

Abstract
Following irradiation (IR), the DNA damage response (DDR) activates p53, which triggers death of cells in which repair cannot be completed. Lost tissue is then replaced and re-patterned through regeneration. We have examined the role of p53 in co-regulation of the DDR and tissue regeneration following IR damage in Drosophila. We find that after IR, p53 is required for imaginal disc cells to repair DNA, and in its absence the damage marker, γ-H2AX is persistently expressed. p53 is also required for the compensatory proliferation and re-patterning of the damaged discs, and our results indicate that cell death is not required to trigger these processes. We identify an IR-induced delay in developmental patterning in wing discs that accompanies an animal-wide delay of the juvenile-adult transition, and demonstrate that both of these delays require p53. In p53 mutants, the lack of developmental delays and of damage resolution leads to anueploidy and tissue defects, and ultimately to morphological abnormalities and adult inviability. We propose that p53 maintains plasticity of imaginal discs by co-regulating the maintenance of genome integrity and disc regeneration, and coordinating these processes with the physiology of the animal. These findings place p53 in a role as master coordinator of DNA and tissue repair following IR.
Introduction
The imaginal discs of Drosophila, larval organs that contain the adult progenitor cells, have remarkable regenerative properties such that loss of over half of disc cells through IR or other tissue damage can be replaced to reestablish an organ of relatively appropriate size (Bergantinos et al.; Bryant and Simpson, 1984; Haynie and Bryant, 1977; Haynie and Bryant, 1976). Damage of imaginal discs induces a continuum of processes with systemic and local components. At the site of damage local events occur including cell death, wound healing, cell cycle arrest, compensatory proliferation and re-patterning. At the same time a systemic developmental delay is activated that ensures that the replacement of damaged tissue is coordinated with the physiology of the animal as a whole. How the local and animal-wide events are regulated and coordinated in time is unknown.
Imaginal discs have long been a powerful experimental system for study of organ development and regeneration. Growth and patterning of the discs are tightly linked and occur primarily during the larval stage where they are fueled by feeding. Terminal differentiation occurs during the pupal stage after cell proliferation has ceased. At the onset of larval development disc cells are multipotent, and over time acquire cell fates in a process regulated by signaling activity from the disc pattern-organizers, Wingless and Dpp. Disc regeneration is dependent upon the activity of the organizers, and can be induced by ectopic expression of Wg and Dpp (Johnston, 2005; Struhl and Basler, 1993; Sustar and Schubiger, 2005). The ability of imaginal discs to regenerate – their plasticity - requires that the cells remain in a state that is permissive to cell proliferation. By the end of the larval feeding period, the growth and patterning of discs are essentially complete and the larvae migrate away from the food to pupariate (PP). In the wing disc, two additional mitoses occur early in the pupal stage prior to metamorphosis (Hartenstein and Posakony, 1989; Schubiger and Palka, 1987). The onset of PP marks the end of the major period of disc proliferation and is controlled by a developmental clock that is regulated by the ring gland and other larval endocrine organs (Bodenstein, 1950). Damage to discs slows the larval developmental clock and the discs are repaired during an extended period of feeding and cell proliferation. Evidence has suggested that it is the continued proliferation of the discs that inhibits progression of the clock (Poodry and Woods, 1990; Simpson et al., 1980), although the mechanism by which this occurs is unknown.
Gamma-irradiation (IR) can cause loss of imaginal disc cells and lead to disc regeneration. In this case, regeneration is preceded by the DNA damage response (DDR), a highly conserved cellular response to DNA damage that is rapidly activated to repair genomic lesions. In the DDR, the DNA damage sensor Ataxia Telangiectasia mutated (ATM) recruits and phosphorylates repair proteins to the DNA break site (Niida and Nakanishi, 2006; Shiloh, 2003; Zhou et al., 2000). Among its targets is the histone H2A variant, H2AX, widely present in nucleosomes (Rogakou et al., 1998) but specifically phosphorylated (γ-H2AX) within minutes of DNA breakage, thus marking the sites of damage (Greer et al., 2003; Srivastava et al., 2009). Drosophila H2Av, the functional homolog of H2AX, is phosphorylated with similar kinetics and specificity after IR and is recognized by mammalian γ-H2AX antibodies (Leach et al., 2000; Madigan et al., 2002). The checkpoint kinase Chk2 is also an ATM target that, upon activation, phosphorylates and activates p53 (Brodsky et al., 2004; Chehab et al., 2000; Flaggs et al., 1997; Guo et al., 2000; Hirao et al., 2000). In Drosophila, p53 mRNA expression is also significantly upregulated after DNA or tissue damage (Brodsky et al., 2000; Wells et al., 2006). In mammalian cells, p53 mediates a cell cycle arrest (Bunz et al., 1998; Kuerbitz et al., 1992), whereas in Drosophila the arrest is mediated by Chk1 and is p53-independent (Brodsky et al., 2000; Fogarty et al., 1997). The arrest in cell division facilitates DNA repair, but if repair is unsuccessful damaged cells are instructed to die by apoptosis. In the Drosophila DDR p53 plays a key role by regulating expression of the proteins that induce apoptosis, and also several DNA repair proteins (Brodsky et al., 2000; Brodsky et al., 2004; Sogame et al., 2003).
Previously, we found that p53 is required for the regeneration response of imaginal disc cells damaged by co-expression of the apoptosis-inducing genes reaper (rpr) or hid and the caspase inhibitor P35 (hereafter referred to as rpr/hid (RH)-induced damage) (Wells et al., 2006). RH damage initiates cell-autonomous apoptosis yet prevents cell elimination and results in a regeneration response that is similar to that induced after IR or surgery. Several processes occur that are autonomous to the damaged tissue, including ectopic expression of Wingless, blastema formation, and compensatory proliferation (Haynie and Bryant, 1976; McClure et al., 2008; Smith-Bolton et al., 2009; Sustar and Schubiger, 2005; Wells et al., 2006). In addition, non-autonomous responses are induced: many non-damaged cells within the disc undergo apoptosis or slow cell division, and the larval developmental clock is slowed, delaying progression to the pupal stage (Sustar and Schubiger, 2005; Wells et al., 2006). p53 is activated upon RH damage and is required for both the autonomous and non-autonomous events (Wells et al., 2006), suggesting that p53 activity might not be restricted to the cells with RH-damage.
In addition to RH-induced tissue damage, blastema formation induced by ectopic expression of Wg requires p53, suggesting that p53 may have a general role in tissue repair (Wells et al., 2006). Moreover, as the DDR and tissue regeneration occur in strict sequence after IR, to promote survival of the organism after tissue damage a role for p53 in their temporal coordination seems likely. To investigate the generality of p53’s requirement in regeneration and to determine the nature of the relationship between the DDR and tissue regeneration we studied the role of p53 in IR-induced regeneration and its coordination with the DDR.
We report here that p53 is also critical for disc regeneration following IR, suggesting that p53 universally promotes regeneration of damaged imaginal discs. Although initially the DDR occurs normally in p53 mutant disc cells, we find that they are unable to repair the DNA lesions. In addition, lack of timely cell death in p53 mutants allows the damaged cells to persist in the epithelium, leading to numerous adult defects and reduced animal survival. However, we find that the absence of the early cell death program in the mutants cannot explain the necessity for p53 in regeneration. In addition, our results suggest that p53 controls a developmental patterning checkpoint in discs and the larval developmental timer. As a whole, our results suggest that p53 maintains plasticity of imaginal discs by integrating the maintenance of genome integrity with disc regeneration and coordinating these processes with the physiology of the animal.
Materials and Methods
Fly Strains
The following strains were used in this study. Unless otherwise indicated, strain descriptions can be found at http://flybase.bio.indiana.edu. All mutant lines were crossed into the background of the yw strain used as a control.
UAS-P35 (Hay et al., 1995)
UAS-hid (Huh et al., 2004)
p53ns (Sogame et al., 2003)
p535a-1-4 (Rong et al., 2002)
chk2P6 (Brodsky et al., 2004)
EnGal4, UAS-GFP (Neufeld et al., 1998)
rpr150-lacZ (Brodsky et al., 2000)
wg-lacZ
ywhsflp122TubGal4, UAS-GFP; FRT82B TubGal80 hsCD2 (de la Cova et al., 2004)
ywhsflp122; FRT82B arm-lacZ
FRT82B TubGal80 hsCD2 p53ns (gift of C. de la Cova)
UAS-p53
yw; TubGal80ts (II)
HhGal4, UAS-GFP
droncI29/TM6B (Xu et al., 2005)
Act>y+>Gal4, UAS-GFP
ywhsflp122; +; +
yw;+;+ (used as control strain).
Fly Husbandry
Eggs from appropriate crosses were collected on yeasted grape plates for 2-3 hours. After hatching, larvae were transferred to standard molasses food vials (≤50/vial to avoid crowding) supplemented with fresh yeast paste and raised at 25 °C for defined periods of time, as described (Johnston and Sanders, 2003; Neufeld et al., 1998).
Clonal induction for autonomous p53RE and ectopic Wg detection
ActGal4 flp-out clones were induced at 48 or 60 hours AEL with an 8 min. heat shock at 37°C. Animals were dissected between 100 and 120 hours AEL.
p53 MARCM clone analysis
The p53ns allele was recombined onto the FRT82B chromosome and used in MARCM experiments (gift of C. de la Cova). For analysis of ectopic Wg expression clones were induced between 30 and 48 hours AEL with a 20 minute heat shock at 37°C. Larvae were raised at 25°C and dissected between 120 and 130 hours AEL.
Gal80ts experiments
Larvae were raised at 18°C, which is permissive for Gal80 expression, for 144 hours to allow growth of the animal without expression of Gal4-driven transgenes. Larvae were then shifted to the restrictive temperature (30°C) to allow transgene expression for 40 hours before dissection.
Irradiation
Larvae were given a dose of 40 Gy while in standard molasses food at specified times AEL using a Nordion gamma-cell 220 irradiation unit with a 60Co source, and dissected at indicated times following IR.
Immunocytochemistry
Fixation and immunocytochemistry of imaginal discs were carried out as described (Johnston and Edgar, 1998). TUNEL assays were carried out using Apoptag Red (Intergen); a detailed protocol is available upon request. Images were acquired using Apotome software and a Zeiss Axioplan 2 microscope with an Orca-100 CCD camera (Hammatsu) and processed with Photoshop (Adobe) software. The following antibodies and dilutions were used: mouse anti-CD2, 1:200; rabbit anti-H3P (Upstate), 1:1000; mouse anti-γ-H2AX (Upstate), 1:250; guinea pig anti-Sens (gift of H. Bellen), 1:1000; mouse anti-Delta (DSHB), 1:200; guinea pig anti-Kni (gift of J. Parker), 1:1000; mouse anti-Nrt (DSHB), 1:30; mouse anti-Wg (DSHB), 1:30; rabbit anti-ß-gal (Cappel), 1:2000; rabbit anti-Hid (gift of B. Hay), 1:1000; rabbit anti-cleaved Caspase 3 (Cell Signaling), 1:100; mouse anti-BrdU (Roche), 1:100; guinea pig anti-Dronc (Xu et al., 2005), 1:1000.
Results
p53 is required cell-autonomously in tissue regeneration and its expression is sufficient to induce a regeneration response in wing imaginal discs
After RH-damage, p53 is required for the regeneration response in the damaged disc and also for the systemic developmental delay that accompanies disc regeneration. However, p53 mRNA expression is induced specifically in the damaged disc cells (Wells et al., 2006). To determine whether p53 activity was autonomous to the damaged tissue we took two approaches. First, we induced RH damage in wing discs and examined the extent of p53 activation with an activity reporter consisting of tandem p53 consensus binding sites in front of the lacZ gene (Brodsky et al., 2000). RH damage created in posterior cells by expressing UAS-hid and UAS-P35 with Engrailed Gal4 induced p53 activity specifically in the damaged cells (data not shown) (Wells et al., 2006). Likewise, RH damage created in small ‘flp-out’ Gal4 clones of cells resulted in cell-autonomous activation of the p53 response element reporter (Figure 1A). We also examined expression of the p53 activity reporter in other tissues in the larvae, including the fat body, ring gland, and Dilp-expressing neurons, but found no change in reporter activity in these tissues in response to RH damage (data not shown).
Figure 1. p53 activity is autonomous to damaged cells and is sufficient to induce a regeneration-like response in the wing disc.
