screening were found. All 147 of the mutants, except the nonreverting one, can be assigned unambiguously to one of the 40 groups. Appropriate crosses have demonstrated that the exceptional nonreverting mutant is in fact a double mutant; each single mutant isolated from the progeny of a cross to wild type can be shown to be a member of one of the two groups with which the original double mutant does not complement.
In studies of other systems, it is often observed that some mutants within a complementation group show partial complementation with some other members of the group. Since in the spot test method many cycles of growth are required before clearing of the spot occurs, it is possible that weakly complementing pairs of am mutants, if they exist, would be scored as noncomplementing. We have examined this possibility in a number of cases with a second, more sensitive test that depends upon the results of only a single cycle of growth. Strain B bacteria are infected at a multiplicity of ∼5 with each of two am mutants, and the yield from those bacteria is compared with yields from bacteria infected with each mutant alone.
This analysis is somewhat complicated by the fact that recombinant genotypes produced during one infectious cycle can express their phenotype during that cycle of growth (Séchaud et al. 1965). However, the contribution to phenotype within the cell by recombinants is very small. For example, we observe that two mutants in the amN58 group, known to give 10% wild-type recombinants, give a yield in B <3% of a wild-type control, while, in some comparisons involving four separate complementation groups, good complementation is observed even though the proportion of wild-type recombinants formed is <1% of the final bacteriophage yield. Thus, it would appear that the phenotype is largely determined by the parental bacteriophage types.
The results of our more sensitive tests involving a single cycle of growth indicate that mutants assigned to the same group by means of the spot test exhibit markedly reduced yields. We do observe in some cases a large increase in the yields from mixed infected complexes as compared to the control infections with each mutant singly, but the yields are very low (seldom more than four or five bacteriophage per cell as compared to ∼200 for the wild-type control), and, further, the frequency of recombination is significantly elevated in the progeny of such complexes in comparison to the values measured for crosses on strain CR63. This latter result suggests that the increase in the yield is in fact due to the functional activity of wild-type recombinants as mentioned above, rather than to complementation due to parental-type interactions. We thus believe that, in most instances, am mutants within the same complementation group do not exhibit the phenomenon of partial complementation.
Mapping of am mutants
The observation that the am mutants can be separated into a large number of groups by the complementation tests strongly suggests that the mutations are located in a large number of genes. If this is so, it follows that the am mutant sites are widely distributed in the genetic structure of T4. For this reason, an extensive series of crosses between am mutants has been performed, and the locations of 48 mutations, each from a different complementation group, have been determined.
While strain B may be used as a host in crosses between complementing mutants, it cannot be employed for mutants in the same group. Strain CR63 (or strain C600) was therefore used for all crosses between noncomplementing mutants and also for most of the crosses between complementing mutants. Some crosses between complementing mutants were also performed in strain B; under the conditions employed, the two hosts gave similar recombination values.
Following the criterion of Doermann and Hill (1953), crosses in which the input ratios of the minority-to-majority parent were <0.67 were rejected. In almost all am × am crosses, the phenotype of the two parental types in a cross could not be distinguished by simple techniques of analysis, and the ratio of the parental types was determined from assays of cross stocks used to prepare the parental mixtures. For 32 am × am crosses, the allele ratios in the progeny were estimated by the spot-test method (see Materials and Methods) and were found to be in good agreement with the input ratios determined from assays of parental bacteriophage stocks. The am double mutant was recovered in all of these crosses and was shown to be am in phenotype. Double-mutant and wild-type recombinants occurred in about equal frequencies.
Wild-type recombinants from am × am crosses were scored by plating upon the selective indicator S/6, and the proportion of recombinant particles in a lysate was taken to be twice the ratio of the plaque counts on S/6 to those on CR63 (since the wild-type recombinants represent half of the total recombinants). The CR63 plating measures the total bacteriophage yield since all genotypes in the cross form plaques on this indicator. A few of the crosses have been analyzed by the double-layer technique (see Materials and Methods) in addition to the usual platings on the selective and nonselective indicators; no disagreements between the two methods were found.
In addition to the two-factor crosses involving only am mutants, we present data from a few crosses between some am mutants and the previously known mutants r48 and eC103. These crosses were performed as described above except that recombinants were scored either on double-layer plates or as wild type among total plaques formed on the indicator S/6. In addition, in the case of crosses involving the eC103 marker, wild-type recombinants were also scored by platings on S/6 plates incubated overnight at 42° to prevent plaque formation by progeny of e genotype.
