

Abstract
Macrophages are terminally differentiated cells of the mononuclear phagocytic lineage and develop under the stimulus of their primary growth and differentiation factor, CSF-1. Although they differentiate into heterogeneous populations, depending upon their tissue of residence, motility is an important aspect of their function. To facilitate their migration through tissues, macrophages express a unique range of adhesion and cytoskeletal proteins. Notably, macrophages do not form large, stable adhesions or actin stress fibers but rely on small, short lived point contacts, focal complexes and podosomes for traction. Thus, macrophages are built to respond rapidly to migratory stimuli. As well as triggering growth and differentiation, CSF-1 is also a chemokine that regulates macrophage migration via activation the CSF-1 receptor tyrosine kinase. CSF-1R autophosphorylation of several intracellular tyrosine residues leads to association and activation of many downstream signaling molecules. However, phosphorylation of just one residue, Y721, mediates association of PI3K with the receptor to activate the major motility signaling pathways in macrophages. Dissection of these pathways will identify drug targets for the inhibition of diseases in which macrophages contribute to adverse outcomes.
1. Introduction
Macrophages reside in almost every tissue of the body and, as a result of their adaptation to the different tissue microenvironments, adopt a diverse range of morphologies and carry out a variety of functions. Despite their heterogeneity, macrophages all originate from the pluripotent hematopoietic stem cell and, under the influence of hematopoietic growth factors, differentiate through several multipotent progenitor stages to lineage committed mononuclear phagocytic precursors in the bone marrow[1–3]. The mononuclear phagocyte system is comprised of the mononuclear phagocyte precursors, monoblasts, and promonocytes, as well as circulating monocytes and fully differentiated, tissue resident macrophages [1–4]. Colony-stimulating factor-1 (CSF-1) has long been recognized as the primary growth factor regulating the survival, proliferation, and differentiation of cells of the mononuclear phagocytic lineage [1, 3, 5]. It is also an essential differentiation factor for the bone resorbing osteoclast [6]. A spontaneously occurring inactivating mutation in the mouse CSF-1 gene (osteopetrotic, Csf-1op) is associated with reduced tissue macrophage numbers and a marked reduction in osteoclasts, and causes osteopetrosis along with other developmental defects [1, 7–9]. CSF-1 signals through the CSF-1 receptor tyrosine kinase (RTK), encoded by the c-fms proto-oncogene [10], to trigger a series of phosphorylation cascades that mediate cellular responses to CSF-1 [1]. While the phenotype of mice nullizygous for the CSF-1R (Csf1r−/Csf1r−) largely recapitulates that seen in the Csf1op/Csf1op mouse, it is more severe and the discrepancy has since been explained by the discovery of a second partially redundant ligand for the CSF-1R, interleukin-34 (IL-34) [11–13].
Macrophages are professionally motile cells that carry out a variety of roles in immune surveillance and normal tissue development by secreting cytokines and growth factors and phagocytosing foreign material and apoptotic cells. Transendothelial and interstitial motility is an essential aspect of their function as they must be able to move to specific sites upon demand. From studies in primary macrophages and CSF-1 dependent macrophage cell lines, it is evident that CSF-1 is not only a mononuclear phagocyte lineage growth factor but is an important regulator of macrophage motility [1, 14–16]. Depletion of specific subsets of tissue macrophages in the Csf1op/Csf1op mouse and their reconstitution upon restoration of CSF-1 expression indicates that CSF-1 regulates the differentiation and migration of trophic and/or scavenger macrophages that are physiologically important for normal development and tissue homeostasis rather than in immune function [3, 9, 11, 17]. CSF-1 or CSF-1R deficient mice demonstrate abnormal neural, skeletal, and glandular development, not only due to reduced macrophage and osteoclast numbers but also through reduced matrix remodeling [3]. Thus, CSF-1-induced motility is likely to be an important element of macrophage function in development. Beyond their critical physiological role, CSF-1 dependent macrophages have also been demonstrated to promote disease progression in conditions ranging from cancer to atherosclerosis and arthritis [1, 3, 18, 19]. Reactivation of developmental macrophage functions may underlie the progression of these pathologies [3]. To participate in the disease process, macrophages must first migrate to the affected tissue. Furthermore, in the case of enhancement of tumor invasion, tumor-associated macrophages and mammary carcinoma cells have been shown to migrate away from the primary tumor together [20]. Yet little is known about how macrophage motility is regulated, how the motility machinery differs from other cell types and whether inhibition of macrophage motility may improve disease outcomes. Moreover, CSF-1 activated signaling pathways activate molecules or protein isoforms selectively expressed in macrophages [1], some of which may be attractive therapeutic targets to specifically inhibit macrophage infiltration into sites of disease. Considering the contribution of macrophages and CSF-1 to tumour dissemination and the progression of several inflammatory disorders [3, 18, 19], this review focuses on our current understanding of macrophage migration and its regulation by CSF-1.
2. Macrophage Motility
Almost all cell types are capable of migration but, in the adult organism, motility is particularly important for cells participating in immune cell function and wound healing. Leukocytes move rapidly compared to other cells, with neutrophils and lymphocytes measured at speeds of up to 25–30 μm/min [21, 22]. While macrophages are slower than other leukocytes, moving at ~1 μm/min in vitro, in vivo they respond rapidly to wounding or inflammatory signals and can migrate over considerable distances. Indeed, their migration speed has been measured at over 10 μm/min when attracted into a wound in a fish model [23]. Compared to fibroblasts and epithelial cells (~0.1–0.5 μm/min) [21], macrophages are considered to be efficient migrators.
The fundamental locomotory mechanisms are broadly similar in most cell types [24, 25]. Motility is a complex and integrated process that has typically been broken down into five components: (1) cell polarization or breaking of symmetry upon designation of the leading edge, (2) actin polymerization-driven protrusion of the leading edge, (3) integrin-mediated adhesion of the extended protrusion to underlying extracellular matrix proteins to provide the necessary traction for (4) actomyosin contractility-based forward translocation of the cell body, and, finally, (5) de-adhesion of the trailing edge to complete the cycle [25, 26]. Nevertheless, this description is a simplification of an integrated process, for example actin polymerization and actomyosin contraction contribute to adhesion structure formation and maturation at the front of the cell and to their disassembly at the rear [27, 28] and adhesion strength affects protrusion and migration [29]. Furthermore, these processes most accurately describe a style of locomotion employed by mesenchymal cells such as fibroblasts and by endothelial cells. Leukocytes more commonly use a migration mode typified by the amoeba, Dictyostelium discoides [22]. The differences in amoeboid and mesenchymal migration are most clearly seen in 3D matrix environments where the interstitial matrix is preserved rather than digested and migrating cells do not appear to adhere to the matrix proteins in amoeboid migration [21, 22]. Indeed, recent work indicates that integrins are not required for interstitial migration of dendritic cells in the dermis or lymph nodes but are indispensable for transendothelial migration [30, 31]. Consistent with their intermediate migration speed, macrophages appear capable of both amoeboid and mesenchymal interstitial migration, depending on the structure and density of the surrounding matrix, as they can either propel themselves through loose connective tissue or actively digest a path through denser interstitial matrix [32, 33]. Moreover, matrix remodeling by tumor-associated macrophages promotes breast cancer cell invasion, indicating that macrophages normally digest extracellular matrix during interstitial migration [34].
Examination in vitro of the actin cytoskeleton and adhesion structures in macrophages and fibroblasts indicates important mechanistic differences in the motility machinery between the two cell types (Figure 1). As the leading edge of a fibroblast extends, it forms small nascent adhesions (Figures 1(a) and 1(c), yellow) that either quickly disappear or, if they are connected to actin microfilaments, cluster into small focal complexes just behind the leading edge (Figures 1(a), arrow; and 1(c), green). Then, as the fibroblast continues to move forward, the focal complexes in their turn either disappear or mature and coalesce into larger focal contacts (1–5 μM) that anchor thick actin bundles or stress fibers (Figures 1(a), arrowhead; 1(c), red) [25, 35–37]. Indeed, the thickness of the bundled actin appears to control the size and shape of the underlying adhesion [36, 37]. In contrast to fibroblasts, macrophages form innumerable dot-like point contacts of varying phosphopaxillin content (Figures 1(b) and 1(d), yellow and red) under the ventral surface, most strikingly in the leading lamellipodium, along with scattered, mostly peripherally located focal complexes (Figure 1(d), green). Point contacts are also found in neuronal growth cones and highly motile cells [38] and resemble the widely distributed nascent adhesions of spreading fibroblasts after replating [39]. Macrophage adhesions do not mature into large focal contacts with attached stress fibers, although some focal complexes do anchor thin actin bundles (Figure 1(b), arrow) [16, 40]. Inverted phosphopaxillin immunofluorescent images clearly demonstrate the strikingly different pattern of adhesion at the leading edges of fibroblasts (Figure 1(c)) and macrophages (Figure 1(d)).