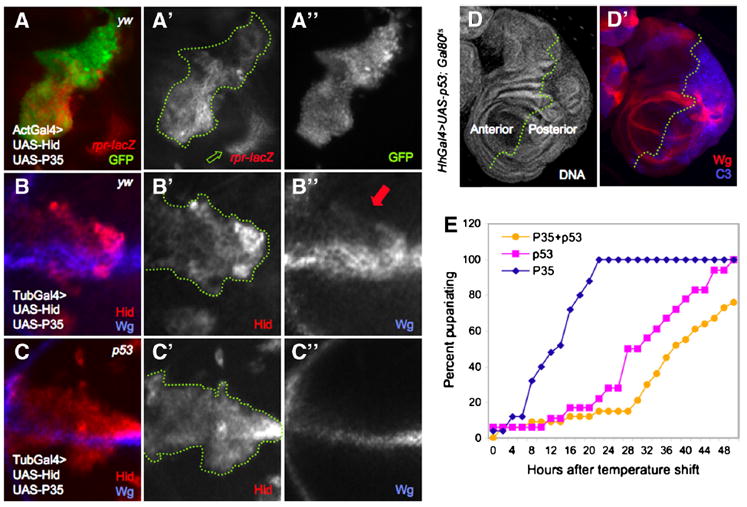
A. Act>y+>Gal4 ‘flp-out’ clones expressing UAS-Hid+UAS-P35, marked by UAS-GFP expression (A”). The p53 activity reporter, rpr150-lacZ, is autonomously activated within the boundaries of the clone (A’, outlined in green). Green arrow indicates p53-independent reporter expression (see Supp. Fig. 1A-D).
B-C. MARCM clones expressing UAS-hid+UAS-P35 in the presence (B) or absence (C) of p53. In a yw background (control), ectopic Wg expression is autonomous to Hid+P35-expressing clones (Hid expression and dotted green line marks the clone, B’). Endogenous Wg expression (clones at the D/V boundary are shown in B and C) is also evident. All cells that express Wg ectopically are also Hid-positive (B”, red arrow). C-C”. Loss of p53 specifically within UAS-hid+UAS-P35-expressing clones do not disturb endogenous expression of Wg but prevents its ectopic expression (Hid expression marks the clone).
D. Expression of p53 for a limited period of time induces tissue repair (see text for details). HhGal4>UAS-p53 induces overgrowth and expression of ectopic Wg and C3 in posterior cells (D’).
E. Developmental timing is delayed after transient induction of UAS-p53 expression. Larvae expressed UAS-P35 ± UAS-p53 under HhGal4 control for 40 hours (see text for details). The presence of Gal80ts in these animals did not affect growth or cell viability in the absence of UAS-p53 (E). Expression of UAS-p53 significantly delayed the time to pupariation relative to controls that expressed P35 alone. Co-expression of p53 and P35 exacerbated the delay. X-axis is expressed as hours after induction of Gal4 activity by Gal80 inactivation at 29°C.
Second, we used the p53ns null allele (Sogame et al., 2003) in the MARCM system (Lee and Luo, 1999) to remove p53 function specifically in RH-damaged cells and looked for ectopic expression of Wg, an early step in regeneration that is p53-dependent (Smith-Bolton et al., 2009; Wells et al., 2006). Of control MARCM clones in which RH damage was created with Hid + P35 expression, 74% (n=87) showed robust and completely autonomous ectopic Wg expression in many cells within the clone (Figure 1B”). The expression of a wg-lacZ reporter was identical to that of Wg protein (data not shown). In contrast, only 12% (n=305) of p53 mutant RH-expressing clones contained cells that expressed ectopic Wg (Figure 1C-C”), and the expression was generally limited to one or two cells. Occasionally wildtype cells with Wg expression in ectopic locations can be found under normal physiological conditions (9% of cells, n=91 discs), thus we infer that the cells expressing ectopic Wg in p53 mutant RH-MARCM clones represented background. The endogenous pattern of Wg expression was unaffected by RH damage in p53 mutant cells (Figure 1C”). As p53 is activated cell-autonomously within RH damaged cells and is required cell-autonomously for the ectopic expression of Wg during regeneration, we conclude that p53 activity and function is restricted to the damaged cells.
The cell autonomous function of p53 in wing disc cells led us to ask whether p53 activation was sufficient to induce a regeneration response in wing discs. Like expression of RH genes, expression of p53 from an early larval stage led to rapid cell death when in the absence of P35 (data not shown) (Huh et al., 2004; Ryoo et al., 2004; Wells et al., 2006). To circumvent this problem we expressed UAS-p53 in posterior cells using Hedgehog (Hh) Gal4 under control of a temperature sensitive Gal80. At low temperature Gal4 is inhibited and expression of UAS-p53 is repressed. Shifting larvae to 29°C allowed expression of UAS-p53 (see methods). Strikingly, when expressed in posterior cells early in the third instar, UAS-p53 cell-autonomously induced processes characteristic of regeneration. Ectopic Wg expression was induced and compensatory proliferation occurred in posterior cells, resulting in overgrowth of the posterior compartment (Figure 1D-D’). The overgrowth arose during a period of delayed larval development, suggesting that the developmental timer was affected (Figure 1E). Apoptosis was also induced, as Caspase-3 (C3) was activated in the p53-expressing cells (Figure 1D’). Interestingly, although the presence of the caspase inhibitor P35 exacerbated the responses to conditional p53 expression, it was not required for them to occur (Figure 1D-E). Taken together, these results provide evidence that p53 is necessary in a cell-autonomous manner after RH damage, and suggest that its expression during the third instar in wing discs is sufficient to induce the regeneration response.
The tissue repair response after IR requires p53
To test the generality of the p53 requirement during regeneration, we examined wing discs after induction of tissue damage by IR. We treated yw (used as a wildtype strain) and p53 mutant larvae with 40 Gray (Gy) of IR, a level sufficient to induce apoptosis in more than 50% of wing disc cells (Haynie and Bryant, 1977; Haynie and Bryant, 1976; Wichmann et al., 2006), at 96 hours after egg laying (AEL). The p53 activity reporter was induced in the wing disc throughout the wing pouch within 3 hours of IR in a p53-dependent manner, demonstrating that p53 was activated (Supp. Figure 1A-C). After IR, p53 induces expression of Rpr and Hid, among other pro-death genes (Brodsky et al., 2004), thus to initially reproduce the conditions of RH damage, we expressed UAS-P35 in posterior cells under control of EnGal4 (Wells et al., 2006). We then examined C3 activation to monitor apoptosis, ectopic Wg expression as a marker of regeneration, and disc overgrowth as a proxy for the regenerative compensatory proliferation (Wells et al., 2006).
In anterior wing disc cells, which did not express P35, C3 activation rapidly spiked after IR and then declined (McNamee and Brodsky, 2009), except for some cells near the anterior-posterior compartment boundary in which C3 staining persisted (Supp. Figure 1E” and data not shown). Posterior cells, which expressed P35, contained high levels of C3 that persisted for at least 72 hours after IR (Supp. Figure 1E”). Patches of posterior cells expressed Wg ectopically, and the posterior compartment grew large and distorted (Supp. Figure 1E’), indicating that the tissue repair program was induced. In contrast, evidence of this program was completely absent from wing discs of p53 mutants after IR (Supp. Figure 1F’-F” and data not shown). Thus, the IR-induced tissue repair response is p53-dependent, as it is after RH damage.
In RH-damaged tissue, a non-apoptotic function of the apical caspase Dronc is required to activate p53 (Wells et al., 2006). To determine whether Dronc was required to activate p53 in IR-induced regeneration, we examined wing discs carrying a dronc null mutation for evidence of regeneration. Loss of dronc prevents C3 activation and cell death in response to IR (Kondo et al., 2006). Interestingly, however, it did not prevent the tissue repair response after IR damage (Supp. Figure 1G and data not shown). This result indicates that Dronc is not required to activate p53 after IR. In contrast, loss of chk2 substantially suppressed the IR-induced response, consistent with its role as a DDR-induced p53 activator (data not shown) (Wichmann et al., 2006). Accordingly, disc regeneration after IR requires DNA checkpoint-dependent, but Dronc-independent activation of p53, suggesting that p53’s function in disc regeneration is proximate to and contingent upon its activation in the DDR. Moreover, the finding that ectopic Wg, compensatory proliferation, and developmental delay (Supp. Figure 1G and data not shown) occurred in IR-treated, dronc mutant discs demonstrates that cell death is not required to induce the tissue repair response.
Differentiation of adult structures after IR is compromised in the absence of p53
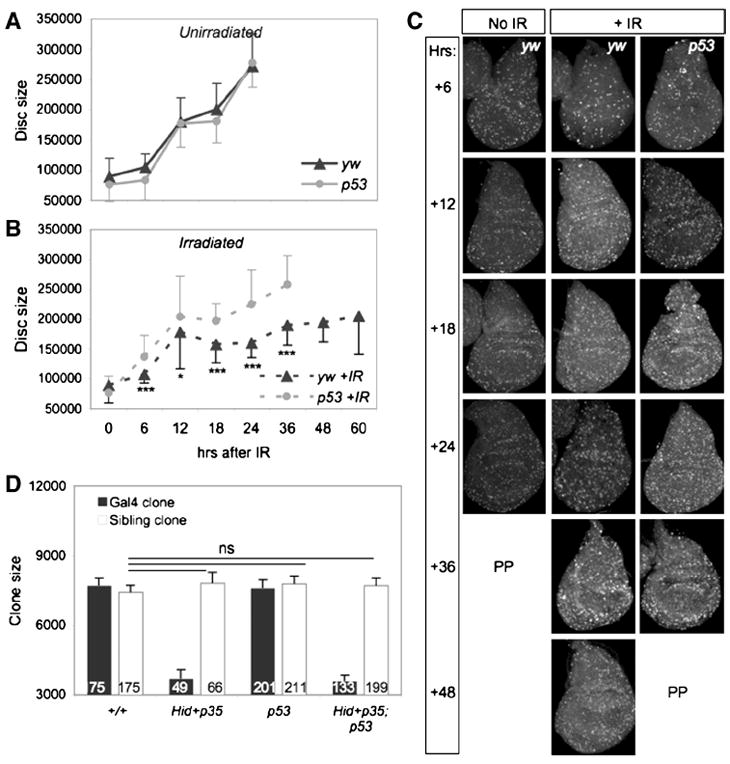
To follow the IR response of the disc in real time we did not express the caspase inhibitor P35 in our subsequent experiments. Larvae were irradiated and examined for developmental timing and for the ability of the wing discs to regenerate. IR at 96 hours AEL induced a significant developmental delay of yw larvae such that pupariation occurred 27 hours later than unirradiated controls (Figure 2A). By contrast, irradiated p53 mutant larvae did not significantly delay pupariation compared to unirradiated controls (Figure 2A), similar to our findings of experiments with RH-induced damage (Wells et al., 2006). These results indicate that the systemic larval delay associated with both types of damage requires p53 function, and suggest that control of developmental timing after disc damage may be generally regulated by p53.
Figure 2. IR induces a p53-dependent delay in developmental timing and leads to morphological defects.
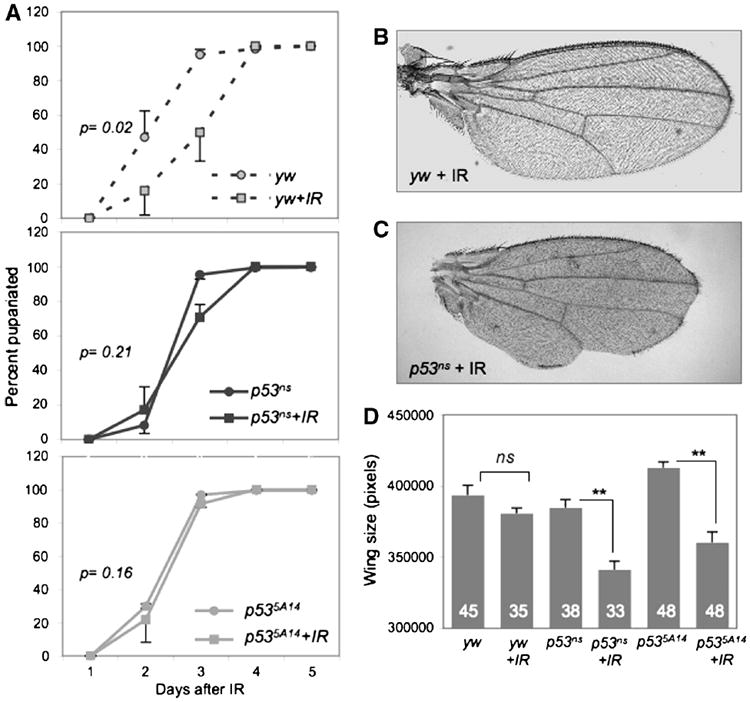
A. Irradiated yw larvae are significantly delayed relative to unirradiated controls. In contrast, irradiated p53ns or p535A14 mutants do not delay relative to unirradiated mutants. Animals were irradiated with 40 Gy at 96 hours AEL. P values represent significance between irradiated and un-irradiated animals when 50% had pupariated. Error bars represent standard error of the mean from 6 independent experiments of at least 200 larvae per genotype.
B-C. Representative wings from adult yw or p53ns animals irradiated at 96 hr AEL. p53 mutant wings are small and morphologically defective.