All crosses have been corrected, using a wild-type control, for differences in the efficiency of plating between S/6 and CR63; in general, this difference is small. In the great majority of our crosses, the recombination values are based on counts of at least 200 recombinant plaques and at least 300 parental plaques. In many crosses, several assays of the cross lysate were made. Recombination values determined by the above method are accurate to ∼25% (Edgar 1958).
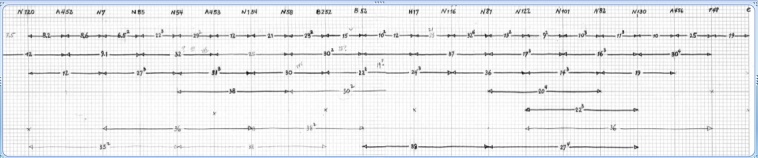
Figure 2 shows a linkage map that has been constructed using data from the crosses described above. The total number of crosses is too large to be represented clearly; therefore, only crosses involving three or fewer elementary intervals are given. The maps have been obtained exclusively from two-factor crosses. Some three-factor crosses have been performed to confirm the order of some mutant sites. While further work is necessary to assure proper ordering, the consistency of the two-factor data suggests that there will be relatively few changes necessary.
Mapping data for mutants in the gene 27–rI–e segment. The data are presented graphically, and the intervals are not proportional in length to recombinational distances. The left-most site (amN120) is in gene 27, and the amN456 mutant is in gene 47. Except for gene 29 (amB7 and amN85), one mutant per complementation group is represented. Mutants amN7 and amB52 are probably mislabeled and correspond to amB7 and amN52.
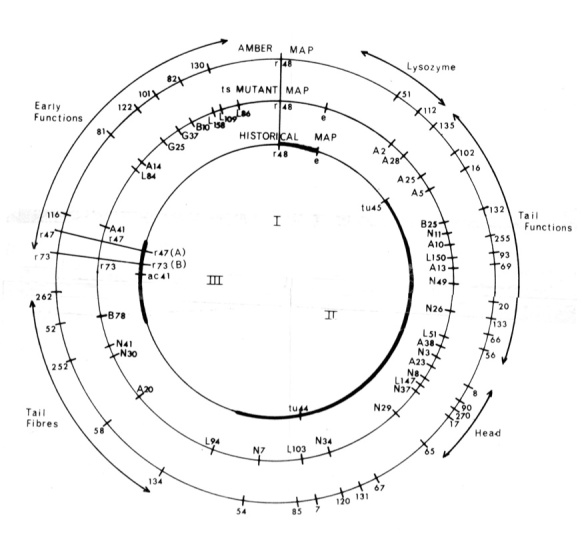
Some of the data represented have been combined in Figure 3, which shows the relative positions of the mutants as the measured distance between adjacent mutant sites. The most distant adjacent sites are linked (32%), and the linkage structure is circular. In numerous crosses, markers that lie “across” the circle from one another show recombination values between 40 and 45%.
The three linkage groups of the historical map are shown in the inner circle. The map of the ts alleles (Edgar and Lielausis 1964) is shown in the middle circle. The map of the am alleles is shown on the outside circle; the prefix of the individual alleles is not indicated but can be found in Epstein et al. (1964); therefore, 51 is amN51 and maps in gene 2. Those am and ts mutants that belong to the same complementation groups are aligned, except for the three gene 29 mutants (tsL103, amB7, and amN85).
With a few exceptions, the map presented has been obtained from crosses between am mutants. However, for those regions that can be tested, the distances that are observed are compatible with determinations made with other genetic markers (Edgar et al. 1964).
The results reported here concern crosses involving am mutations in different functional groups. The detailed results of fine-structure mapping will be presented elsewhere. To date, we have performed mapping experiments with non-allelic mutants in 17 complementation groups. These crosses have revealed no obvious clustering of sites, but in all cases the sites of mutants within a given complementation group fall between the sites from adjacent groups. Thus, the mutants that are grouped together by functional relatedness lie in a restricted segment of the genome. The distance between terminal markers in a group has a considerable range (4–20% recombination), suggesting that these segments vary greatly in length.