Figure 1.
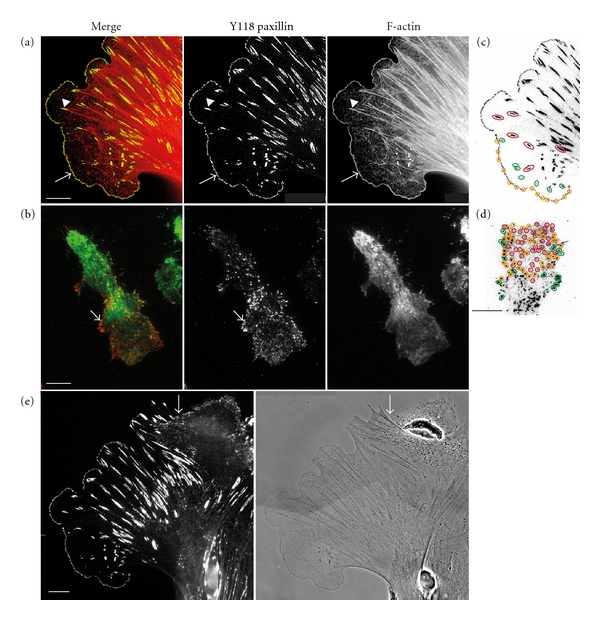
Macrophages are built for rapidly responsive migration. (a) A primary fibroblast, grown on a fibronectin coated coverslip in α+MEM, 10% FCS, and 120 ng/ml recombinant CSF-1, was fixed and stained for phosphoY118 paxillin (green) and F-actin (red). Phospho-Y118 paxillin staining eliminates background cytoplasmic staining of unphosphorylated paxillin [1, 16]. The arrow indicates focal complexes at the leading edge and the arrowhead indicates a focal adhesion giving rise to a stress fiber. (b) MacCsf1r−/−. WT macrophage, grown on a fibronectin coated coverslip in α+MEM, 10% FCS, and 120 ng/ml recombinant CSF-1, was fixed and stained for phosphoY118 paxillin (red) and F-actin (green) and examined by TIRF microscopy. The arrow indicates a focal complex giving rise to an F-actin cable. (c) Inverted image of phosphoY118 paxillin IF staining at the leading edge of the primary fibroblast, yellow circles indicate several nascent adhesions, green ovals highlight focal complexes and red ovals outline some focal adhesions. (d) Inverted image of phospho-Y118 paxillin IF staining in the leading lamellipodium of the macrophage, yellow circles indicate point contacts with strong phosphopaxillin staining, red circles indicate point contacts with moderate phosphopaxillin staining and green ovals outline the linear focal complexes. (e) A larger view of the fibroblast stained for pY118 paxillin (left) and shown by phase contrast (right) to demonstrate the co-cultured primary macrophage migrating underneath the fibroblast (arrow) and disrupting its focal adhesions. Note the lack of macrophage focal adhesions. Scale bars = 10 μM.
3. Macrophage Adhesions
Adhesions are multiprotein complexes that not only structurally link the cell adhesion receptors, integrins, to the actin cytoskeleton but also integrate and regulate a range of signals important for cell motility and growth [41]. The molecular associations and movement of individual components in the complexes are highly dynamic, allowing rapid responses to environmental and cellular cues [28, 42]. Tyrosine phosphorylation is an important regulatory mechanism for dynamic interplay of these components [43–45]. A number of tyrosine kinases localize to adhesions, including Src family kinases (SFK) and the adhesion kinases, focal adhesion kinase (FAK) and Pyk2, where they phosphorylate many adhesion proteins upon integrin engagement [41, 46]. Prominent among the phosphorylated adhesion proteins are the kinases themselves and paxillin, a highly phosphorylated multidomain scaffold protein that integrates and coordinates the regulation of adhesion signaling molecules, many of which control actin polymerization and actomyosin contractility [47]. Indeed, fluorescently tagged paxillin has been widely used to examine adhesion formation in living and fixed cells. Due to their small size and extensive ventral surface distribution, macrophage adhesion structures are difficult to visualize, as cytoplasmic paxillin almost completely obscures them. However, paxillin translocates to the plasma membrane and to adhesions when phosphorylated on Y31/Y118, so the use of phosphospecific paxillin antibodies and total internal reflection fluorescence microscopy (TIRF) greatly enhances our capacity to examine macrophage adhesions (Figure 1(b)) [16, 25, 48]. Despite their lack of large, longer lived adhesions, macrophages can be successfully cultured on glass and bacterial plastic. Moreover, the combined adhesive capacity of their collected point contacts and focal complexes enables macrophages to push underneath cocultured fibroblasts, disrupting the fibroblast's focal contacts in the process (Figure 1(e), arrow). Consistent with this observation, adhesion strength is positively correlated with the adhesive area of a cell and leading edge focal complexes have been shown to support a stronger traction force than mature focal contacts [35, 49]. Thus, it appears that individually weak adhesions in macrophages collectively give rise to robust but dynamic adhesion and, in combination with a readily remodeled actin cytoskeleton, permit rapidly responsive migration in macrophages.
In addition to point contacts and focal complexes, macrophages also form podosomes, which are short-lived adhesion/motility organelles that consist of a dense core of actin surrounded by a collar of adhesion proteins (Figure 2(a)) [50, 51]. Cytoskeletal transmission EM studies indicate that podosomes have a distinctive hub and spoke microfilament architecture (Figure 2(b), arrow) [52]. In contrast to focal complexes, they are able to digest extracellular matrix and so are thought to be important for interstitial migration of macrophages and other myeloid cells [51, 53, 54]. It is not clear how podosomes contribute to motility but individual, short-lived podosomes often coalesce into higher order, more stable structures such as rosettes that efficiently digest the underlying matrix (Figure 2(c)) [55, 56]. Osteoclasts form large podosomal arrays or belts within which separate podosomes may become indistinct and which create the sealing zone necessary for effective bone resorption within the perimeter of this gasket [52]. Podosomes may also be important for leukocyte diapedesis, either between or through endothelial cells [57, 58]. Importantly, matrix digesting actin-rich rosette-like structures have been imaged in human primary macrophages within a 3D gelled collagen matrix, strongly suggesting podosomes play a role in macrophage migration in vivo [33].
Figure 2.
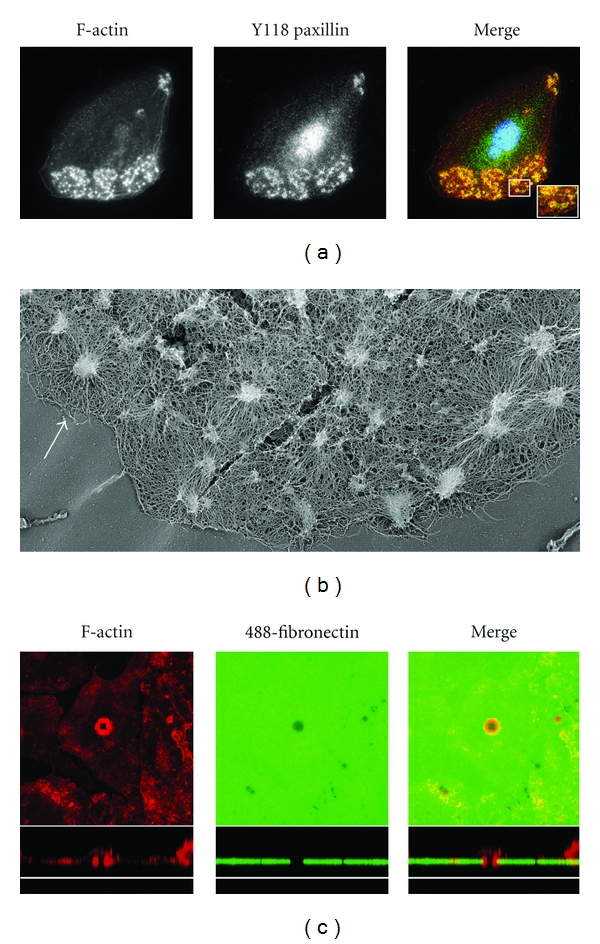
Macrophages form podosomes. (a) A human monocyte-derived macrophage, grown on a fibronectin coated coverslip in RPMI, 10% FCS, and 120 ng/ml of recombinant CSF-1, was fixed and stained for F-actin (red) and pY118 paxillin (green). Inset in the merge panel demonstrates the central F-actin-rich stud and the surrounding pY118 paxillin collar. (b) Cytoskeletal TEM preparation of a human monocyte-derived macrophage, grown on a glass coverslip, demonstrating many podosomes at the presumed leading edge. The arrow highlights a good example of a podosome containing a central dense actin column surrounded by radially orientated actin filament spokes. (c) Mouse bone marrow-derived macrophages were plated on Alexa-488 labeled fibronectin overlaying gelatin for 24 hours prior to fixation, staining for F-actin (red) and examination by confocal microscopy to demonstrate a podosomal rosette digesting the underlying matrix.