D. Adult wing size of irradiated and unirradiated yw and p53ns and p535A14mutants. Wings of unirradiated and irradiated yw controls are similar in size, but wings of irradiated p53 mutants were significantly smaller. Error bars denote standard error of the mean; n values for each genotype are listed. Larvae were irradiated with 40 Gy at 96 hours AEL.
To determine how IR affected the formation of adults we examined the morphology and eclosion rates of adult flies that had been IR-treated at 96 hours AEL. The vast majority of IR-treated yw animals differentiated appropriately and eclosed (Table 1). Of these, only 19% had minor morphological abnormalities, such as occasional missing thoracic machrochaetae (Table 2). As a proxy for regenerative growth we measured the size of the adult wings and found that after IR at 96 hour AEL, wings of yw control flies grew to a similar size as unirradiated controls, indicating that regeneration had occurred successfully (Figure 2D).
Table 1. Irradiated p53 mutants have significantly reduced survival rates.
| yw (1086) | yw+IR (572) | p53 (952) | p53+IR (1679) | |
|---|---|---|---|---|
| % differentiated | 99±1 | 99±1 | 99±1 | 78±5 |
| % eclosed | 99±1 | 83±3 | 99±1 | 11±7 |
Both irradiated (40 Gy at 96 hours AEL) and unirradiated yw controls and p53 mutant animals were assayed for differentiation during pupal stages (determined by cuticle deposition), as well as successful eclosion from pupal cases. Unirradiated larvae differentiate and eclose with nearly a 100% success rate and irradiated yw larvae differentiate and eclose with a very high level of success. Percentages of differentiating p53 mutants are lower following IR (78%) and irradiated mutants are highly deficient in eclosion (11%). Values represent percentage ± standard deviation, number of animals scored are in parentheses.
Table 2. p53 mutant adults exhibit numerous morphological defects following IR.
| Phenotype (n) | Eclosed yw (69) | Eclosed yw+IR (51) | Eclosed p53 (72) | Eclosed p53+IR (69) | Pharate p53+IR (24) |
|---|---|---|---|---|---|
| Notched wings | 0 | 3±1 | 0 | 45±14 | 21 |
| Blistered wings | 0 | 1±0 | 0 | 13±0 | nd |
| Missing thoracic bristles | 0 | 19±3 | 0 | 54±13 | 63 |
| Rough eyes | 0 | 0 | 0 | 100±0 | 100 |
| Leg abnormalities | 0 | 1±1 | 0 | 29±14 | 29 |
Animals were assayed for gross morphological defects. Unirradiated animals exhibited no defects and irradiated yw animals had a low percentage of defects. In contrast, both eclosed and uneclosed (pharate) p53ns mutant adults exhibited similar and significant defects AIR. Values represent percentage ± standard deviation (when available); number of animals scored are in parentheses.
In striking contrast, although all IR-treated p53 mutant animals pupariated and 78% of pupae differentiated and secreted adult cuticle, only 11% of these animals eclosed (Table 1 and data not shown). Eclosed p53 mutant adults exhibited little movement and were unbalanced, shaky, and uncoordinated, suggesting damaged sensory and/or motor systems, and typically survived less than one day after eclosion (Supp. Figure 2A). Moreover, IR resulted in numerous external defects of p53 mutant adults, including notched and blistered wings, missing thoracic macro- and microchaetae, rough eyes and leg abnormalities (Table 2, Figure 2C). In addition, wings from IR-treated p53 mutants were 12% smaller than unirradiated p53 mutants, a significant reduction in size (Figure 2C-D). The range of morphological defects in uneclosed, irradiated p53 mutant pharate adults extracted from the pupal case was similar to that of eclosed adults, suggesting that successful eclosion was not simply due to less severe damage (Table 2). Thus in the absence of p53, regeneration was significantly impaired.
Resolution of DNA repair following IR requires p53
IR-induced DNA damage is rapidly recognized by disc cells and the DDR is initiated, leading to a DNA repair program and apoptosis mediated by p53 (Brodsky et al., 2000; Rong et al., 2002; Sogame et al., 2003). The morphological defects in irradiated p53 mutant adults prompted us to carry out a detailed examination of the DDR response in wing discs of p53 mutant and control larvae. To this end we monitored the recognition of DNA damage, its repair, and apoptosis of IR-damaged cells in wing discs from yw controls and p53 mutants at short intervals after IR. Phosphorylation of H2AX (γ-H2AX) is an early event in the recognition of DNA damage and antibodies against the mammalian form recognize γ-H2Av, its functional equivalent in Drosophila (Madigan et al., 2002), which was highly elevated in yw discs immediately after IR (Figure 3H). γ-H2AX staining persisted in the yw disc cells for approximately 18 hours but was cleared from the discs by 24 hours after IR (Figure 3B-B”, H). Activated C3 increased and declined with similar kinetics (data not shown). Likewise, a steep increase in TUNEL-positive cells immediately followed IR, however in contrast to activated C3 and γ-H2AX, TUNEL-positive cells remained high for at least 48 hours after IR (Figure 3C-C”, I). In addition, cell division arrested in control wing disc cells within 1 hour of IR and remained arrested for approximately 5 hours (Figure 3A-A”, G).
Figure 3. DNA damage persists and apoptosis is delayed in irradiated p53 mutants.
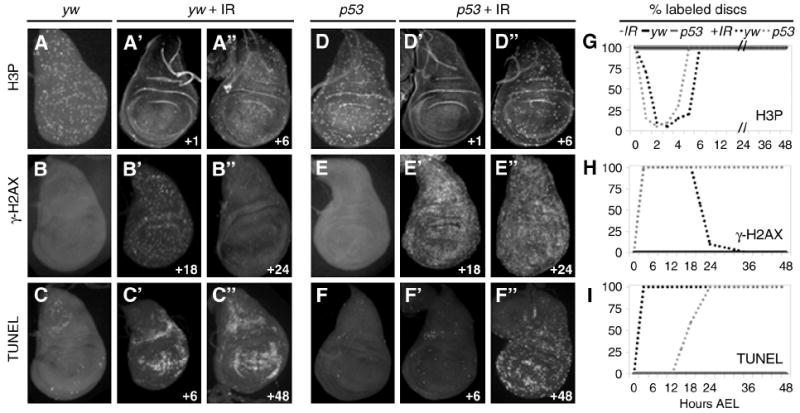
A-A”, D-D”, G. IR-induced transient cell cycle arrest. Discs from yw larvae (A-A”) and p53ns larvae (D-D”) show decreased H3P labeling at 1 hour AIR, which returns to normal by 6 hours AIR. A and D. Unirradiated yw and p53ns disc, respectively, at 97 hours AEL. G. Both yw and p53 wing discs show transient cell cycle arrest between 0 and 6 hours AIR (dashed lines). Unirradiated yw and p53 discs do not arrest (solid lines).
B-B”, E-E”, H. DNA damage persists in p53 mutant wing discs. B’-B”. Irradiated control animals exhibit elevated levels of γ-H2AX from the onset of damage until 18 hours AIR (B’). γ-H2AX is largely absent in control discs 24 hours AIR (B”). B. Control disc - No IR, 114 hours AEL. E’-E”. γ-H2AX is elevated in p53 mutant discs at 18 hours AIR (E’) and still at 24 hours AIR (E”). E. Unirradiated p53 mutant disc at 114 hours AEL. H. γ-H2AX appears immediately after irradiation in yw and p53 mutant discs. γ-H2AX disappears from yw discs after 18 hours, but persist in p53ns wing discs. γ-H2AX was not detected in discs from unirradiated animals.
C-C”, F-F”, I. p53 mutants delay apoptosis AIR. C. yw disc - No IR, 102 hours AEL. C’-C”. yw animals upregulate TUNEL by 6 hours AIR (C’), which persists more than 48 hours AIR (C”). F. Unirradiated p53 mutant disc at 102 hours AEL. F’-F”. p53 wing discs do not label with TUNEL at 6 hours AIR (F’) but TUNEL increases later and is maintained until after 48 hours AIR (F”). I. TUNEL labeling in yw animals AIR persists until pupariation. TUNEL is absent from p53 mutant discs until approximately 18 hours AIR.
G-I. Graphs depict percent of discs expressing markers at indicated time AIR. Black lines denote yw animals, grey lines denote p53 mutants, solid lines are unirradiated, dashed lines are irradiated.
Animals were given 40 Gy IR at 96 hours AEL. Time of dissection of irradiated animals is shown as hours following IR (bottom right of each panel). Discs were dissected at 30-minute intervals until after 5 hours; subsequently 6-hour time points were taken.
Cells in p53 mutant wing discs also transiently arrested cell division after IR with similar kinetics as yw control disc cells (Figure 3D-D”, G) (Brodsky et al., 2000; Rong et al., 2002; Sogame et al., 2003). In addition, γ-H2AX staining was upregulated within minutes of IR in p53 mutant wing discs (Figure 3E-E”, H), demonstrating that the recognition of DNA damage does not require p53. In contrast to controls, however, γ-H2AX staining persisted in 100% of wing discs of p53 mutants, even far into the pupal stage (Figure 3E-E”, H; Figure 6E”, G”). This result differs from a report citing that γ-H2Av declined within a few hours in p53 mutants, as in irradiated yw discs (see Supp. Figure 3) (McNamee and Brodsky, 2009). Antibodies made against γ-H2Av recognized a subset of cells recognized by γ-H2AX antibodies (Supp. Figure 3). The presence of γ-H2AX on DNA reflects ongoing DNA repair, and completion of repair leads to its dephosphorylation (Hromas et al., 2008; MacPhail et al., 2003; Olive, 2004), thus the persistence of γ-H2AX in the p53 mutant disc cells suggests that p53 is required to resolve IR-induced DNA damage. These results are consistent with previous findings that p53 regulates expression of DNA repair genes and its loss leads to aneuploidy after IR (Brodsky et al., 2004; McNamee and Brodsky, 2009; Titen and Golic, 2008).
Figure 6. Damaged cells persist into pupal development in p53 mutants.
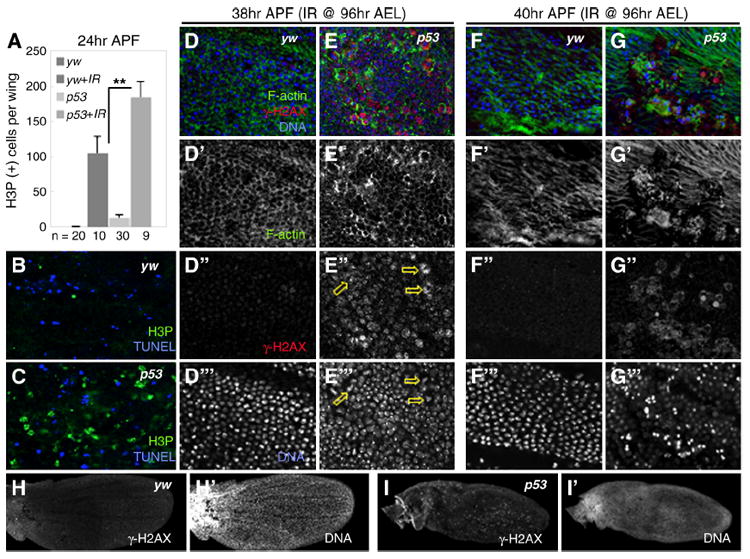
A. Pupal wings have additional rounds of division that normally cease by 24 hours APF. Pupal wings of irradiated yw and p53 mutant animals continue to divide after this 24 hour period. Irradiated p53 mutants contain twice as many dividing cells as irradiated controls (**p=0.02).
B-C. At 24 hours APF, pupal wings from irradiated yw controls and p53 mutants have elevated levels of H3P and TUNEL. B. yw. C. p53 mutant.
D-D”’. Irradiated pupal wings from yw controls have cleared damaged cells by 38 hours APF (D”), cells are hexagonal in shape and regularly packed (D’, D”’).
E-E”’. Irradiated p53 mutant pupal wings at 38 hours APF contain many γ-H2AX-positive cells, indicating that DNA damage persists (E”, arrows). DNA in many γ-H2AX-positive cells is aberrant (E”’, arrows). Cells have become hexagonal in shape (E’), suggesting they are able to undergo some metamorphic processes but are frequently large and irregularly packed.
F-F”’. By 40 hours APF, pupal wing cells from irradiated yw animals secrete a pre-hair (F’) and are free of damage (F”-F”’).
G-G”’. Irradiated p53 mutant pupal wings at 40 hours APF begin forming pre-hairs (G’), but these remain absent in most γ-H2AX positive cells, which are still prevalent (G”). Many damaged cells appear to contain fragmented DNA or continue to attempt cell division (G”’).
H-I’. At 38 hours APF, pupal wings from irradiated yw animals have cleared nearly all damaged cells (H) and have begun vein formation (H’). p53 mutant pupal wings still contain many damaged cells (I) and are much smaller and delayed in development relative to yw (I’; note lack of vein formation).