Discussion
There is a superficial resemblance between the amber mutants and the well-known rII mutants in that they are both conditional lethal mutants defined by whether or not they are capable of growing in certain bacterial hosts: they are able to grow in a permissive host (B for rII mutants, CR63 for am mutants) but are unable to grow in a restrictive host [K(λ) for rII mutants, B for am mutants]. There is, however, a fundamental distinction between these two classes of mutants. The rII phenotype arises from a spectrum of mutational events—ranging from presumed single-base substitutions to gross deletions—occurring in a small segment of the T4 genome comprising two genes. The am phenotype, on the other hand, arises from a restricted type of mutational event that may occur in many genes that are widely distributed over the T4 genome. Furthermore, it has been shown (Epstein et al. 1964) that the genes in which amber mutations have been isolated control a variety of physiological functions.
The ability of a bacterial host to support the growth of amber mutants is controlled by a genetic determinant that can be localized in the bacterial genome (Anna Reale, unpublished results). Since the effect of the bacterial gene is to reverse the mutant phenotype of the infecting bacteriophage, it is formally a suppressor. That the suppression is not always complete, however, is demonstrated by the fact that about one-fifth of the amber mutants show a temperature-sensitive phenotype when growing in the permissive host. The same suppressor that reverses the phenotype of amber mutants also suppresses mutants of bacteriophage λ as well as mutations in the bacterium itself (Campbell 1961). Other suppressors in E. coli that control the expression of various bacterial mutants (Garen and Siddiqi 1962; Brody and Yanofsky 1963) are known, some of which also reverse the phenotype of amber mutants.
There are several lines of evidence that amber mutants arise from a restricted class of mutational event. They are rarely, if ever, induced by proflavine. Since most spontaneous mutations in T4 are of the proflavine type (Freese 1959), it is easy to understand the low frequency of spontaneous mutation to amber. However, amber mutants are readily induced by such agents as 2-AP, 5-BdU, nitrous acid, EMS, and hydroxylamine. This evidence concerning forward mutation strongly suggests that amber mutants are of the base-substitution type. Reversion studies provide further insight into the mutational event that gives rise to amber mutants. All mutants studied, regardless of how they were induced, are very much more revertible with 2-AP than with 5-BdU, and they are not revertible with hydroxylamine. According to Champe and Benzer (1962), these are diagnostic criteria that the mutant site is AT and, hence, that the original forward mutation was a GC-to-AT transition. Quite aside from the validity of the specific base pair assignment, the homogeneity of the mutants in reversion studies indicates that they arose from a restricted class of mutagenic events.
With all mutagens used, fewer amber than rII mutants were induced. But all rII mutants lie within two genes while amber mutants can occur in at least 56 genes. We conclude that potential sites for amber mutations are quite limited in number.
We have used complementation, a test for functional allelism, to assign the amber mutants to different genes. When such an analysis is attempted with other types of mutants, intragenic complementation has been found to occur. This is the case, for example, with ts mutants of bacteriophage T4 (Edgar and Lielausis 1964). With the amber mutants, however, there is no evidence for intragenic complementation, and the mutants can all be assigned unambiguously to one or another gene. Since many genes contain both ts and am mutants (Edgar et al. 1964), the absence of intragenic complementation must be a property of the mutations and not of the genes in which they occur.
The currently accepted explanation for intragenic complementation (Woodward 1959; Fincham and Coddington 1963) involves the interaction of the protein products of genes. Hence, the most obvious explanation for the systematic absence of intragenic complementation among amber mutants is that such mutations lead either to no polypeptide chain synthesis or to the synthesis of a drastically altered one. That is, the amber mutants are nonsense mutants. The correctness of this explanation in the case of the gene determining the bacteriophage head protein has been demonstrated by Sarabhai et al. (1964). These authors found that amber mutants of this gene produced polypeptide-chain products in the restrictive host that corresponded to fragments of the wild-type protein. While the nonsense nature of amber mutants has been definitively demonstrated for the head-protein gene only, the absence of intragenic complementation indicates that fragment synthesis is a general property of amber mutants.
Acknowledgments
This work was supported by the U.S. Public Health Service (grant E 4267) and by a U.S. Public Health Postdoctoral grant (RG–6965).