Compared to actin-rich rosettes, imaging adhesion structures in living cells in 3D culture is difficult, leading to some doubt that they exist in vivo. However, by lowering the expression levels of genetically encoded fluorescently tagged adhesion proteins to reduce background cytoplasmic fluorescence, dynamic paxillin-rich 1 μM cell-matrix adhesions were observed in the protrusions of U2OS osteosarcoma cells grown in 3D collagen gels [59]. These adhesions were demonstrated to form in contact with collagen fibers, suggesting adhesions are likely to be found in vivo in mesenchymally migrating cells. However, in vivo detection of adhesion structures will be extremely difficult, particularly in macrophages, which form such small adhesions in 2D culture systems.
4. CSF-1 Regulation of Macrophage Motility
Although CSF-1 was initially identified as a macrophage growth and differentiation factor [5], it was subsequently demonstrated to stimulate monocyte migration [14] and later studies confirmed that CSF-1 is a potent chemokinetic and chemotactic factor for macrophages [15]. Indeed, the pathophysiological importance of CSF-1-stimulated macrophage migration has recently been demonstrated in several diseases, including tumour invasion and metastasis [20, 60], inflammatory arthritis [61–63] and atherosclerosis [64, 65]. Tumor-associated macrophages secrete epidermal growth factor (EGF) and carcinoma cells secrete CSF-1 to set up a paracrine chemotactic loop that induces comigration of both cell types and promotion of invasion and metastasis [20, 59]. Inhibition of either EGF receptor or CSF-1R signaling prevents tumour cell motility in vivo [20]. Synovial macrophages have long been known to play a critical role in chronic rheumatoid arthritis and conventional therapies all reduce macrophage numbers in the synovium [66]. CSF-1, which is secreted by synoviocytes and endothelial cells, attracts monocytes to arthritic joints and stimulates their differentiation into inflammatory cytokine secreting macrophages and bone resorbing osteoclasts [66]. Importantly, selective CSF-1R inhibition significantly reduced joint infiltration and differentiation of macrophages in several autoimmune arthritis models with subsequent improvement in arthritis severity [63]. Downstream of the CSF-1R, a mutation causing reduced expression of PSTPIP2, a signaling protein that is selectively expressed in macrophages, results in an autoinflammatory disease [67, 68]. PSTPIP2 is tyrosine phoshorylated in response to CSF-1 and regulates ruffling, filopodia formation, and CSF-1-induced motility [67]. Thus CSF-1 regulation of macrophage migration is important in the development and progression of several diseases and elucidation of CSF-1-stimulated motility pathways is likely to identify possible therapeutic targets to modulate macrophage infiltrative capacity.
5. CSF-1R Signaling to Macrophage Motility
CSF-1 initially triggers membrane ruffling and spreading followed by increased formation of phosphotyrosine-rich adhesions and finally the macrophages polarize and begin to move [16, 40, 69]. CSF-1-stimulated actin polymerization is very rapid, with a sharp peak at 30 sec followed by a longer lasting wave at 3 min [69]. Polymerization is regulated by Rho family GTPases, Rac, Rho, and Cdc42, whose effectors include the Wiskott Aldrich syndrome protein (WASP)/WASP-family verprolin homologous (WAVE) family of actin nucleators [70, 71]. Increased focal complex and point contact formation is visible by 5 minutes but does not peak until 15 min after CSF-1 stimulation, coincident with maximal phosphorylation of paxillin by its adhesion kinases, Pyk2 and FAK [40]. Consistent with the importance of actin polymerization and adhesion formation in macrophage migration, macrophages deficient in Pyk2, FAK, WASP, or WAVE2 are poorly motile [48, 72–74]. However, the mechanisms by which CSF-1 stimulates actin polymerization and adhesion formation are not well understood and require careful dissection of the signaling pathways triggered by CSF-1R activation.
The effects of CSF-1 are mediated by the CSF-1R, a RTK of the platelet derived growth factor receptor (PDGFR) family. Upon binding of homodimeric CSF-1, the CSF-1R dimerizes, becomes activated and autophosphorylates at least 7 of its 20 intracellular tyrosine residues [1]. Phosphorylation of these tyrosine residues creates specific binding sites for phosphotyrosine (pTyr) binding domain-containing molecules and initiates a series of signaling cascades, leading to rapid stimulation of cytoskeletal remodeling and adhesion as well as gene transcription and protein translation [1, 75]. To identify pTyr CSF-1R-associated molecules and examine the specific pathways that mediate the various effects of CSF-1, earlier studies either ectopically expressed wild-type or tyrosine-to-phenylalanine (Y→F) mutant CSF-1Rs in fibroblasts or expressed chimeric receptors composed of a non-CSF-1R extracellular domain and Y→F mutated CSF-1R intracellular domains in myeloid cells. Results differed between fibroblast and myeloid cell studies, in part because mature macrophages selectively express specific proteins, isoforms or splice variants important for CSF-1R signaling [1, 67, 76]. To overcome these problems, we developed a system to express a single species of CSF-1R in a mature macrophage context. Immortalised macrophages derived from the CSF-1R−/− mouse were transduced with either a wild-type or a tyrosine mutant CSF-1R [11, 77]. The Y-Eight-F (YEF) mutant CSF-1R, with eight tyrosine residues mutated to phenylalanine, is not phosphorylated in response to CSF-1 and macrophages expressing this receptor cannot survive in CSF-1 [77]. The system was used to examine loss-of-function effects of a panel of individual Y→F CSF-1R molecules and we have shown that phosphorylation of Y706 and Y721 in the kinase insert and Y974 at the C-terminus of the CSF-1R are important for normal macrophage morphology while juxta-membrane Y559 and activation loop Y807 are critical for macrophage proliferation and differentiation [77]. The YEF CSF-1R can be used as a backbone on which to add-back individual tyrosine residues. Add-back to the YEF CSF-1R of two known phosphotyrosine residues, Y559 and Y807, and a third, Y544, that may not be phosphorylated but is thought to be important for CSF-1R conformation, restores full proliferation in response to CSF-1 (unpublished results). Add-back of single tyrosine residues to the YEF receptor has been used to demonstrate that Y559 is the first residue phosphorylated in response to CSF-1 [78] and that it is necessary and sufficient for c-Cbl-mediated receptor ubiquitylation, full activation, and subsequent degradation of the receptor [78]. Individual tyrosine residues can also be added back to minimal proliferation competent add-back (AB) receptor, YEF. Y544, Y509, Y807 AB CSF-1R, to examine return-of-function signaling for the remaining pTyr residues.
Macrophages elongate when they polarize and begin to move [15, 79] so a loss of elongation can indicate reduced motility [40, 80]. Cells expressing the Y721F mutant receptor were apolar and previous studies in other cell lines had demonstrated a pY721-dependent association with the CSF-1R of two proteins known to signal to cell motility, phosphoinositide 3-kinase (PI3K) and phospholipase C (PLC)γ2 [81–83]. A detailed examination of the Y721F CSF-1R macrophages revealed a significant reduction in motility in vitro and, perhaps more importantly, these macrophages moved less well in vivo and there was a significant reduction in their capacity to enhance tumour cell invasion in vitro [69]. Underlying the reduction in motility was a loss of the first peak of CSF-1-stimulated actin polymerization and reduced paxillin phosphorylation and incorporation into adhesions. Add-back of Y721 to the YEF. Y544, Y559, Y807 AB CSF-1R restored actin polymerization and cell motility, indicating that pY721-based signaling regulates CSF-1-induced macrophage motility [69]. This system was also used to identify which of the two possible effectors, PI3K or PLCγ2, was responsible for initiating pY721-based motility signaling. While PLCγ2 associated with the activated CSF-1R in a pY721-independent manner, CSF-1 rapidly stimulated a prolonged Y721-dependent association of PI3K with the receptor, which resulted in PIP3 production [69]. Thus the primary mediator of CSF-1-stimulated motility in macrophages is PI3K.