C3 activation and TUNEL-positive cells were largely absent from p53 mutant wing discs up to 18 hours following IR (Figure 3F’, I and data not shown), consistent with the requirement for p53 in death induced by the DDR (Brodsky et al., 2000; Rong et al., 2002; Sogame et al., 2003). Subsequently TUNEL positive cells increased dramatically and persisted into pupal development (Figure 3F”, I; Figure 6C). This late cell death corroborates previous reports of p53-independent late apoptosis after IR (McNamee and Brodsky, 2009; Wichmann et al., 2006), which may be activated in response to aneuploidy (Titen and Golic, 2008). Interestingly, both C3-positive and TUNEL-positive cells in irradiated yw wing discs were patterned and accumulated in particular around the boundaries of developmental compartments, suggesting input from the patterning system (Figure 3C’-C”) (Adachi-Yamada et al.). By contrast, the late appearance of TUNEL positive cells in irradiated p53 mutant discs was comparatively unpatterned (Figure 3F”). We also observed TUNEL-positive cells with no obvious pattern in late larval p53 mutant wing discs containing RH-induced tissue damage (data not shown). As JNK signaling is induced by and requires p53 after IR (data not shown) (McEwen and Peifer, 2005), and is also activated by distortions in patterning organizing activity (Adachi-Yamada et al., 1999), these observations indicate that p53 activity couples patterning information to JNK activation and apoptosis in IR damaged cells.
Growth of p53 mutant wing discs is not impaired by IR
The major proliferative phase of imaginal disc development occurs during the larval stage and is prolonged during tissue repair (Figure 2A). The length of larval delay after disc injury generally correlates with the extent of damage, implying that it is related to the amount of growth needed to regenerate lost tissue (Poodry and Woods, 1990; Schubiger and Palka; Simpson et al., 1980). Our findings that irradiated p53 mutants did not delay larval development and were also defective in aspects of the DDR suggested the possibility that disc growth was impaired in the mutants. To determine if this was the case, we followed the growth of wing discs by measuring their size at regular intervals after IR, from 96 hours AEL until just prior to pupariation.
We first followed growth in our wildtype control strain. During the first 12 hours after IR, wing discs from yw larvae grew at a similar rate as unirradiated controls. Subsequently, the rate of increase in disc size slowed significantly and remained slow during the extended larval period (Figure 4A-B). The slow growth of the discs occurred at the same time as the massive and ongoing apoptosis during this time (Figure 3C’-C”, I). Between 48 and 60 hours after IR all of the irradiated yw larvae reached the wandering stage, yet their wing discs remained significantly smaller than those of unirradiated yw controls, which had wandered almost a day earlier (Figure 2A, Figure 4A-B).
Figure 4. Wing disc cell proliferation progresses at a developmentally programmed rate after IR and RH damage.
A-B. Wing disc growth slows after irradiation (AIR). Animals were irradiated with 40 Gy at 96 hours AEL (= 0 hour). Black lines denote yw animals, grey lines denote p53ns mutants; solid lines are unirradiated, dashed lines are irradiated. A. Unirradiated yw and p53ns wing discs reach final size approximately 24 hours AIR (120 hours AEL). B. Wing discs from irradiated controls fail to reach final size by 50% PP (60 hours AIR,156 hours AEL) while wing discs of damaged p53ns animals reach final size by 50% PP (36 hours AIR, 132 hours AEL). 100 % of larvae had wandered at the last point graphed for each genotype. p53ns mutant discs grew significantly more than yw discs at every timepoint indicated (* = <0.05, ***= < 0.001). Error bars denote standard deviation.
C. Panels across each row show H3P-positive cells at 6-hour intervals after IR; intervals are noted as +hours post IR. At +6 hours both irradiated yw and p53ns disc cells re-enter the cell cycle after the IR-induced arrest (see Figure 3). H3P staining is distributed similarly in the discs regardless of genotype or irradiation. PP indicates entry into pupariation.
D. MARCM experiment to measure cell proliferation in Gal4 clones with RH damage and their siblings. Black bars, Gal4 clones that express UAS-GFP alone (+/+ or p53ns), and Gal4 clones that express UAS-Hid + UAS-P35 and are either +/+ or p53 mutant. White bars, wildtype (+/+) sibling clones growing next to RH expressing clones. All of the sibling clones grow at the same rate, indicating that proximate dying cells do not alter their rate of proliferation. Numbers of clones examined are indicated in each bar. Ns, not significant.
By contrast, growth of p53 mutant discs followed a different trend. In two independent experiments, p53 mutant wing discs increased in size during the first 12 hours after IR, slightly outpacing unirradiated controls in rate. After this period the growth of the mutant discs slowed; however, they reached the same size as unirradiated controls by late wandering stage (Figure 4A-B). The slowing of disc growth coincided with the increase in late cell death in the p53 mutants (Figure 3F”), suggesting that an abrupt loss of cells led to a reduction in disc mass. Nevertheless, the average size of p53 mutant discs was significantly larger than IR-treated yw discs at every timepoint examined after IR, even at the point when the larvae had entered wandering stage (Figure 4B). Thus, p53 mutant wing discs in irradiated larvae reached a normal size during larval development in the absence of additional time, suggesting that disc cells continue to proliferate at a relatively normal pace even after the onset of late apoptosis. We conclude that the small adult wing size of irradiated p53 mutant larvae is not due to impaired growth during the larval stage.
Cell proliferation progresses at a developmentally programmed rate during regeneration
We then examined cell proliferation at 6 or 12 hour intervals in the regenerating discs. After the resolution of the DDR-induced cell cycle arrest, mitotic, H3P-positive cells were similar in number and distribution in wing discs in both yw and p53 mutants, and also similar to unirradiated discs (Figure 4C, no IR versus +IR). This pattern in those discs continued with little variation for the next 36-48 hours, although IR-treated discs continued to proliferate longer than unirradiated discs during the extended larval phase. H3P-positive cells became sparse as the larvae from both genotypes neared the pupariation stage, in keeping with the temporary arrest of cell division near the larval-pupal transition (Schubiger and Palka, 1987). These results suggest that cell division during regeneration occurs at a developmentally programmed rate that is not altered by IR. Moreover, they indicate that the persistence of DNA damage in p53 mutant disc cells did not interfere with their ability to proliferate.
To directly measure cell proliferation of the damaged cells, as well as non-damaged cells in their vicinity during regeneration, we generated RH-damage in wing discs via the MARCM system of mitotic recombination. This method avoids the stochastic nature of IR damage, and provides a powerful method of measuring compensatory cell proliferation stimulated by cellular loss or damage. We expressed UAS-Hid and UAS-P35 specifically in marked Gal4 clones of cells and measured the growth of the Gal4 clones and their independently marked sibling clones after 40 hours of growth (de la Cova et al., 2004). Control Gal4 and p53 mutant Gal4 clones (Figure 4D, black bars) and their siblings (white bars) grew at the same rate, and Gal4 clones expressing Hid + P35 grew poorly regardless of their genetic background (Figure 4D, +/+ vs p53), as expected. Strikingly, sibling clones of either +/+ or p53 mutant Gal4, UAS-Hid UAS-P35 -expressing clones grew at the same rate as control sibling clones in discs with no RH damage (Figure 4D).
All together, these results demonstrate that disc cell division progresses at a developmentally programmed rate during regeneration that is independent of p53 status, does not require repair of DNA damage, does not require cell death for its stimulation, and occurs at the same rate regardless of a developmental delay.
A p53-dependent patterning checkpoint in wing discs after IR
Our experiments indicate that IR induces two p53-dependent processes that contribute to disc regeneration: rapid induction of massive apoptosis, and slowed progression of the developmental clock to delay PP, allowing cell division - at a developmentally programmed pace - to replace lost cells. Since disc pattern formation and disc growth (increase in disc size) are tightly coupled, a delay in patterning might be expected in wing discs from IR-treated larvae. We therefore tracked the patterning process during regeneration by examining the expression of several patterning genes in wing discs from larvae treated with IR. Wg expression undergoes dynamic changes as development proceeds and is expressed ectopically during regeneration after concentrated tissue damage (Huh et al., 2004; Perez-Garijo et al., 2004; Ryoo et al., 2004; Wells et al., 2006). At 96 hours AEL, the time of IR treatment, wing discs have a mature pattern of Wg expression: two rings encircling the central region with a bisecting line across the middle (Figure 5E). We examined Wg expression at several timepoints after IR, and found no evidence of ectopic expression in controls or in p53 mutants (Figure 5E, J). This contrasts with the widespread, p53-dependent ectopic Wg seen after RH damage (Figure 1B-C) (Huh et al., 2004; Ryoo et al., 2004; Smith-Bolton et al., 2009; Wells et al., 2006), or IR-induced damage when cell death is prevented by P35 or the dronc mutation (Supp. Figure 1E-G) (Perez-Garijo et al., 2004). We attribute this difference to the stochastic and transient nature of IR damage. In addition, we did not detect a major loss of the mature pattern of Wg expression, suggesting that the cells maintained Wg expression in an appropriate pattern after IR and did not revert to an earlier developmental stage.
Figure 5. A p53-dependent wing patterning checkpoint occurs after IR.
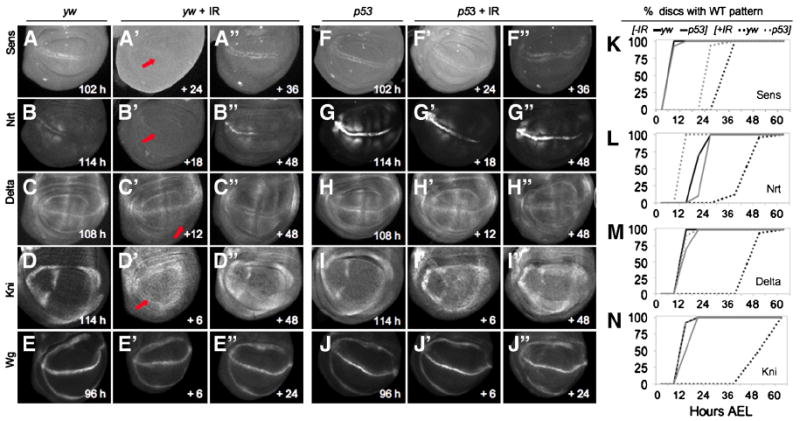
A-J. Wing discs expressing the indicated patterning genes. Panels A-E and F-J represent the time when the mature pattern of each gene is observed in unirradiated yw and p53ns mutant discs, respectively. The age of unirradiated animals is shown in the bottom right of panels; for irradiated discs time is represented as hours after IR.
A-A”, F-F”, K. Sens expression is delayed by IR. A. Sens expression at 102 hours AEL in unirradiated discs. Following IR, Sens is absent (arrow) at 120 hours AEL (A’) and expression is delayed until ~132 hours AEL (A”). F. Unirradiated p53 mutants also express Sens at 102 hours AEL. Following IR, p53 mutants already express Sens at 120 hours AEL (F’); the pattern is still present at 132 hours AEL (F”). K. Quantification of mature Sens expression pattern in each genotype.
B-B”, G-G”, L. Nrt protein expression is delayed by IR. B. Nrt expression at 114 hours AEL in unirradiated discs. Following IR, Nrt is absent (arrow) at 114 hours AEL (B’) and expression is delayed until 144 hours AEL (B”). G. Unirradiated p53 mutants express Nrt at 114 hours AEL, however AIR, p53 mutants do not delay Nrt expression (G’ -G”). L. Quantification of mature Nrt pattern in each genotype.
C-C”, H-H”, M. Delta expression is delayed by IR. C. Delta is expressed at 108 hours AEL in unirradiated discs. Following IR, Delta is absent (arrow) at 108 hours AEL (C’) and expression is delayed until 144 hours AEL (C”). H. Unirradiated p53 mutants express Delta at 108 hours AEL. Following IR, p53 mutants express the mature pattern of Delta by 108 hrs, which continues past 144 hrs AEL (H’-H”). M. Quantification of mature Delta pattern in each genotype.
D-D”, I-I”, N. Kni expression is delayed by IR. D. Kni expression at 114 hours AEL in unirradiated discs. Following IR, Kni is absent (arrow) at 120 hours AEL (D’) and expression is delayed until 144 hours AEL (D”). I. Unirradiated p53 mutants also express Kni at 114 hours AEL. Following IR, p53 mutants do not delay Kni expression (I’) and expression continues past 144 hours AEL (I”). N. Quantification of mature Kni pattern in each genotype.
E-E”, J-J”. Wg is expressed in wing discs from the 2nd instar on and is mature by 96 hours AEL in both unirradiated yw and p53 mutants (E, J). Irradiating either yw or p53 mutants does not grossly alter Wg expression (E’-E”, J’-J”).