Literature Cited
Appleyard R. K., 1954. Segregation of new lysogenic types during growth of a doubly lysogenic strain derived from Escherichia coli K12. Genetics
39: 440–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
Appleyard R. K., McGregor J. F., Baird K. M., 1956. Mutation to extended host range and the occurrence of phenotypic mixing in the temperate coliphage lambda. Virology
2: 565–574. [DOI] [PubMed] [Google Scholar]
Benzer S., 1955. Fine structure of a genetic region in bacteriophage. Proc. Natl. Acad. Sci. USA
41: 344–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
Benzer S., Champe S. P., 1961. Ambivalent rII mutants of phage T4. Proc. Natl. Acad. Sci. USA
47: 1025–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Benzer S., Champe S. P., 1962. A change from nonsense to sense in the genetic code. Proc. Natl. Acad. Sci. USA
48: 1114–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
Brody S., Yanofsky C., 1963. Suppressor gene alteration of protein primary structure. Proc. Natl. Acad. Sci. USA
50: 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Campbell A., 1961. Sensitive mutants of bacteriophage lambda. Virology
14: 22–32. [DOI] [PubMed] [Google Scholar]
Champe S. P., Benzer S., 1962. Reversal of mutant phenotypes by 5-fluorouracil: an approach to nucleotide sequences in messenger-RNA. Proc. Natl. Acad. Sci. USA
48: 532–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
Doermann A. H., Hill M. B., 1953. Genetic structure of bacteriophage T4 as described by recombination studies of factors influencing plaque morphology. Genetics
38: 79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
Edgar R. S., 1958. Mapping experiments with rII and h mutants of bacteriophage T4D. Virology
6: 215–225. [DOI] [PubMed] [Google Scholar]
Edgar R. S., Epstein R. H., 1965. The genetics of a bacterial virus. Sci. Am.
212: 70–78. [DOI] [PubMed] [Google Scholar]
Edgar R. S., Lielausis I., 1964. Temperature-sensitive mutants of bacteriophage T4D: their isolation and genetic characterization. Genetics
49: 649–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
Edgar R. S., Denhardt G. H., Epstein R. H., 1964. A comparative genetic study of conditional lethal mutations of bacteriophage T4D. Genetics
49: 635–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
Epstein R. H., Bolle A., Steinberg C. M., Kellenberger E., Boy de la Tour E., et al. , 1964. Physiological studies on conditional lethal mutants of bacteriophage T4D. Cold Spring Harb. Symp. Quant. Biol.
28: 375–394. [Google Scholar]
Fincham J. R., Coddington A., 1963. Complementation at the am locus of Neurospora crassa: a reaction between different mutant forms of glutamate dehydrogenase. J. Mol. Biol.
6: 361–373. [DOI] [PubMed] [Google Scholar]
Freese E., 1959. The difference between spontaneous and base-analogue induced mutations of phage T4. Proc. Natl. Acad. Sci. USA
45: 622–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
Garen A., Siddiqi O., 1962. Suppression of mutations in the alkaline phosphatase structural cistron of E. coli. Proc. Natl. Acad. Sci. USA
48: 1121–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
Jacob F., Fuerst C. R., Wollman E. L., 1957. Recherches sur les bactéries lysogènes défectives. II. Les types physiologiques liés aux mutations du prophage. Ann. Inst. Pasteur (Paris)
93: 724–753. [PubMed] [Google Scholar]
Loveless A., Howarth S., 1959. Mutation of bacteria at high levels of survival by ethyl methane sulphonate. Nature
184: 1780–1782. [DOI] [PubMed] [Google Scholar]
Sarabhai A. S., Stretton A. O., Brenner S., Bolle A., 1964. Co-linearity of the gene with the polypeptide chain. Nature
201: 13–17. [DOI] [PubMed] [Google Scholar]
Séchaud J., Streisinger G., Emrich J., Newton J., Lanford H., et al. , 1965. Chromosome structure in phage T4, II. Terminal redundancy and heterozygosis. Proc. Natl. Acad. Sci. USA
54: 1333–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
Steinberg C. M., Edgar R. S., 1962. A critical test of a current theory of genetic recombination in bacteriophage. Genetics
47: 187–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
Vielmetter W., Wieder C. M., 1959. Mutagenic and inactivating effect of nitrous acid on free particles of the phage T2. Z. Naturforsch. B
14b: 312–317 (in German). [Google Scholar]
Woodward D. O., 1959. Enzyme complementation in vitro between adenylosuccinaseless mutants of Neurospora crassa. Proc. Natl. Acad. Sci. USA
45: 846–850. [DOI] [PMC free article] [PubMed] [Google Scholar]