Class IA PI3Ks consist of a p110 catalytic subunit bound to a p85 regulatory subunit that translocates to activated RTKs upon interaction of its SH2 domains with pYXXM motifs, including Y721VEM in the CSF-1R [69, 84]. Upon binding, p85 activates p110 to produce phosphatidylinositol 3, 4, 5-trisphosphate (PIP3) from PI 4, 5-bisphosphate (PIP2) at the cell membrane [85]. An accumulation of PIP3 at the leading edge stimulates migration by inducing plasma membrane translocation of pleckstrin homology (PH) domain-containing molecules, PDK1, Akt, and Rho family GTPase regulators, or molecules with other PIP3 binding motifs such as WASP and its homologues [86, 87]. RTK-induced PIP3 levels are rapidly returned to baseline levels by the phospholipid phosphatase, PTEN [84]. While p110α and β are ubiquitously expressed, p110δ expression is highly enriched in hematopoietic cells, including macrophages. The three PI3K p110 isoforms have nonredundant biological roles and their function differs between primary and immortalised macrophages such that while p110δ is the main isoform recruited to the CSF-1R in bone marrow-derived macrophages, all three are recruited to the receptor in BAC1.2F5 macrophages [84]. However, PI3K p110δ appears to be the main regulator of migration in both primary macrophages and BAC1.2F5 cells, in which it triggers actin polymerization, cytoskeletal remodeling, and cell adhesion [84]. The exact pathways by which induction of PIP3 mediates these disparate effects of CSF-1 stimulation have yet to be identified, but regulation of individual elements further downstream in macrophage migration are becoming clearer and appear to converge on the Rho family GTPases.
6. Rho Family GTPases in Macrophage Motility
Rho family GTPases are well-known regulators of actin polymerization and cell adhesion downstream of RTKs in many different cell types [88]. The main Rho family proteins found in macrophages are RhoA, RhoB, Rac1, Rac2, and Cdc42 [70]. Rho GTPases are activated by guanine-nucleotide exchange factors (GEFs), which stimulate the exchange of GDP for GTP, and inactivated by GTPase activating proteins (GAPs), which stimulate GTP hydrolysis (Figure 3). Upon activation, Rho family GTPases interact with effector proteins, including actin polymerization activators and protein kinases. Selective expression of GEFs, GAPs, and effector proteins plus spatiotemporal regulation of activation and regulatory crosstalk between Rho family proteins results in highly complex and dynamic coordination of cytoskeletal remodeling in response to RTK stimulation [88, 89]. Fluorescence resonance energy transfer (FRET) biosensors have been used to demonstrate in real time that the three ubiquitously expressed Rho family proteins, RhoA, Rac1, and Cdc42, are all activated at the leading edge of cells with very small differences in time and space [90, 91]. Rho family biosensors have yet to be used in CSF-1-dependent macrophage cell lines, which are difficult to transfect, but recent use of a WASP biosensor in RAW264.7/L5 macrophages demonstrated Cdc42-dependent activation of WASP in CSF-1-induced protrusions [92]. Early research into the role of Rho family proteins in macrophages used microinjection of constitutively active or dominant negative RhoA, Rac1, or Cdc42 to show that Cdc42 promoted filopodia formation while Rac stimulated ruffling and lamellipodial spreading and Rho triggered actomyosin contractility and retraction of the trailing edge in response to CSF-1 [93]. In addition, Rac and Cdc42 stimulated focal complex formation [94]. A subsequent study indicated that Rac and Rho were important for macrophage migration while Cdc42 regulated polarization and chemotactic sensing [94]. However, dominant negative proteins, particularly when overexpressed, may not be specific for their GEFs, and conditional knock-out and knock-down approaches have been used more recently to examine the role of Rho family proteins in macrophage actin remodeling and motility [70]. Although little is known about the loss of Cdc42 function, the effects of deletion of Rac1, which is ubiquitously expressed, and the hematopoietically restricted Rac2 have been reported [95–97]. Surprisingly, loss of both Rac1 and Rac2 did not decrease CSF-1-induced 2D motility, although Rac1/2−/− macrophages did not form ruffles, normal lamellipodia, or podosomes [97]. Moreover, loss of Rac1 reduced invasive capacity of macrophages in Matrigel while loss of Rac2 reduced peritoneal macrophage infiltration in response to an inflammatory stimulus, suggesting both Rac proteins may be important for macrophage interstitial migration in vivo [96, 97]. In contrast to the unexpectedly mild loss-of-function phenotype of Rac1/2, C3 transferase-induced inhibition of RhoA-C inhibited CSF-1-stimulated macrophage migration and actomyosin contractility [40, 94]. Interestingly, global activation of Rac, Rho, or Cdc42 is not detected in CSF-1 stimulated macrophages at the time of the first wave of actin polymerization, suggesting local changes may be subtle [69]. Thus, the complexities and redundancies of Rho family GTPase signaling makes it difficult to tease apart the finer aspects of their role in CSF-1-induced motility. Production and examination of Cdc42−/− and RhoA−/− macrophages as well as FRET studies should prove illuminating. Nevertheless, further mechanistic insights into Rho family GTPase regulation of macrophage motility have been gained through examination of their main downstream effectors for actin polymerization, the WASP and WAVE complexes [71, 98].
Figure 3.
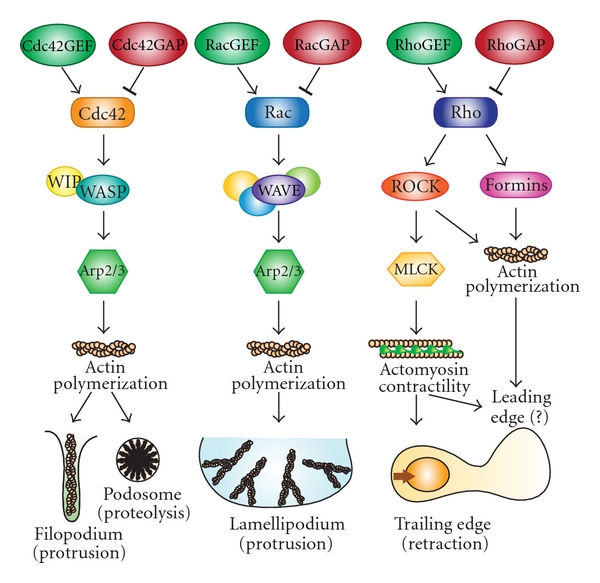
A schematic of signaling pathways activated by the Rho GTPases in macrophages. Generic GEFs and GAPs are designated as upstream regulators of Cdc42, Rac, and Rho activity. Downstream signaling molecules specifically labeled in the schematic are those that are described in the text. Signaling pathway outcomes are depicted for actin cytoskeleton responses only. Regulation of adhesion by Rho GTPases in macrophages is not shown as the signaling pathways are not yet elucidated.
In macrophages, Cdc42 and Rac stimulate actin polymerization by activating the Arp2/3 complex through their GTP-dependent association with the Arp2/3 activating scaffold proteins, WASP and WAVE, respectively [98, 99]. WASP was originally discovered as the hematopoietically expressed product of the gene mutated in the X-linked immunological disorder Wiskott Aldrich Syndrome (WAS) and is an important downstream effector of Cdc42 [92, 98, 99]. WAS myeloid cells display marked cytoskeletal abnormalities and cannot form podosomes, and WAS macrophages fail to chemotax towards CSF-1 [53, 73, 99]. WASP does not act in isolation and requires its N-terminal binding protein, WASP interacting protein (WIP), to form a functional unit that activates Arp2/3 in podosome formation and transendothelial migration in macrophages [99, 100]. Consistent with the requirement for both proteins to activate Arp2/3, WIP−/− dendritic cells also fail to form podosomes, instead forming longer lived focal contacts [101]. The WAVE family proteins, WAVE1, 2, and 3, each stably complex with several other proteins in order to mediate the effects of Rac [71, 98]. WAVE2, which is the major WAVE isoform expressed in macrophages, is important for CSF-1 stimulation of ruffling and migration [74]. Thus, both WASP and WAVE2 activate the Arp2/3 complex to stimulate dendritic or branched actin polymerization, but WASP mediates regulation of chemotaxis by Cdc42, while WAVE2 mediates the regulation of ruffling and motility by Rac. Rather than activating Arp2/3, Rho promotes actomyosin contractility through activation of Rho-kinase(ROCK)1 and ROCK2. Unexpectedly however, ROCK1−/− macrophages were more motile towards CSF-1 in vitro and responded to an inflammatory stimulus more readily in vivo [102]. Underlying their increased migration, ROCK1−/− macrophages demonstrated increased adhesion on fibronectin and increased CSF-1-stimulated F-actin levels in association with increased PIP3 levels. ROCK1 was shown to negatively regulate CSF-1-induced migration through regulation of PTEN activity [102]. Rho also mediates its effects through activation of the formin family of actin nucleators, which assemble linear rather than branched actin filaments, but their role in macrophage motility is currently unknown [103].