K-N. Graphical representation of gene expression. Graphs depict the percent of discs with the mature pattern of expression of each gene at 6-hour intervals following IR. Black lines denote yw control animals, grey lines denote p53ns mutants; solid lines are unirradiated, dashed lines are irradiated. IR was induced with 40 Gy at 96 hours AEL and time points (bottom scale) are taken from the time of IR (0 point = 96 hours AEL).
We next addressed whether the onset of expression of patterning genes that initiate later in larval development was altered. A number of genes required for sensory organ precursor (SOP) and vein formation are initially expressed in mid-third instar wing discs, including senseless (sens), (Nolo et al., 2000, 2001), Delta (Dl) (O’Brochta and Bryant, 1987), knirps (kni) (Lunde et al., 1998) and neurotactin (nrt) (Speicher et al., 1998). Each of these genes has a dynamic expression pattern in the wing disc that either begins or changes after 96 hours AEL, the time at which larvae were subjected to IR in our experiments. In unirradiated yw animals sens was expressed in SOP cells along the D/V boundary of wing discs at 102 hours AEL (100% of discs; Figure 5A, K). Similarly, Dl was expressed in the presumptive L5 vein region at 108 hours AEL (100%; Figure 5C, M), kni was expressed in the presumptive L2 vein region by 114 hours AEL (100%; Figure 5D, N), and nrt was expressed at the D/V boundary by 120 hours AEL in 100% of unirradiated yw wing discs (Figure 5B, L). Following IR of yw animals, expression of each of these genes was delayed: sens expression by 30 hours (100% of discs); Dl (100%) by 48 hours, kni (100%) by 42 hours, and nrt (100%) by 32 hours (Figure 5A’-D’, K-N). The delayed expression patterns of these genes correlated well with the delay in larval development after IR. However, despite the temporal delay, the spatial pattern of expression of each gene in irradiated animals was identical to unirradiated controls.
In unirradiated p53 mutants, these genes were expressed at similar times: sens by 108 hours (100% of discs), Dl by 114 hours (100%), kni by 114 hours (100%) and nrt by 120 hours AEL (100%; (Figure 5F-I, K-N). However, in striking contrast to yw, in IR-treated p53 mutants most of the genes were expressed with normal timing: Dl (100%) and kni (100%) expression occurred with normal timing, and nrt expression was induced at 108 hours AEL (100% of discs), prior to its onset in unirradiated yw animals (Figure 5G-I, L-N). Only sens expression was delayed in the mutants, although the delay was reduced relative to irradiated yw controls (Figure 5F-K). Thus, in wildtype animals the IR-induced delay of the larval stage is accompanied by a delay in the onset of expression of several late patterning genes in the wing disc, and both of these delays are p53-dependent.
Cell proliferation continues during pupal development after IR
Our finding that wandering stage wing discs from IR-treated yw larvae were smaller than comparably aged discs from unirradiated controls (Figure 4A-B) suggested that the delay to PP was not sufficient to regenerate wing discs to an appropriate size. Nevertheless, adult wings arising from irradiated larvae were similar in size to unirradiated larvae, indicating that ultimately, regeneration was complete (Figure 2B, D). These results suggested that IR-treated wing disc cells continue to divide beyond the time cell division normally ceases in pupal development. Supporting this idea, the pupal stage was extended by 29 hours in IR-treated yw control larvae (Supp. Fig. 2C). Interestingly, irradiated p53 mutants also extended the pupal stage (Supp. Fig. 2C), indicating that in contrast to the larval delay, the pupal extension was not p53-dependent. To determine its consequences on wing regeneration we examined pupal wing discs, developed from irradiated larvae, for markers of cell division, cell death and DNA damage. Cell proliferation normally ceases by 24 hours after puparium formation (APF), thus few cells were positive for H3P in unirradiated wing discs from yw controls at this time. However, pupal discs from IR-treated yw larvae contained significant numbers of H3P positive cells at 24 hours APF (Figure 6A). Cell division continued for approximately 14 additional hours in these discs, finally ending by 38 hours APF (data not shown). Coupled with our findings that regenerating yw larval wing discs were small at the larval-pupal transition but reached a normal adult size, these results suggest that the proliferation during the pupal stage was necessary to complete regeneration of the damaged tissue.
We then examined pupal wing discs from p53 mutants. In the absence of IR, cells in p53 mutant discs stopped dividing with normal kinetics, and few H3P positive cells were observed in wing discs 24 hours APF (Figure 6A). However, many cells in IR-treated p53 mutant pupal wing discs were mitotic, and at 24 hours APF we observed twice the number of H3P positive cells relative to IR-treated yw pupal discs (p=0.02; Figure 6A-C). Cell division in the p53 mutant discs also continued longer than in yw discs: at 40 hours APF, p53 mutant pupal wings still contained numerous H3P positive cells (data not shown).
Unrepaired, damaged cells have limited differentiation potential
The additional cell proliferation in pupae helped to explain how wings of control flies irradiated during L3 reached an appropriate final size. In contrast, despite similar extensions of the pupal stage (Supp. Figure 2C) and continued cell proliferation (Figure 6A), p53 mutant pupal wing discs (Figure 6H-I) and adult wings (Figure 2C-D) remained significantly smaller than controls. The persistent γ-H2AX in larval discs implied that DNA damage was not repaired, thus we examined pupal wing discs from IR-treated yw and p53 mutant animals in more detail. At 24 hours APF, pupal wing discs from yw and p53 mutant were undersized relative to unirradiated controls, and discs of both genotypes had numerous apoptotic cells (Figure 6B-C and data not shown). However, although the yw pupal discs contained numerous TUNEL positive cells, very few contained γ-H2AX positive cells at 24 hours APF (data not shown) (Figure 6B). Apoptosis subsided by 40 hours APF and few cells in yw pupal wing discs were TUNEL positive at this time (data not shown).
In contrast, cells with DNA damage persisted in the p53 mutant pupal discs. 24 hours APF, p53 mutant cells frequently contained γ-H2AX (data not shown). TUNEL-positive cells were also common in these discs but they rarely coincided with γ-H2AX-positive cells, suggesting that damaged cells were not always eliminated (Figure 6C and data not shown). We also found that p53 mutant cells were frequently substantially larger than yw cells of the same chronological age (Figure 6E’ vs D’), and some appeared to be aneuploid, suggesting aberrant mitoses had occurred (Figure 6E”’, arrows; Supp. Figure 4C-C’ and data not shown). Consistent with this idea, many cells contained both H3P and γ-H2AX (data not shown). At 38 and 40 hours APF, p53 mutants still maintained high levels of γ-H2AX and many TUNEL positive cells, but again these markers rarely overlapped (Figure 6E”, G” and data not shown). Collectively, these results indicate that the small pupal wing discs and adult wings of IR-treated p53 mutants result from a combination of a late wave of apoptosis that eliminates many damaged cells, and continued, unproductive cell divisions due to persistent and unresolved DNA damage.
By 38 hours APF, IR-treated wing disc cells from yw animals became hexagonal in shape (Figure 6D) and each formed a pre-hair before 40 hours APF (Figure 6F’). These processes are associated with cell differentiation and are connected in time during wing morphogenesis (Classen et al., 2005; Waddington, 1941). However, in IR-treated discs they occurred approximately 10 hours later than normal, consistent with the overall delay in the pupal stage. Interestingly, even γ-H2AX-positive cells in p53 mutant pupal wings become hexagonal (Figure 6E’ and data not shown). By contrast, although non-damaged cells formed pre-hairs, most of the cells with DNA damage (γ-H2AX-positive) did not (Figure 6G’), suggesting that differentiation of the cells was limited. Each pre-hair forms a trichome, which is displayed with proximal to distal polarity in a regularly-spaced pattern across the adult wing, and trichome density is an indicator of cell size prior to cuticle secretion (Dobzhansky, 1929). Regenerated wings from IR-treated yw animals were patterned with correct trichome polarity (Figure 2B). Although the overall wing size of IR-treated p53 mutants was reduced and wings frequently had defects (Figure 2C-D; Table 2), trichome density and polarity of eclosed p53 mutants was similar to regenerated yw controls (Supp. Figure 2B). The trichome density indicates that the small wing size in the IR-treated p53 mutants was due to fewer, rather than smaller cells. Together, these observations suggest that the IR-treated p53 mutant cells had limited differentiation potential due to the persistence of damage, and that many damaged cells were ultimately eliminated from the epithelium.
Discussion
In this work, we make several key findings about imaginal disc regeneration. First, we find that both RH- and IR- induced tissue damage require p53 function for regeneration, but each process utilizes a different genetic mechanism to activate p53, suggesting that p53 plays a universal role in regulating regeneration. Second, we demonstrate that cell death is not necessary for regeneration to be induced, indicating that the lack of the early cell death program in p53 mutant wing discs cannot explain their inability to regenerate. Third, we find that cell proliferation is not stimulated during regeneration, but progresses at a developmentally programmed pace. Fourth, we identify a developmental checkpoint that controls the timing of disc patterning during the regeneration process. Finally, our data indicate that compensatory proliferation is not restricted to the larval stage of development. We discuss these findings below.
p53 functionally coordinates the DDR with tissue regeneration
Our results show that tissue regeneration subsequent to the DDR also requires p53. They add to previous work indicating that a continuum of events follows IR that culminates in regeneration of damaged tissue and survival of the animal (Figure 7). The process initiates with a stereotypical DDR in damaged imaginal disc cells within minutes of IR: damage is sensed and H2AX is phosphorylated, caspases are activated, and cell division in the disc transiently arrests. After approximately 5 hrs disc cells re-enter the cell cycle and continue to divide at apparently normal rates. Repair of DNA damage leads to loss of γ-H2AX, while ongoing apoptosis eliminates unrepaired cells. Our results indicate that the high level of cell death significantly slows the net growth of wing discs, compelling continuous cell division. This is facilitated by a delay of pupariation; in parallel, the expression of late patterning genes is delayed in wing discs. Interestingly, we found that after 40 Gy of IR, tissue damage was severe enough to require disc cell proliferation to continue not only during the extension of the larval developmental timer, but also into the pupal stage. Thus in contrast to what has been generally believed, disc regeneration is not restricted to the larval “growth phase” of development, but can continue in the early stages of pupal development. The ability of discs to continue regenerative growth after the hormonal cues that stimulate pupariation suggests that disc cell proliferation is only loosely regulated at the juvenile-adult transition.
Figure 7. Timeline of events induced after IR.
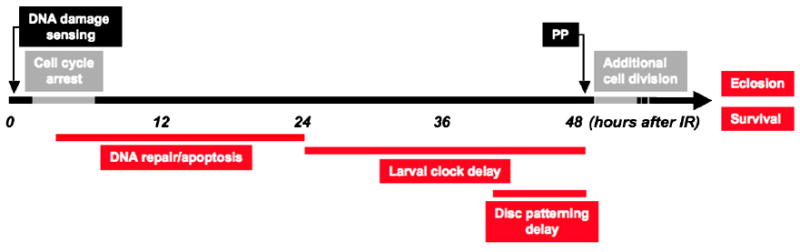
IR interrupts the normal larval and imaginal disc developmental programs to induce a stereotypical cell and tissue repair response. The response initiates when DNA damage is sensed, a cell cycle arrest is enacted, followed by cell cycle re-entry. These initial processes of the response are p53-independent (time scale is approximate and reflects hours AIR). The onset of cell death is followed by a systemic delay that allows for DNA and tissue damage repair. Massive apoptosis occurs in wing discs, eliminating many cells, while after reentry into the cell cycle cell division continues at normal rates over a prolonged period of growth. Disc cells delay expression of late patterning genes. DNA repair is essentially complete within 24 hours AIR. Disc patterning resumes despite small overall disc size, and the animals ultimately make the larval-pupal transition. Cell division continues longer than normal in pupal discs and discs reach appropriate final size. Animals eclose as healthy adults after an extended pupal period.
p53 is required for many subsequent events to occur (indicated by red lines). p53 mutants do not significantly delay development. DNA repair is unsuccessful and damaged DNA persists. Continued cell division at the normally programmed rate and a lack of apoptosis results in temporarily appropriate wing disc growth, but most late patterning occurs on the normal developmental schedule. 18 hours AIR cells begin to die, probably due to aneuploidy. Despite this, larvae make the larval-pupal transition; expression of γ-H2AX persists in disc cells leading to defects in cell shape and size. Disc cells continue to divide but many apoptose, reducing overall wing size; pupal development is extended for approximately 36 hours beyond the norm. Animals differentiate with morphological defects. Few adults eclose, and viability is severely reduced.
We find that in p53 mutants the events following IR are initially identical to wildtype, but subsequently show several differences. Cells lacking p53 recognize DNA damage and H2AX is phosphorylated, and the cell cycle checkpoint transiently arrests p53 mutant disc cells. p53 mutant cells also reenter the cell cycle with the same kinetics as controls. However, γ-H2AX persists at high levels in mutant discs, indicating that DNA damage is lingering, but the cells are unable to undergo apoptosis. Moreover, the mutant larvae do not significantly delay development, suggesting that p53 is required to regulate the developmental timer. Despite these differences, we found that disc cells divide at a normal rate and thus the size of the wing disc initially increases after IR. Later, the persistence of damaged DNA coupled with cell division creates aneuploidy, which, as been noted previously (Titen and Golic, 2008), may contribute to a late wave of apoptosis that we find continues late into the pupal stage.
Cell division continues beyond the normal cessation time in both our wildtype strain and the p53 mutants. In wildtype these additional pupal cell divisions are productive. In contrast, the late pupal divisions of p53 mutant disc cells appear to be largely futile since cell death is still prevalent; surviving cells appear to be aneuploid. Perhaps as a result, wing morphogenesis is delayed in irradiated p53 mutants relative to controls, although the mechanisms that pack wing disc cells and re-shape the wing disc ultimately do occur. The lack of DNA repair impairs cell differentiation and/or function throughout the pupa and leads to defects that prevent most animals from eclosing. This is interesting in light of our finding that the persistent DNA damage in p53 mutants did not appear to interfere with cell division during the larval phase of regeneration.
We found that p53 functions cell autonomously during disc regeneration, and that conditional expression of p53 in the wing disc is sufficient to induce ectopic expression of Wg, compensatory tissue growth, and a systemic developmental delay, all common aspects of regeneration. These results suggest that p53 is activated and operates cell-autonomously in damaged cells to promote regeneration. However, p53 also regulates the larval developmental clock, with the result that it coordinates control of disc regeneration with the physiology of the whole animal. Collectively, our results indicate that p53 functions to ensure repair of damaged DNA, to regulate the developmental timing of the animal, and to coordinate disc and animal maturation via a patterning checkpoint that delays cell fate acquisition in the disc. This linkage provides a mechanism that coordinates the two processes in time and thus facilitates the survival of the animal after DNA and tissue damage.
Repair of damaged DNA is critical for tissue regeneration
Our experiments show that in the absence of p53, DNA damage remains unrepaired, rendering cells incapable of completing the differentiation process. This is exacerbated by the absence of apoptosis immediately after IR in the p53 mutants, allowing cells with DNA damage to persist. Later, some of the persisting damaged cells in the mutants are eliminated by a late surge of disc cell death that continues into the pupal stage, in accordance with previous reports (McNamee and Brodsky, 2009; Wichmann et al., 2006). However, although this rids the disc of many damaged cells, it is not induced within a time frame that allows replacement of lost tissue, leading to small pupal wing disc size and small adult wings. Shortly after the onset of the late wave of apoptosis in p53 mutant discs the larval-pupal transition is crossed and metamorphosis is initiated. Although p53 mutant pupal wing disc cells continue to proliferate long after their wildtype counterparts have exited the cell cycle, our results suggest that the juvenile-to-adult transition – the commitment to produce adult traits - prevents critical developmental and patterning cues or render cells incapable of responding to them. However, damaged, aneuploid cells can differentiate trichomes (Wichmann et al., 2006), and we observed that some damaged cells did carry out aspects of trichome differentiation, including prehair formation. In addition, it is possible that most of the severely damaged cells in p53 mutants were ultimately eliminated.
Our results are strikingly similar to observations made in mouse and human cells. Loss of the murine DNA damage checkpoint protein Hus1 in a p53-deficient background results in accumulation of damaged cells after IR and prevents the compensatory responses in mammary epithelium (Yazinski et al., 2009). In serial transplantation experiments, self-renewal of irradiated human hematopoietic stem cells (HSCs) is compromised when they are deficient for p53 (Milyavsky et al., 2010), and, like our experiments with wing disc cells, γ-H2AX persisted in the HSCs. Collectively the data indicate that p53’s role in Drosophila disc regeneration is analogous to its role in tissue remodeling and stem cell renewal in vertebrates, and suggest that these functions of p53 are conserved.
p53 and systemic versus local effects during regeneration
The argument can be made that Drosophila imaginal discs merely take advantage of and extend developmental programs to repair and re-pattern lost tissue. This requires that the appropriate hormonal milieu be maintained by prolonging the juvenile, larval stage. We irradiated animals late during larval development but still within the disc growth period, and found that p53 function is required for the delay of the developmental timer that controls the juvenile-adult transition. Likewise, delay of the timer after RH-damage requires p53 (Wells et al., 2006). There is a strong correlation between delay of the timer and continued proliferation of discs (Bryant and Simpson, 1984; Cruz et al., 2009; Ramet et al., 2002). Although this relationship remains mysterious it is generally thought that negative feedback from proliferating discs inhibits a neural or humoral target (Poodry and Woods, 1990). In contrast to imaginal discs, the polyploid larval cells are relatively insensitive to IR (Halme et al.; Poodry and Woods, 1990). We were unable to detect induction of p53 activity after either RH damage or IR in tissues known to play key roles in developmental timing, such as the fat body, Dilp-2 expressing neurons, the prothoracic gland, and the corpora allota (data not shown). The p53 activity reporter contains 2 consensus p53 binding sites and is thus expected to report accurately in many tissues. Although we cannot exclude more trivial possibilities, the absence of induction in these tissues suggests that p53 function in imaginal discs is sufficient for the developmental delay induced upon IR as well as for the disc-autonomous responses. Thus, our data support the view that imaginal discs “signal” to the developmental clock to delay pupariation, and indicates that the putative signal requires p53 for its production.
Compensatory cell proliferation proceeds at a developmentally programmed rate during regeneration
We have presented two independent lines of evidence that argue that, in contrast to previous reports (Huh et al., 2004; Perez-Garijo et al., 2004; Ryoo et al., 2004), cell proliferation occurs at the same rate during regeneration as it does under normal developmental conditions. First, we found the number and distribution of mitotic cells to be similar in yw and in p53 mutant discs following IR at every examination from the cell cycle reentry at +6 hours until pupariation, despite the significant differences in the length of this period between the two genotypes. Second, RH-damage in clonal experiments showed that undamaged cells in the vicinity of RH-damaged cells proliferate at the same rate as cells in control discs without damage (Figure 4D) (Wells et al., 2006). Our results agree with Haynie and Bryant (Haynie and Bryant, 1977), who observed an IR dose-dependent lengthening of the developmental timer that correlated with an increase in clone size in adult wings, and concluded that the remaining cells “undergo additional divisions to compensate for this loss”. As a whole the data indicate that cell divisions occur at the normal rate, with additional divisions that occur during a p53-dependent slowing of the larval timer.
In addition, our results indicate that some aspects of disc patterning are delayed while the disc regenerates. This delay is also p53-dependent. One interpretation of these results is that the early, p53-dependent cell death program, by eliminating massive numbers of cells, directly delays ongoing the patterning process. However, our finding that dronc mutant animals, which are unable to induce cell death, exhibit the same regeneration responses as wildtype after IR argues against this idea. An alternative possibility arises from the observation that the disc patterning delay and the animal-wide delay are correlated in time, and thus could be inter-dependent. A third possibility is that p53 induces a disc-wide developmental checkpoint, directly dependent upon its role in the DDR but independent of the disc-produced “signal” that delays the larval timer, which couples regenerative growth to stage-appropriate cell fate specification. Further experiments are required to distinguish between these alternatives. Regardless of the mechanism, however, our finding that cell division proceeds at a similar rate during the delay of patterning regardless of p53 status implies that cell division and late patterning gene expression are independently regulated under these conditions.
What is the regeneration trigger?
It has been hypothesized that dying cells emit information that stimulates proliferation of surviving cells to regenerate the damaged tissue (Huh et al., 2004; Perez-Garijo et al., 2004; Ryoo et al., 2004). The regulation of expression of pro-apoptotic genes and pathways such as Rpr, Hid, Eiger/TNF and JNK by p53 (Brodsky et al., 2004; McEwen and Peifer, 2005), is consistent with the idea that p53 induces apoptosis, which in turn stimulates regeneration. However, our results indicate that this is not the case: the regeneration response is induced after IR even in cells rendered incapable of inducing apoptosis because of a null mutation in dronc. Since these dronc mutant cells remain wildtype for p53, these data support an apoptosis-independent role of p53 in provoking regeneration. Indeed, p53 is required for the tissue repair response even when RH genes are expressed (Wells et al., 2006). Thus, at a minimum, the data indicate that expression of pro-death genes and caspase activation are not sufficient to trigger regeneration.
As regeneration does not occur in its absence, p53 appears to be upstream of the signal that triggers regeneration. We suggest three scenarios for the regeneration trigger downstream of p53 activation. First, we find that p53 functions cell-autonomously to promote the ectopic induction of Wg, an early event in regeneration induced by a variety of methods in numerous animals (De Robertis; Galliot and Chera, 2010; McClure and Schubiger, 2008; Ryoo et al., 2004; Smith-Bolton et al., 2009; Wells et al., 2006). Moreover, expression of p53 under conditional Gal4 control induces ectopic Wg in the wing disc. Thus, the re-organization of disc patterning of the damaged tissue by ectopic expression of Wg, which necessitates a developmental delay for the completion of patterning and growth, may serve to trigger regeneration. However, in contrast to previous models invoking Wg as a mitogen, our findings indicate that cell proliferation continues at its developmentally programmed pace during this extended period of time.
Since JNK activation after tissue damage is p53-dependent (our unpublished data) (McEwen and Peifer, 2005), a second candidate for the regeneration trigger is the JNK signaling pathway. JNK is activated early after tissue damage and is important for wound healing (Bergantinos et al.; de Celis et al., 1996a; de Celis et al., 1996b; Galko and Krasnow, 2004; Hay et al., 1995; Mattila et al., 2005). JNK signaling is also activated upon disruptions of Wg and Dpp in the wing disc (Adachi-Yamada et al., 1999), and can itself lead to activation of Wg and Dpp expression (Perez-Garijo et al., 2009). JNK activity appears to be upstream of Wg expression, since hep null mutations, which eliminate JNK activity, prevent ectopic expression of Wg after RH damage (our unpublished data). A third possibility is that regeneration is triggered via a distinct program of gene expression directed by p53, which is independent of JNK or Wg.
Overall, our work suggests that p53 acts as a master regulator of tissue plasticity through its roles in the DDR, in tissue repair, and in coordinating these events with the animal’s physiology. In addition to its role in the initiation of regeneration, our results argue that p53 is responsible for regulating the expression of a signal(s) from discs that prolongs larval development to allow regeneration after either RH or IR damage. Studies that identify this signal, that determine from which tissue it arises, and that delineate the mechanism by which p53 controls each aspect of the regeneration process are important goals for the future.
Supplementary Material
Supplementary Figure 1. p53 activity is increased throughout the wing pouch and is required for tissue regeneration following IR
A-D. Animals were given a dose of 40 Gy at 96 hours AEL and monitored for expression of the p53 activity reporter, rpr150-lacZ. All images were taken with the same exposure parameters. A-B. B-gal expression in unirradiated WT (A) or p53ns (B) wing discs. The three regions of expression (marked by red asterisks) do not correspond to p53 activity, as they are still present in p53 mutant discs (B). C. Wing discs from yw larvae show an increase in B-gal expression throughout the wing pouch AIR, indicating increased p53 activity (arrow). D. p53 activity is absent in p53ns mutant wing discs AIR.
E-G. p53-dependent ectopic Wg expression and compensatory proliferation following IR (40 Gy at 72 hours AEL). E-E”’. Irradiated wing discs expressing UAS-P35 under EnGal4 control. The posterior compartment is marked by GFP (E”’); the A/P compartment boundary is marked by a dotted green line in E’-E”’). The posterior compartment becomes overgrown and expressed ectopic Wg (E’) and C3 (E”). C3 is also expressed in a small stripe of cells anterior to the A/P boundary. In wing discs from irradiated p53ns mutants, all posterior overgrowth, ectopic Wg and C3 expression is absent (F-F”’) G-G”’. Irradiated dronc mutant wing discs do not activate C3 (G”), but still overgrow and express ectopic Wg (G’).
Supplementary Figure 2. After IR, the absence of p53 function lengthens pupal development, leads to small wings with fewer cells and reduced adult viability
A. Following eclosion, 100% of unirradiated yw and p53ns animals survived more than 15 days. After irradiation, whereas 20% of yw animals survived for at least 15 days following eclosion, 100% of p53ns mutants died during the first day after eclosion. Larvae were irradiated with 40 Gy at 96 hours AEL.
B. Wings of irradiated yw and p53ns animals contain similar numbers of trichomes per unit area as unirradiated controls. Error bars, sem.
C. Following 40 Gy at 96 hours AEL, yw animals extended pupal development by 29 hours (p=0.06). p53ns mutants extend pupal development by 36 hours (p=0.03). yw vs p53 (p=3.3E-06). yw +IR vs p53+IR (p=0.03). Error bars, sem. Larvae were irradiated with 40 Gy at 96 hours AEL.