The specific roles of individual Rho family proteins in the regulation of macrophage adhesion is less well understood than their roles in actin polymerization and actomyosin contractility and is made more complex by the fact that there is crosstalk between actin polymerization, actomyosin contractility, and adhesion formation and turnover [37, 104]. Nevertheless, CSF-1 stimulates the incorporation of the adhesion kinases, FAK and Pyk2, and their substrate, paxillin, into focal complexes and point contacts a few minutes after it stimulates actin polymerization, ruffling, and spreading in macrophages [40, 48, 69]. FAK is known to regulate adhesion formation and disassembly and both FAK and Pyk2 regulate macrophage migration in vitro and in vivo, apparently via the same pathway, as loss of Pyk2 does not further reduce migration and invasion in FAK−/− macrophages [48, 72]. The precise mechanism by which CSF-1R signaling activates Pyk2 and FAK to regulate adhesion formation and turnover is not understood but CSF-1-stimulated FAK−/− macrophages demonstrated high levels of Rac activity in association with hyperprotrusiveness [48] while Pyk2−/− macrophages showed reduced integrin-mediated Rho activation [72]. These suggestions that adhesion signaling feeds back on actin polymerization and actomyosin contractility are not unexpected as many Rho family GEFs and GAPs are recruited to adhesions where they can activate or inhibit Rho family proteins [41]. Moreover, activated and autophosphorylated FAK and Pyk2 associate with SFKs in adhesions, facilitating SFK-based phosphorylation and regulation of nearby Rho family GEFs and GAPs as well as other adhesion proteins [104]. Phosphorylation of paxillin triggers its translocation to adhesions and brings along associated Rac effectors that are critical for leading edge formation and adhesion turnover [43, 47]. Thus, adhesions themselves are important platforms for the regulation of Rho family proteins [37, 104].
An area where the role of individual Rho GTPases has been more clearly defined in macrophages is phagocytosis, which is a highly ordered process of membrane protrusion and actin polymerization that uses many of the same elements of cellular machinery as locomotion [105]. FRET studies of Fcγ receptor-mediated phagocytosis in RAW264.7 macrophages reveal distinct spatiotemporally regulated patterns of Rac1, Rac2, and Cdc42 activation underlying actin polymerization in the phagocytic cup [106]. In addition, RhoG, which is more closely related to Rac than to Rho [107], is recruited to phagocytic cups in J774 macrophages [108]. Rho GTPases activate many of the same downstream effectors in phagocytosis as they do in motility, with both WASP and Arp2/3 being required for normal Fcγ receptor-mediated phagocytosis in macrophages [105]. Further refinements in the application of FRET to CSF-1-dependent mature macrophages will reveal the specific spatiotemporally regulated roles of individual Rho GTPases in adhesion, motility, and phagocytosis.
7. Concluding Remarks
It is clear that the interplay between the different elements of the adhesion and motility apparatus, coordinated in large part by Rho family GTPases, is complex and will require the use of many different approaches to unpack these complexities. Mature macrophages have proven difficult to adapt to some approaches as, not only do they selectively express a number of important adhesion and motility proteins but they are difficult to transfect [1]. Nevertheless, motile macrophages contribute to the progression of a number of important diseases and elucidation of how CSF-1 regulates polarization, protrusion, adhesion, actomyosin contractility, and trailing edge retraction to stimulate migration is important in the development of therapies to treat these diseases. The CSF-1R-deficient mouse macrophage cell line (MacCsf1r−/−), when transduced with individual wild-type or tyrosine mutant CSF-1Rs, allows examination of specific signaling pathways triggered by individual tyrosine residues in mature macrophages [77]. Using this system, Y721 was recently identified as the major CSF-1R phosphotyrosine residue triggering ruffling, adhesion, and motility in response to CSF-1 [69]. Furthermore, the primary mediator of pY721-based signaling to motility was demonstrated to be PI3K [69], and work is now focused in identifying the specific PI3K p110 isoform and PI3K-activated pathways that regulate actin polymerization, adhesion formation, and migration in macrophages. CSF-1R and isoform specific PI3K inhibitors are available and may prove useful in the treatment of disseminated tumors and chronic inflammatory arthritides.
Acknowledgments
This paper was supported by a National Health and Medical Research Grant 513817. The author thanks Michael Cammer, Frank Macaluso, and Paul Rigby for their technical assistance with the microscopy and Natalia Sampaio for her critical reading of the paper.
Abbreviations
- CSF-1:
Colony-stimulating factor-1
- CSF-1R:
Colony-stimulating factor-1 receptor
- EGF:
Epidermal growth factor
- EM:
Electron microscopy
- FAK:
Focal adhesion kinase
- FRET:
Fluorescence resonance energy transfer
- GEF:
Guanine-nucleotide exchange factor
- GAP:
GTPase activating protein
- IL-34:
interleukin-34
- MLCK:
Myosin light chain kinase
- op:
Osteopetrotic
- PDGFR:
Platelet derived growth factor receptor
- PH:
Pleckstrin homology
- PI3K:
Phosphoinositol 3′-kinase
- PIP3:
Phosphatidylinositol 3, 4, 5, trisphosphate
- PLCγ:
Phospholipase Cγ
- pTyr:
Phosphotyrosine
- ROCK:
Rho kinase
- RTK:
Receptor tyrosine kinase
- SFK:
Src family kinase
- TIRF:
Total internal reflection fluorescence microscopy
- WASP:
Wiskott Aldrich Syndrome protein
- WAVE:
WASP-family verprolin homologous
- WIP:
WASP-interacting protein.
References
- 1.Pixley FJ, Stanley ER. CSF-1 regulation of the wandering macrophage: complexity in action. Trends in Cell Biology. 2004;14(11):628–638. doi: 10.1016/j.tcb.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 2.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nature Reviews Immunology. 2005;5(12):953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 3.Pollard JW. Trophic macrophages in development and disease. Nature Reviews Immunology. 2009;9(4):259–270. doi: 10.1038/nri2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Furth R, Cohn ZA, Hirsch JG, Humphrey JH, Spector WG, Langevoort HL. The mononuclear phagocyte system: a new classification of macrophages, monocytes, and their precursor cells. Bulletin of the World Health Organization. 1972;46(6):845–852. [PMC free article] [PubMed] [Google Scholar]
- 5.Stanley ER, Heard PM. Factors regulating macrophage production and growth. Purification and some properties of the colony stimulating factor from medium conditioned by mouse L cells. Journal of Biological Chemistry. 1977;252(12):4305–4312. [PubMed] [Google Scholar]
- 6.Asagiri M, Takayanagi H. The molecular understanding of osteoclast differentiation. Bone. 2007;40(2):251–264. doi: 10.1016/j.bone.2006.09.023. [DOI] [PubMed] [Google Scholar]
- 7.Yoshida H, Hayashi SI, Kunisada T, et al. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature. 1990;345(6274):442–444. doi: 10.1038/345442a0. [DOI] [PubMed] [Google Scholar]
- 8.Wiktor-Jedrzejczak W, Bartocci A, Ferrante AW, Jr., et al. Total absence of colony-stimulating factor 1 in the macrophage-deficient osteopetrotic (op/op) mouse. Proceedings of the National Academy of Sciences of the United States of America. 1990;87(12):4828–4832. doi: 10.1073/pnas.87.12.4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cecchini MG, Dominguez MG, Mocci S, et al. Role of colony stimulating factor-1 in the establishment and regulation of tissue macrophages during postnatal development of the mouse. Development. 1994;120(6):1357–1372. doi: 10.1242/dev.120.6.1357. [DOI] [PubMed] [Google Scholar]