Supplementary Figure 3. γ-H2AX but not H2Av persists in irradiated p53 mutants
Double labeling of wing discs with antibodies against both γ-H2Av and γ-H2AX showed complete overlap of expression in yw and p53ns mutant discs (Supp. Figure 3A, B). However, while γ-H2Av expression declined over time in p53ns mutant discs (Supp. Figure 3G-J), γ-H2AX persisted into the pupal stage (Figure 3E”, Figure 6E”, G”).
A-A”’. At 24 hours following IR, both H2Av (A’) and γ-H2AX (A”) are present in yw wing discs. They are also completely overlapping (A).
B-B”’. At 24 hours following IR, H2Av (B’) but not γ-H2AX (B”) has largely cleared from p53ns mutant wing discs. However, they are co-localized when present (B).
C-F. Time course of H2Av staining in irradiated yw larvae show that it comes on shortly following IR (D) and persists until after 24 hours following IR (E-F). C. No IR. Time (hours) AIR is noted in upper right corner of images.
G-J. Time course of H2Av staining in irradiated p53ns larvae show that it comes on slowly following IR (H) before peaking around 12 hours following IR (I) and then begins to disappear around 18 hours following IR (J). G. No IR. Hours AIR are noted in upper right corner of image.
Animals were given 40 Gy IR at 96 hours AEL.
Supplementary Figure 4. Irradiated p53 mutant pupal wing disc cells contain foci of persistent γ-H2AX
A. Wing disc cells of irradiated p53ns mutant at 120 hr AEL contain numerous γ-H2AX foci (red). Hoechst-stained DNA is shown in blue. B. γ-H2AX – positive cells in pupal wing disc at 38hr APF from irradiated p53ns animal (IR at 96 hr AEL). Box indicates region magnified in C and C’. C. Merged channels (DNA, blue and γ-H2AX, red) of region boxed in B. C’. Single channel of C showing DNA. Arrows point to γ-H2AX-positive mitotic and/or aneuploid cells.
Acknowledgments
We thank Claire de la Cova for providing the FRT p53ns flies, the Bloomington Stock Center and Developmental Studies Hybridoma bank for Drosophila strains and antibodies, respectively, and members of the Johnston lab for advice and Cora Bergantinos-Crespo for additional experiments and stimulating discussions. Supported by a pre-doctoral traineeship from NIH T32 CA09503 to BSW and NIH grants R01HD042770 and R01GM078464 to LAJ.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adachi-Yamada T, Fujimura-Kamada K, Nishida Y, Matsumoto K. Distortion of proximodistal information causes JNK-dependent apoptosis in Drosophila wing. Nature. 1999;400:166–169. doi: 10.1038/22112. [DOI] [PubMed] [Google Scholar]
- Bergantinos C, Corominas M, Serras F. Cell death-induced regeneration in wing imaginal discs requires JNK signalling. Development. 137:1169–1179. doi: 10.1242/dev.045559. [DOI] [PubMed] [Google Scholar]
- Bergantinos C, Vilana X, Corominas M, Serras F. Imaginal discs: Renaissance of a model for regenerative biology. Bioessays. 32:207–217. doi: 10.1002/bies.200900105. [DOI] [PubMed] [Google Scholar]
- Bodenstein D. The postembryonic development of Drosophila. In: Demerec M, editor. Biology of Drosophila. Wiley; New York: 1950. pp. 275–367. [Google Scholar]
- Brodsky MH, Nordstrom W, Tsang G, Kwan E, Rubin GM, Abrams JM. Drosophila p53 binds a damage response element at the reaper locus. Cell. 2000;101:103–113. doi: 10.1016/S0092-8674(00)80627-3. [DOI] [PubMed] [Google Scholar]
- Brodsky MH, Weinert BT, Tsang G, Rong YS, McGinnis NM, Golic KG, Rio DC, Rubin GM. Drosophila melanogaster MNK/ Chk2 and p53 regulate multiple DNA repair and apoptotic pathways following DNA damage. Mol Cell Biol. 2004;24:1219–1231. doi: 10.1128/MCB.24.3.1219-1231.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant PJ, Simpson P. Intrinsic and extrinsic control of growth in developing organs. Q Rev Biol. 1984;59:387–415. doi: 10.1086/414040. [DOI] [PubMed] [Google Scholar]
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- Chehab NH, Malikzay A, Appel M, Halazonetis TD. Chk2/ hCds1 functions as a DNA damage checkpoint in G(1) by stabilizing p53. Genes Dev. 2000;14:278–288. [PMC free article] [PubMed] [Google Scholar]
- Classen AK, Anderson KI, Marois E, Eaton S. Hexagonal packing of Drosophila wing epithelial cells by the planar cell polarity pathway. Dev Cell. 2005;9:805–817. doi: 10.1016/j.devcel.2005.10.016. [DOI] [PubMed] [Google Scholar]
- Cruz C, Glavic A, Casado M, de Celis JF. A gain-of-function screen identifying genes required for growth and pattern formation of the Drosophila melanogaster wing. Genetics. 2009;183:1005–1026. doi: 10.1534/genetics.109.107748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Celis JF, de Celis J, Ligoxygakis P, Preiss A, Delidakis C, Bray S. Functional relationships between Notch, Su(H) and the bHLH genes of the E(spl) complex: the E(spl) genes mediate only a subset of Notch activities during imaginal development. Development. 1996a;122:2719–2728. doi: 10.1242/dev.122.9.2719. [DOI] [PubMed] [Google Scholar]
- de Celis JF, Garcia-Bellido A, Bray SJ. Activation and function of Notch at the dorsal-ventral boundary of the wing imaginal disc. Development. 1996b;122:359–369. doi: 10.1242/dev.122.1.359. [DOI] [PubMed] [Google Scholar]
- de la Cova C, Abril M, Bellosta P, Gallant P, Johnston LA. Drosophila myc regulates organ size by inducing cell competition. Cell. 2004;117:107–116. doi: 10.1016/s0092-8674(04)00214-4. [DOI] [PubMed] [Google Scholar]
- De Robertis EM. Wnt signaling in axial patterning and regeneration: lessons from planaria. Sci Signal. 3:pe21. doi: 10.1126/scisignal.3127pe21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobzhansky T. The influence of the quantity and quality of chromosomal material on the size of the cells in Drosophila melanogaster. Roux Arch. EntwMech Organ. 1929;115:363–379. doi: 10.1007/BF02078996. [DOI] [PubMed] [Google Scholar]
- Flaggs G, Plug AW, Dunks KM, Mundt KE, Ford JC, Quiggle MR, Taylor EM, Westphal CH, Ashley T, Hoekstra MF, Carr AM. Atm-dependent interactions of a mammalian chk1 homolog with meiotic chromosomes. Curr Biol. 1997;7:977–986. doi: 10.1016/s0960-9822(06)00417-9. [DOI] [PubMed] [Google Scholar]
- Fogarty P, Campbell SD, Abu-Shumays R, Phalle BS, Yu KR, Uy GL, Goldberg ML, Sullivan W. The Drosophila grapes gene is related to checkpoint gene chk1/ rad27 and is required for late syncytial division fidelity. Curr Biol. 1997;7:418–426. doi: 10.1016/s0960-9822(06)00189-8. [DOI] [PubMed] [Google Scholar]
- Galko MJ, Krasnow MA. Cellular and genetic analysis of wound healing in Drosophila larvae. PLoS Biol. 2004;2:E239. doi: 10.1371/journal.pbio.0020239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galliot B, Chera S. The Hydra model: disclosing an apoptosis-driven generator of Wnt-based regeneration. Trends Cell Biol. 2010;20:514–523. doi: 10.1016/j.tcb.2010.05.006. [DOI] [PubMed] [Google Scholar]
- Greer DA, Besley BD, Kennedy KB, Davey S. hRad9 rapidly binds DNA containing double-strand breaks and is required for damage-dependent topoisomerase II beta binding protein 1 focus formation. Cancer Res. 2003;63:4829–4835. [PubMed] [Google Scholar]
- Guo N, Faller DV, Vaziri C. A novel DNA damage checkpoint involving post-transcriptional regulation of cyclin A expression. J Biol Chem. 2000;275:1715–1722. doi: 10.1074/jbc.275.3.1715. [DOI] [PubMed] [Google Scholar]
- Halme A, Cheng M, Hariharan IK. Retinoids regulate a developmental checkpoint for tissue regeneration in Drosophila. Curr Biol. 20:458–463. doi: 10.1016/j.cub.2010.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartenstein V, Posakony JW. Development of adult sensilla on the wing and notum of Drosophila melanogaster. Development. 1989;107:389–405. doi: 10.1242/dev.107.2.389. [DOI] [PubMed] [Google Scholar]
- Hay BA, Wassarman DA, Rubin GM. Drosophila homologs of baculovirus inhibitor of apoptosis proteins function to block cell death. Cell. 1995;83:1253–1262. doi: 10.1016/0092-8674(95)90150-7. [DOI] [PubMed] [Google Scholar]
- Haynie J, Bryant PJ. The effects of X-rays on the proliferation dynamics of cells in the imaginal wing disc of Drosophila melanogaster. Wilhelm Roux’s Arch. 1977;183:85–100. doi: 10.1007/BF00848779. [DOI] [PubMed] [Google Scholar]
- Haynie JL, Bryant PJ. Intercalary regeneration in imaginal wing disk of Drosophila melanogaster. Nature. 1976;259:659–662. doi: 10.1038/259659b0. [DOI] [PubMed] [Google Scholar]
- Hirao A, Kong YY, Matsuoka S, Wakeham A, Ruland J, Yoshida H, Liu D, Elledge SJ, Mak TW. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science. 2000;287:1824–1827. doi: 10.1126/science.287.5459.1824. [DOI] [PubMed] [Google Scholar]
- Hromas R, Wray J, Lee SH, Martinez L, Farrington J, Corwin LK, Ramsey H, Nickoloff JA, Williamson EA. The human set and transposase domain protein Metnase interacts with DNA Ligase IV and enhances the efficiency and accuracy of non-homologous end-joining. DNA Repair (Amst) 2008;7:1927–1937. doi: 10.1016/j.dnarep.2008.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh JR, Guo M, Hay BA. Compensatory proliferation induced by cell death in the Drosophila wing disc requires activity of the apical cell death caspase Dronc in a nonapoptotic role. Curr Biol. 2004;14:1262–1266. doi: 10.1016/j.cub.2004.06.015. [DOI] [PubMed] [Google Scholar]
- Johnston LA. Regeneration and transdetermination: new tricks from old cells. Cell. 2005;120:288–290. doi: 10.1016/j.cell.2005.01.022. [DOI] [PubMed] [Google Scholar]
- Johnston LA, Edgar BA. Wingless and Notch regulate cell-cycle arrest in the developing Drosophila wing. Nature. 1998;394:82–84. doi: 10.1038/27925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston LA, Sanders AL. Wingless promotes cell survival but constrains growth during Drosophila wing development. Nat Cell Biol. 2003;5:827–833. doi: 10.1038/ncb1041. [DOI] [PubMed] [Google Scholar]
- Kondo S, Senoo-Matsuda N, Hiromi Y, Miura M. DRONC coordinates cell death and compensatory proliferation. Mol Cell Biol. 2006;26:7258–7268. doi: 10.1128/MCB.00183-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuerbitz SJ, Plunkett BS, Walsh WV, Kastan MB. Wild-type p53 is a cell cycle checkpoint determinant following irradiation. Proc Natl Acad Sci U S A. 1992;89:7491–7495. doi: 10.1073/pnas.89.16.7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach TJ, Mazzeo M, Chotkowski HL, Madigan JP, Wotring MG, Glaser RL. Histone H2A.Z is widely but nonrandomly distributed in chromosomes of Drosophila melanogaster. J Biol Chem. 2000;275:23267–23272. doi: 10.1074/jbc.M910206199. [DOI] [PubMed] [Google Scholar]
- Lee T, Luo L. Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron. 1999;22:451–461. doi: 10.1016/s0896-6273(00)80701-1. [DOI] [PubMed] [Google Scholar]
- Lunde K, Biehs B, Nauber U, Bier E. The knirps and knirps-related genes organize development of the second wing vein in Drosophila. Development. 1998;125:4145–4154. doi: 10.1242/dev.125.21.4145. [DOI] [PubMed] [Google Scholar]
- MacPhail SH, Banath JP, Yu TY, Chu EH, Lambur H, Olive PL. Expression of phosphorylated histone H2AX in cultured cell lines following exposure to X-rays. Int J Radiat Biol. 2003;79:351–358. doi: 10.1080/0955300032000093128. [DOI] [PubMed] [Google Scholar]
- Madigan JP, Chotkowski HL, Glaser RL. DNA double-strand break-induced phosphorylation of Drosophila histone variant H2Av helps prevent radiation-induced apoptosis. Nucleic Acids Res. 2002;30:3698–3705. doi: 10.1093/nar/gkf496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattila J, Omelyanchuk L, Kyttala S, Turunen H, Nokkala S. Role of Jun N-terminal Kinase (JNK) signaling in the wound healing and regeneration of a Drosophila melanogaster wing imaginal disc. Int J Dev Biol. 2005;49:391–399. doi: 10.1387/ijdb.052006jm. [DOI] [PubMed] [Google Scholar]