- 10.Sherr CJ, Rettenmier CW, Sacca R, Roussel MF, Look AT, Stanley ER. The c-fms proto-oncogene product is related to the receptor for the mononuclear phagocyte growth factor, CSF-1. Cell. 1985;41(3):665–676. doi: 10.1016/s0092-8674(85)80047-7. [DOI] [PubMed] [Google Scholar]
- 11.Dai XM, Ryan GR, Hapel AJ, et al. Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood. 2002;99(1):111–120. doi: 10.1182/blood.v99.1.111. [DOI] [PubMed] [Google Scholar]
- 12.Lin H, Lee E, Hestir K, et al. Discovery of a cytokine and its receptor by functional screening of the extracellular proteome. Science. 2008;320(5877):807–811. doi: 10.1126/science.1154370. [DOI] [PubMed] [Google Scholar]
- 13.Wei S, Nandi S, Chitu V, et al. Functional overlap but differential expression of CSF-1 and IL-34 in their CSF-1 receptor-mediated regulation of myeloid cells. Journal of Leukocyte Biology. 2010;88(3):495–505. doi: 10.1189/jlb.1209822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang JM, Griffin JD, Rambaldi A, Chen ZG, Mantovani A. Induction of monocyte migration by recombinant macrophage colony-stimulating factor. Journal of Immunology. 1988;141(2):575–579. [PubMed] [Google Scholar]
- 15.Webb SE, Pollard JW, Jones GE. Direct observation and quantification of macrophage chemoattraction to the growth factor CSF-1. Journal of Cell Science. 1996;110:707–720. doi: 10.1242/jcs.109.4.793. [DOI] [PubMed] [Google Scholar]
- 16.Pixley FJ, Lee PSW, Condeelis JS, Stanley ER. Protein tyrosine phosphatase φ regulates paxillin tyrosine phosphorylation and mediates colony-stimulating factor 1-induced morphological changes in macrophages. Molecular and Cellular Biology. 2001;21(5):1795–1809. doi: 10.1128/MCB.21.5.1795-1809.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ryan GR, Dai XM, Dominguez MG, et al. Rescue of the colony-stimulating factor 1 (CSF-1)-nullizygous mouse (Csf1op/Csf1op) phenotype with a CSF-1 transgene and identification of sites of local CSF-1 synthesis. Blood. 2001;98(1):74–84. doi: 10.1182/blood.v98.1.74. [DOI] [PubMed] [Google Scholar]
- 18.Chitu V, Stanley ER. Colony-stimulating factor-1 in immunity and inflammation. Current Opinion in Immunology. 2006;18(1):39–48. doi: 10.1016/j.coi.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 19.Hamilton JA. Colony-stimulating factors in inflammation and autoimmunity. Nature Reviews Immunology. 2008;8(7):533–544. doi: 10.1038/nri2356. [DOI] [PubMed] [Google Scholar]
- 20.Wyckoff J, Wang W, Lin EY, et al. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Research. 2004;64(19):7022–7029. doi: 10.1158/0008-5472.CAN-04-1449. [DOI] [PubMed] [Google Scholar]
- 21.Friedl P, Zänker KS, Bröcker EB. Cell migration strategies in 3-D extracellular matrix: differences in morphology, cell matrix interactions, and integrin function. Microscopy Research and Technique. 1998;43(5):369–378. doi: 10.1002/(SICI)1097-0029(19981201)43:5<369::AID-JEMT3>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 22.Friedl P, Weigelin B. Interstitial leukocyte migration and immune function. Nature Immunology. 2008;9(9):960–969. doi: 10.1038/ni.f.212. [DOI] [PubMed] [Google Scholar]
- 23.Grabher C, Cliffe A, Miura K, et al. Birth and life of tissue macrophages and their migration in embryogenesis and inflammation in medaka. Journal of Leukocyte Biology. 2007;81(1):263–271. doi: 10.1189/jlb.0806526. [DOI] [PubMed] [Google Scholar]
- 24.Rafelski SM, Theriot JA. Crawling toward a unified model of cell motility: spatial and temporal regulation of actin dynamics. Annual Review of Biochemistry. 2004;73:209–239. doi: 10.1146/annurev.biochem.73.011303.073844. [DOI] [PubMed] [Google Scholar]
- 25.Parsons JT, Horwitz AR, Schwartz MA. Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nature Reviews Molecular Cell Biology. 2010;11(9):633–643. doi: 10.1038/nrm2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lauffenburger DA, Horwitz AF. Cell migration: a physically integrated molecular process. Cell. 1996;84(3):359–369. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- 27.Giannone G, Dubin-Thaler BJ, Rossier O, et al. Lamellipodial actin mechanically links myosin activity with adhesion-site formation. Cell. 2007;128(3):561–575. doi: 10.1016/j.cell.2006.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wolfenson H, Bershadsky A, Henis YI, Geiger B. Actomyosin-generated tension controls the molecular kinetics of focal adhesions. Journal of Cell Science. 2011;124(9):1425–1432. doi: 10.1242/jcs.077388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barnhart EL, Lee KC, Keren K, Mogilner A, Theriot JA. An adhesion-dependent switch between mechanisms that determine motile cell shape. PLoS Biology. 2011;9(5) doi: 10.1371/journal.pbio.1001059. Article ID e1001059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lämmermann T, Bader BL, Monkley SJ, et al. Rapid leukocyte migration by integrin-independent flowing and squeezing. Nature. 2008;453(7191):51–55. doi: 10.1038/nature06887. [DOI] [PubMed] [Google Scholar]
- 31.Lämmermann T, Sixt M. Mechanical modes of “amoeboid” cell migration. Current Opinion in Cell Biology. 2009;21(5):636–644. doi: 10.1016/j.ceb.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 32.van Goethem E, Poincloux R, Gauffre F, Maridonneau-Parini I, le Cabec V. Matrix architecture dictates three-dimensional migration modes of human macrophages: differential involvement of proteases and podosome-like structures. Journal of Immunology. 2010;184(2):1049–1061. doi: 10.4049/jimmunol.0902223. [DOI] [PubMed] [Google Scholar]
- 33.van Goethem E, Guiet R, Balor S, et al. Macrophage podosomes go 3D. European Journal of Cell Biology. 2011;90(2-3):224–236. doi: 10.1016/j.ejcb.2010.07.011. [DOI] [PubMed] [Google Scholar]
- 34.Guiet R, van Goethem E, Cougoule C, et al. The process of macrophage migration promotes matrix metalloproteinase-independent invasion by tumor cells. Journal of Immunology. 2011;187(7):3806–3814. doi: 10.4049/jimmunol.1101245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beningo KA, Dembo M, Kaverina I, Small JV, Wang YL. Nascent focal adhesions are responsible for the generation of strong propulsive forces in migrating fibroblasts. Journal of Cell Biology. 2001;153(4):881–887. doi: 10.1083/jcb.153.4.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zamir E, Geiger B. Molecular complexity and dynamics of cell-matrix adhesions. Journal of Cell Science. 2001;114(20):3583–3590. doi: 10.1242/jcs.114.20.3583. [DOI] [PubMed] [Google Scholar]
- 37.Vicente-Manzanares M, Choi CK, Horwitz AR. Integrins in cell migration—the actin connection. Journal of Cell Science. 2009;122(2):199–206. doi: 10.1242/jcs.018564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Renaudin A, Lehmann M, Girault JA, McKerracher L. Organization of point contacts in neuronal growth cones. Journal of Neuroscience Research. 1999;55(4):458–471. doi: 10.1002/(SICI)1097-4547(19990215)55:4<458::AID-JNR6>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 39.Zimerman B, Volberg T, Geiger B. Early molecular events in the assembly of the focal adhesion-stress fiber complex during fibroblast spreading. Cell Motility and the Cytoskeleton. 2004;58(3):143–159. doi: 10.1002/cm.20005. [DOI] [PubMed] [Google Scholar]
- 40.Pixley FJ, Xiong Y, Yu RYL, Sahai EA, Stanley ER, Ye BH. BCL6 suppresses RhoA activity to alter macrophage morphology and motility. Journal of Cell Science. 2005;118(9):1873–1883. doi: 10.1242/jcs.02314. [DOI] [PubMed] [Google Scholar]
- 41.Zaidel-Bar R, Geiger B. The switchable integrin adhesome. Journal of Cell Science. 2010;123(9):1385–1388. doi: 10.1242/jcs.066183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zaidel-Bar R, Ballestrem C, Kam Z, Geiger B. Early molecular events in the assembly of matrix adhesions at the leading edge of migrating cells. Journal of Cell Science. 2003;116(22):4605–4613. doi: 10.1242/jcs.00792. [DOI] [PubMed] [Google Scholar]
- 43.Zaidel-Bar R, Milo R, Kam Z, Geiger B. A paxillin tyrosine phosphorylation switch regulates the assembly and form of cell-matrix adhesions. Journal of Cell Science. 2007;120(1):137–148. doi: 10.1242/jcs.03314. [DOI] [PubMed] [Google Scholar]
- 44.Kirchner J, Kam Z, Tzur G, Bershadsky AD, Geiger B. Live-cell monitoring of tyrosine phosphorylation in focal adhesions following microtubule disruption. Journal of Cell Science. 2003;116(6):975–986. doi: 10.1242/jcs.00284. [DOI] [PubMed] [Google Scholar]
- 45.Ballestrem C, Erez N, Kirchner J, Kam Z, Bershadsky A, Geiger B. Molecular mapping of tyrosine-phosphorylated proteins in focal adhesion using flourescence resonance energy transfer. Journal of Cell Science. 2006;119(5):866–875. doi: 10.1242/jcs.02794. [DOI] [PubMed] [Google Scholar]
- 46.Volberg T, Romer L, Zamir E, Geiger B. pp60c-src and related tyrosine kinases: a role in the assembly and reorganization of matrix adhesions. Journal of Cell Science. 2001;114(12):2279–2289. doi: 10.1242/jcs.114.12.2279. [DOI] [PubMed] [Google Scholar]
- 47.Deakin NO, Turner CE. Paxillin comes of age. Journal of Cell Science. 2008;121(15):2435–2444. doi: 10.1242/jcs.018044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Owen KA, Pixley FJ, Thomas KS, et al. Regulation of lamellipodial persistence, adhesion turnover, and motility in macrophages by focal adhesion kinase. Journal of Cell Biology. 2007;179(6):1275–1287. doi: 10.1083/jcb.200708093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gallant ND, Michael KE, García AJ. Cell adhesion strengthening: contributions of adhesive area, integrin binding, and focal adhesion assembly. Molecular Biology of the Cell. 2005;16(9):4329–4340. doi: 10.1091/mbc.E05-02-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marchisio PC, Cirillo D, Teti A, Zambonin-Zallone A, Tarone G. Rous Sarcoma virus-transformed fibroblasts and cells of monocytic origin display a peculiar dot-like organization of cytoskeletal proteins involved in microfilament-membrane interactions. Experimental Cell Research. 1987;169(1):202–214. doi: 10.1016/0014-4827(87)90238-2. [DOI] [PubMed] [Google Scholar]
- 51.Linder S, Aepfelbacher M. Podosomes: adhesion hot-spots of invasive cells. Trends in Cell Biology. 2003;13(7):376–385. doi: 10.1016/s0962-8924(03)00128-4. [DOI] [PubMed] [Google Scholar]
- 52.Luxenburg C, Geblinger D, Klein E, et al. The architecture of the adhesive apparatus of cultured osteoclasts: from podosome formation to sealing zone assembly. PLoS ONE. 2007;2(1, article e179) doi: 10.1371/journal.pone.0000179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Calle Y, Burns S, Thrasher AJ, Jones GE. The leukocyte podosome. European Journal of Cell Biology. 2006;85(3-4):151–157. doi: 10.1016/j.ejcb.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 54.Murphy DA, Courtneidge SA. The “ins” and “outs” of podosomes and invadopodia: characteristics, formation and function. Nature Reviews Molecular Cell Biology. 2011;12(7):413–426. doi: 10.1038/nrm3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yamaguchi H, Pixley F, Condeelis J. Invadopodia and podosomes in tumor invasion. European Journal of Cell Biology. 2006;85(3-4):213–218. doi: 10.1016/j.ejcb.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 56.Evans JG, Correia I, Krasavina O, Watson N, Matsudaira P. Macrophage podosomes assemble at the leading lamella by growth and fragmentation. Journal of Cell Biology. 2003;161(4):697–705. doi: 10.1083/jcb.200212037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Carman CV, Sage PT, Sciuto TE, et al. Transcellular diapedesis is initiated by invasive podosomes. Immunity. 2007;26(6):784–797. doi: 10.1016/j.immuni.2007.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hidalgo A, Frenette PS. Leukocyte podosomes sense their way through the endothelium. Immunity. 2007;26(6):753–755. doi: 10.1016/j.immuni.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 59.Kubow KE, Horwitz AR. Reducing background fluorescence reveals adhesions in 3D matrices. Nature Cell Biology. 2011;13(1):3–5. doi: 10.1038/ncb0111-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Goswami S, Sahai E, Wyckoff JB, et al. Macrophages promote the invasion of breast carcinoma cells via a colony-stimulating factor-1/epidermal growth factor paracrine loop. Cancer Research. 2005;65(12):5278–5283. doi: 10.1158/0008-5472.CAN-04-1853. [DOI] [PubMed] [Google Scholar]
- 61.Bischof RJ, Zafiropoulos D, Hamilton JA, Campbell IK. Exacerbation of acute inflammatory arthritis by the colony-stimulating factors CSF-1 and granulocyte macrophage (GM)-CSF: evidence of macrophage infiltration and local proliferation. Clinical and Experimental Immunology. 2000;119(2):361–367. doi: 10.1046/j.1365-2249.2000.01125.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang PT, Kasai H, Xiao WG, et al. Increased expression of macrophage colony-stimulating factor in ankylosing spondylitis and rheumatoid arthritis. Annals of the Rheumatic Diseases. 2006;65(12):1671–1672. doi: 10.1136/ard.2006.054874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Paniagua RT, Chang A, Mariano MM, et al. c-Fms-mediated differentiation and priming of monocyte lineage cells play a central role in autoimmune arthritis. Arthritis Research & Therapy. 2010;12(1):p. R32. doi: 10.1186/ar2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kleemann R, Zadelaar S, Kooistra T. Cytokines and atherosclerosis: a comprehensive review of studies in mice. Cardiovascular Research. 2008;79(3):360–376. doi: 10.1093/cvr/cvn120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145(3):341–355. doi: 10.1016/j.cell.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gierut A, Perlman H, Pope RM. Innate immunity and rheumatoid arthritis. Rheumatic Disease Clinics of North America. 2010;36(2):271–296. doi: 10.1016/j.rdc.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chitu V, Pixley FJ, Macaluso F, et al. The PCH family member MAYP/PSTPIP2 directly regulates F-actin bundling and enhances filopodia formation and motility in macrophages. Molecular Biology of the Cell. 2005;16(6):2947–2959. doi: 10.1091/mbc.E04-10-0914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Grosse J, Chitu V, Marquardt A, et al. Mutation of mouse Mayp/Pstpip2 causes a macrophage autoinflammatory disease. Blood. 2006;107(8):3350–3358. doi: 10.1182/blood-2005-09-3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sampaio NG, Yu W, Cox D, et al. Phosphorylation of CSF-1R Y721 mediates its association with PI3K to regulate macrophage motility and enhancement of tumor cell invasion. Journal of Cell Science. 2011;124(12):2021–2031. doi: 10.1242/jcs.075309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ridley AJ. Regulation of macrophage adhesion and migration by Rho GTP-binding proteins. Journal of Microscopy. 2008;231(3):518–523. doi: 10.1111/j.1365-2818.2008.02064.x. [DOI] [PubMed] [Google Scholar]
- 71.Park H, Chan MM, Iritani BM. Hem-1: putting the “WAVE” into actin polymerization during an immune response. FEBS Letters. 2010;584(24):4923–4932. doi: 10.1016/j.febslet.2010.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Okigaki M, Davis C, Falascat M, et al. Pyk2 regulates multiple signaling events crucial for macrophage morphology and migration. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(19):10740–10745. doi: 10.1073/pnas.1834348100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jones GE, Zicha D, Dunn GA, Blundell M, Thrasher A. Restoration of podosomes and chemotaxis in Wiskott-Aldrich syndrome macrophages following induced expression of WASp. International Journal of Biochemistry and Cell Biology. 2002;34(7):806–815. doi: 10.1016/s1357-2725(01)00162-5. [DOI] [PubMed] [Google Scholar]
- 74.Kheir WA, Gevrey JC, Yamaguchi H, Isaac B, Cox D. A WAVE2-Abi1 complex mediates CSF-1-induced F-actin-rich membrane protrusions and migration in macrophages. Journal of Cell Science. 2005;118(22):5369–5379. doi: 10.1242/jcs.02638. [DOI] [PubMed] [Google Scholar]
- 75.Yeung YG, Stanley ER. Proteomic approaches to the analysis of early events in colony-stimulating factor-1 signal transduction. Molecular & Cellular Proteomics. 2003;2(11):1143–1155. doi: 10.1074/mcp.R300009-MCP200. [DOI] [PubMed] [Google Scholar]