- McClure KD, Schubiger G. A screen for genes that function in leg disc regeneration in Drosophila melanogaster. Mech Dev. 2008;125:67–80. doi: 10.1016/j.mod.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClure KD, Sustar A, Schubiger G. Three genes control the timing, the site and the size of blastema formation in Drosophila. Dev Biol. 2008;319:68–77. doi: 10.1016/j.ydbio.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen DG, Peifer M. Puckered, a Drosophila MAPK phosphatase, ensures cell viability by antagonizing JNK-induced apoptosis. Development. 2005;132:3935–3946. doi: 10.1242/dev.01949. [DOI] [PubMed] [Google Scholar]
- McNamee LM, Brodsky MH. p53-independent apoptosis limits DNA damage-induced aneuploidy. Genetics. 2009;182:423–435. doi: 10.1534/genetics.109.102327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milyavsky M, Gan OI, Trottier M, Komosa M, Tabach O, Notta F, Lechman E, Hermans KG, Eppert K, Konovalova Z, Ornatsky O, Domany E, Meyn MS, Dick JE. A distinctive DNA damage response in human hematopoietic stem cells reveals an apoptosis-independent role for p53 in self-renewal. Cell Stem Cell. 2010;7:186–197. doi: 10.1016/j.stem.2010.05.016. [DOI] [PubMed] [Google Scholar]
- Neufeld TP, de la Cruz AF, Johnston LA, Edgar BA. Coordination of growth and cell division in the Drosophila wing. Cell. 1998;93:1183–1193. doi: 10.1016/s0092-8674(00)81462-2. [DOI] [PubMed] [Google Scholar]
- Niida H, Nakanishi M. DNA damage checkpoints in mammals. Mutagenesis. 2006;21:3–9. doi: 10.1093/mutage/gei063. [DOI] [PubMed] [Google Scholar]
- Nolo R, Abbott LA, Bellen HJ. Senseless, a Zn finger transcription factor, is necessary and sufficient for sensory organ development in Drosophila. Cell. 2000;102:349–362. doi: 10.1016/s0092-8674(00)00040-4. [DOI] [PubMed] [Google Scholar]
- Nolo R, Abbott LA, Bellen HJ. Drosophila Lyra mutations are gain-of-function mutations of senseless. Genetics. 2001;157:307–315. doi: 10.1093/genetics/157.1.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brochta DA, Bryant PJ. Distribution of S-phase cells during the regeneration of Drosophila imaginal wing discs. Dev Biol. 1987;119:137–142. doi: 10.1016/0012-1606(87)90215-6. [DOI] [PubMed] [Google Scholar]
- Olive PL. Detection of DNA damage in individual cells by analysis of histone H2AX phosphorylation. Methods Cell Biol. 2004;75:355–373. doi: 10.1016/s0091-679x(04)75014-1. [DOI] [PubMed] [Google Scholar]
- Perez-Garijo A, Martin FA, Morata G. Caspase inhibition during apoptosis causes abnormal signalling and developmental aberrations in Drosophila. Development. 2004 doi: 10.1242/dev.01432. [DOI] [PubMed] [Google Scholar]
- Perez-Garijo A, Shlevkov E, Morata G. The role of Dpp and Wg in compensatory proliferation and in the formation of hyperplastic overgrowths caused by apoptotic cells in the Drosophila wing disc. Development. 2009;136:1169–1177. doi: 10.1242/dev.034017. [DOI] [PubMed] [Google Scholar]
- Poodry C, Woods D. Conrol of the developmental timer for Drosophila pupariation. Roux’s Arch Dev Biol. 1990;199:219–227. doi: 10.1007/BF01682081. [DOI] [PubMed] [Google Scholar]
- Ramet M, Lanot R, Zachary D, Manfruelli P. JNK signaling pathway is required for efficient wound healing in Drosophila. Dev Biol. 2002;241:145–156. doi: 10.1006/dbio.2001.0502. [DOI] [PubMed] [Google Scholar]
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- Rong YS, Titen SW, Xie HB, Golic MM, Bastiani M, Bandyopadhyay P, Olivera BM, Brodsky M, Rubin GM, Golic KG. Targeted mutagenesis by homologous recombination in D. melanogaster. Genes Dev. 2002;16:1568–1581. doi: 10.1101/gad.986602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryoo HD, Gorenc T, Steller H. Apoptotic cells can induce compensatory cell proliferation through the JNK and the Wingless signaling pathways. Dev Cell. 2004;7:491–501. doi: 10.1016/j.devcel.2004.08.019. [DOI] [PubMed] [Google Scholar]
- Schubiger M, Palka J. Changing spatial patterns of DNA replication in the developing wing of Drosophila. Dev Biol. 1987;123:145–153. doi: 10.1016/0012-1606(87)90436-2. [DOI] [PubMed] [Google Scholar]
- Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer. 2003;3:155–168. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- Simpson P, Berreur P, Berreur-Bonnenfant J. The initiation of pupariation in Drosophila: dependence on growth of the imaginal discs. J Embryol Exp Morphol. 1980;57:155–165. [PubMed] [Google Scholar]
- Smith-Bolton RK, Worley MI, Kanda H, Hariharan IK. Regenerative growth in Drosophila imaginal discs is regulated by Wingless and Myc. Dev Cell. 2009;16:797–809. doi: 10.1016/j.devcel.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogame N, Kim M, Abrams JM. Drosophila p53 preserves genomic stability by regulating cell death. Proc Natl Acad Sci U S A. 2003;100:4696–4701. doi: 10.1073/pnas.0736384100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speicher S, Garcia-Alonso L, Carmena A, Martin-Bermudo MD, de la Escalera S, Jimenez F. Neurotactin functions in concert with other identified CAMs in growth cone guidance in Drosophila. Neuron. 1998;20:221–233. doi: 10.1016/s0896-6273(00)80451-1. [DOI] [PubMed] [Google Scholar]
- Srivastava N, Gochhait S, de Boer P, Bamezai RN. Role of H2AX in DNA damage response and human cancers. Mutat Res. 2009;681:180–188. doi: 10.1016/j.mrrev.2008.08.003. [DOI] [PubMed] [Google Scholar]
- Struhl G, Basler K. Organizing activity of wingless protein in Drosophila. Cell. 1993;72:527–540. doi: 10.1016/0092-8674(93)90072-x. [DOI] [PubMed] [Google Scholar]
- Sustar A, Schubiger G. A transient cell cycle shift in Drosophila imaginal disc cells precedes multipotency. Cell. 2005;120:383–393. doi: 10.1016/j.cell.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Titen SW, Golic KG. Telomere loss provokes multiple pathways to apoptosis and produces genomic instability in Drosophila melanogaster. Genetics. 2008;180:1821–1832. doi: 10.1534/genetics.108.093625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddington CH. The genetic control of wing development in Drosophila. J Genet. 1941;41:75–139. [Google Scholar]
- Wells BS, Yoshida E, Johnston LA. Compensatory proliferation in Drosophila imaginal discs requires Dronc-dependent p53 activity. Curr Biol. 2006;16:1606–1615. doi: 10.1016/j.cub2006.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichmann A, Jaklevic B, Su TT. Ionizing radiation induces caspase-dependent but Chk2- and p53-independent cell death in Drosophila melanogaster. Proc Natl Acad Sci U S A. 2006;103:9952–9957. doi: 10.1073/pnas.0510528103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Li Y, Arcaro M, Lackey M, Bergmann A. The CARD-carrying caspase Dronc is essential for most, but not all, developmental cell death in Drosophila. Development. 2005;132:2125–2134. doi: 10.1242/dev.01790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazinski SA, Westcott PM, Ong K, Pinkas J, Peters RM, Weiss RS. Dual inactivation of Hus1 and p53 in the mouse mammary gland results in accumulation of damaged cells and impaired tissue regeneration. Proc Natl Acad Sci U S A. 2009;106:21282–21287. doi: 10.1073/pnas.0904965106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou BB, Chaturvedi P, Spring K, Scott SP, Johanson RA, Mishra R, Mattern MR, Winkler JD, Khanna KK. Caffeine abolishes the mammalian G(2)/ M DNA damage checkpoint by inhibiting ataxia-telangiectasia-mutated kinase activity. J Biol Chem. 2000;275:10342–10348. doi: 10.1074/jbc.275.14.10342. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. p53 activity is increased throughout the wing pouch and is required for tissue regeneration following IR
A-D. Animals were given a dose of 40 Gy at 96 hours AEL and monitored for expression of the p53 activity reporter, rpr150-lacZ. All images were taken with the same exposure parameters. A-B. B-gal expression in unirradiated WT (A) or p53ns (B) wing discs. The three regions of expression (marked by red asterisks) do not correspond to p53 activity, as they are still present in p53 mutant discs (B). C. Wing discs from yw larvae show an increase in B-gal expression throughout the wing pouch AIR, indicating increased p53 activity (arrow). D. p53 activity is absent in p53ns mutant wing discs AIR.
E-G. p53-dependent ectopic Wg expression and compensatory proliferation following IR (40 Gy at 72 hours AEL). E-E”’. Irradiated wing discs expressing UAS-P35 under EnGal4 control. The posterior compartment is marked by GFP (E”’); the A/P compartment boundary is marked by a dotted green line in E’-E”’). The posterior compartment becomes overgrown and expressed ectopic Wg (E’) and C3 (E”). C3 is also expressed in a small stripe of cells anterior to the A/P boundary. In wing discs from irradiated p53ns mutants, all posterior overgrowth, ectopic Wg and C3 expression is absent (F-F”’) G-G”’. Irradiated dronc mutant wing discs do not activate C3 (G”), but still overgrow and express ectopic Wg (G’).
Supplementary Figure 2. After IR, the absence of p53 function lengthens pupal development, leads to small wings with fewer cells and reduced adult viability
A. Following eclosion, 100% of unirradiated yw and p53ns animals survived more than 15 days. After irradiation, whereas 20% of yw animals survived for at least 15 days following eclosion, 100% of p53ns mutants died during the first day after eclosion. Larvae were irradiated with 40 Gy at 96 hours AEL.
B. Wings of irradiated yw and p53ns animals contain similar numbers of trichomes per unit area as unirradiated controls. Error bars, sem.
C. Following 40 Gy at 96 hours AEL, yw animals extended pupal development by 29 hours (p=0.06). p53ns mutants extend pupal development by 36 hours (p=0.03). yw vs p53 (p=3.3E-06). yw +IR vs p53+IR (p=0.03). Error bars, sem. Larvae were irradiated with 40 Gy at 96 hours AEL.
Supplementary Figure 3. γ-H2AX but not H2Av persists in irradiated p53 mutants
Double labeling of wing discs with antibodies against both γ-H2Av and γ-H2AX showed complete overlap of expression in yw and p53ns mutant discs (Supp. Figure 3A, B). However, while γ-H2Av expression declined over time in p53ns mutant discs (Supp. Figure 3G-J), γ-H2AX persisted into the pupal stage (Figure 3E”, Figure 6E”, G”).
A-A”’. At 24 hours following IR, both H2Av (A’) and γ-H2AX (A”) are present in yw wing discs. They are also completely overlapping (A).
B-B”’. At 24 hours following IR, H2Av (B’) but not γ-H2AX (B”) has largely cleared from p53ns mutant wing discs. However, they are co-localized when present (B).
C-F. Time course of H2Av staining in irradiated yw larvae show that it comes on shortly following IR (D) and persists until after 24 hours following IR (E-F). C. No IR. Time (hours) AIR is noted in upper right corner of images.
G-J. Time course of H2Av staining in irradiated p53ns larvae show that it comes on slowly following IR (H) before peaking around 12 hours following IR (I) and then begins to disappear around 18 hours following IR (J). G. No IR. Hours AIR are noted in upper right corner of image.
Animals were given 40 Gy IR at 96 hours AEL.
Supplementary Figure 4. Irradiated p53 mutant pupal wing disc cells contain foci of persistent γ-H2AX
A. Wing disc cells of irradiated p53ns mutant at 120 hr AEL contain numerous γ-H2AX foci (red). Hoechst-stained DNA is shown in blue. B. γ-H2AX – positive cells in pupal wing disc at 38hr APF from irradiated p53ns animal (IR at 96 hr AEL). Box indicates region magnified in C and C’. C. Merged channels (DNA, blue and γ-H2AX, red) of region boxed in B. C’. Single channel of C showing DNA. Arrows point to γ-H2AX-positive mitotic and/or aneuploid cells.