- 76.Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. Journal of Immunology. 2006;177(10):7303–7311. doi: 10.4049/jimmunol.177.10.7303. [DOI] [PubMed] [Google Scholar]
- 77.Yu W, Chen J, Xiong Y, et al. CSF-1 receptor structure/function in MacCsf1r-/-macrophages: regulation of proliferation, differentiation, and morphology. Journal of Leukocyte Biology. 2008;84(3):852–863. doi: 10.1189/jlb.0308171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Xiong Y, Song D, Cai Y, Yu W, Yeung YG, Stanley ER. A CSF-1 receptor phosphotyrosine 559 signaling pathway regulates receptor ubiquitination and tyrosine phosphorylation. Journal of Biological Chemistry. 2011;286(2):952–960. doi: 10.1074/jbc.M110.166702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Boocock CA, Jones GE, Stanley ER, Pollard JW. Colony-stimulating factor-1 induces rapid behavioural responses in the mouse macrophage cell line, BAC1.2F5. Journal of Cell Science. 1989;93(3):447–456. doi: 10.1242/jcs.93.3.447. [DOI] [PubMed] [Google Scholar]
- 80.Neumeister P, Pixley FJ, Xiong Y, et al. Cyclin D1 governs adhesion and motility of macrophages. Molecular Biology of the Cell. 2003;14(5):2005–2015. doi: 10.1091/mbc.02-07-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Reedijk M, Liu X, van der Geer P, et al. Tyr721 regulates specific binding of the CSF-1 receptor kinase insert to PI 3′-kinase SH2 domains: a model for SH2-mediated receptor-target interactions. EMBO Journal. 1992;11(4):1365–1372. doi: 10.1002/j.1460-2075.1992.tb05181.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Faccio R, Takeshita S, Colaianni G, et al. M-CSF regulates the cytoskeleton via recruitment of a multimeric signaling complex to c-Fms Tyr-559/697/721. Journal of Biological Chemistry. 2007;282(26):18991–18999. doi: 10.1074/jbc.M610937200. [DOI] [PubMed] [Google Scholar]
- 83.Bourette RP, Myles GM, Choi JL, Rohrschneider LR. Sequential activation of phoshatidylinositol 3-kinase and phospholipase C-γ2 by the M-CSF receptor is necessary for differentiation signaling. EMBO Journal. 1997;16(19):5880–5893. doi: 10.1093/emboj/16.19.5880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Papakonstanti EA, Zwaenepoel O, Bilancio A, et al. Distinct roles of class IA PI3K isoforms in primary and immortalised macrophages. Journal of Cell Science. 2008;121(24):4124–4133. doi: 10.1242/jcs.032763. [DOI] [PubMed] [Google Scholar]
- 85.Wu H, Yan Y, Backer JM. Regulation of class IA PI3Ks. Biochemical Society Transactions. 2007;35, part 2:242–244. doi: 10.1042/BST0350242. [DOI] [PubMed] [Google Scholar]
- 86.Charest PG, Firtel RA. Feedback signaling controls leading-edge formation during chemotaxis. Current Opinion in Genetics and Development. 2006;16(4):339–347. doi: 10.1016/j.gde.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 87.Hawkins PT, Anderson KE, Davidson K, Stephens LR. Signalling through class I PI3Ks in mammalian cells. Biochemical Society Transactions. 2006;34, part 5:647–662. doi: 10.1042/BST0340647. [DOI] [PubMed] [Google Scholar]
- 88.Ridley AJ. Life at the leading edge. Cell. 2011;145(7):1012–1022. doi: 10.1016/j.cell.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 89.Spiering D, Hodgson L. Dynamics of the rho-family small GTPases in actin regulation and motility. Cell Adhesion and Migration. 2011;5(2):170–180. doi: 10.4161/cam.5.2.14403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.MacHacek M, Hodgson L, Welch C, et al. Coordination of Rho GTPase activities during cell protrusion. Nature. 2009;461(7260):99–103. doi: 10.1038/nature08242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pertz O. Spatio-temporal Rho GTPase signaling—where are we now? Journal of Cell Science. 2010;123(11):1841–1850. doi: 10.1242/jcs.064345. [DOI] [PubMed] [Google Scholar]
- 92.Cammer M, Gevrey JC, Lorenz M, Dovas A, Condeelis J, Cox D. The mechanism of CSF-1-induced Wiskott-Aldrich syndrome protein activation in vivo. A role for phosphatidylinositol 3-kinase and Cdc42. Journal of Biological Chemistry. 2009;284(35):23302–23311. doi: 10.1074/jbc.M109.036384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Allen WE, Jones GE, Pollard JW, Ridley AJ. Rho, Rac and Cdc42 regulate actin organization and cell adhesion in macrophages. Journal of Cell Science. 1997;110(6):707–720. doi: 10.1242/jcs.110.6.707. [DOI] [PubMed] [Google Scholar]
- 94.Allen WE, Zicha D, Ridley AJ, Jones GE. A role for Cdc42 in macrophage chemotaxis. Journal of Cell Biology. 1998;141(5):1147–1157. doi: 10.1083/jcb.141.5.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wells CM, Walmsley M, Ooi S, Tybulewicz V, Ridley AJ. Rac1-deficient macrophages exhibit defects in cell spreading and membrane ruffling but not migration. Journal of Cell Science. 2004;117(7):1259–1268. doi: 10.1242/jcs.00997. [DOI] [PubMed] [Google Scholar]
- 96.Yamauchi A, Kim C, Li S, et al. Rac2-deficient murine macrophages have selective defects in superoxide production and phagocytosis of opsonized particles. Journal of Immunology. 2004;173(10):5971–5979. doi: 10.4049/jimmunol.173.10.5971. [DOI] [PubMed] [Google Scholar]
- 97.Wheeler AP, Wells CM, Smith SD, et al. Rac1 and Rac2 regulate macrophage morphology but are not essential for migration. Journal of Cell Science. 2006;119(13):2749–2757. doi: 10.1242/jcs.03024. [DOI] [PubMed] [Google Scholar]
- 98.Takenawa T, Suetsugu S. The WASP-WAVE protein network: connecting the membrane to the cytoskeleton. Nature Reviews Molecular Cell Biology. 2007;8(1):37–48. doi: 10.1038/nrm2069. [DOI] [PubMed] [Google Scholar]
- 99.Monypenny J, Chou HC, Bañón-Rodríguez I, et al. Role of WASP in cell polarity and podosome dynamics of myeloid cells. European Journal of Cell Biology. 2011;90(2-3):198–204. doi: 10.1016/j.ejcb.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tsuboi S. Requirement for a complex of Wiskott-Aldrich syndrome protein (WASP) with WASP interacting protein in podosome formation in macrophages. Journal of Immunology. 2007;178(5):2987–2995. doi: 10.4049/jimmunol.178.5.2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chou HC, Antón IM, Holt MR, et al. WIP regulates the stability and localization of WASP to podosomes in migrating dendritic cells. Current Biology. 2006;16(23):2337–2344. doi: 10.1016/j.cub.2006.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Vemula S, Shi J, Hanneman P, Wei L, Kapur R. ROCK1 functions as a suppressor of inflammatory cell migration by regulating PTEN phosphorylation and stability. Blood. 2010;115(9):1785–1796. doi: 10.1182/blood-2009-08-237222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Goode BL, Eck MJ. Mechanism and function of formins in the control of actin assembly. Annual Review of Biochemistry. 2007;76:593–627. doi: 10.1146/annurev.biochem.75.103004.142647. [DOI] [PubMed] [Google Scholar]
- 104.Brunton VG, MacPherson IRJ, Frame MC. Cell adhesion receptors, tyrosine kinases and actin modulators: a complex three-way circuitry. Biochimica et Biophysica Acta. 2004;1692(2-3):121–144. doi: 10.1016/j.bbamcr.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 105.Park H, Ishihara D, Cox D. Regulation of tyrosine phosphorylation in macrophage phagocytosis and chemotaxis. Archives of Biochemistry and Biophysics. 2011;510(2):101–111. doi: 10.1016/j.abb.2011.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Beemiller P, Zhang Y, Mohan S, et al. A Cdc42 activation cycle coordinated by PI 3-kinase during Fc receptor-mediated phagocytosis. Molecular Biology of the Cell. 2010;21(3):470–480. doi: 10.1091/mbc.E08-05-0494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Vega FM, Ridley AJ. Rho GTPases in cancer cell biology. FEBS Letters. 2008;582(14):2093–2101. doi: 10.1016/j.febslet.2008.04.039. [DOI] [PubMed] [Google Scholar]
- 108.Tzircotis G, Braga VMM, Caron E. RhoG is required for both FcgammaR-and CR3-mediated phagocytosis. Journal of Cell Science. 2011;124:2897–2902. doi: 10.1242/jcs.084269. [DOI] [PubMed] [Google Scholar]