

Abstract
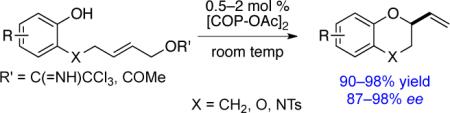
2-Vinylchromanes (1), 2-vinyl-1,4-benzodioxanes (2), and 2,3-dihydro-2-vinyl-2H-1,4-benzoxazines (3) can be prepared in high yields (90–98%) and excellent enantiomeric purities (87–98% ee) by [COP-OAc]2-catalyzed cyclization of phenolic (E)-allylic trichloroacetimidate precursors. Deuterium-labeling and computational experiments are consistent with these cyclization reactions taking place by an anti-oxypalladation/syn-deoxypalladation mechanism. 2-Vinylchromanes can be prepared also in good yields and high enantiomeric purities from analogous (E)-allylic acetate precursors, which constitutes the first report that acetate is a competent leaving group in COP-catalyzed enantioselective SN2′ substitution reactions.
Introduction
2-Alkenyl and 2-alkyl chromanes (4),1 1,4-benzodioxanes (5), and 2,3-dihydro-2H-1,4-benzoxazines (6) are heterocyclic fragments found in many molecules exhibiting useful therapeutic properties.2 The corresponding enantioenriched 2-vinyl heterocycles—2-vinylchromanes (1), 2-vinyl-1,4-benzodioxanes (2), and 2,3-dihydro-2-vinyl-2H-1,4-benzoxazines (3)—would be attractive precursors for the enantioselective synthesis of many heterocycles 4–6 (eq 1).3 Previously reported catalytic enantioselective methods for preparing 2-vinyl heterocycles 1–3 involve Pd(0)- or Ir(I)-catalyzed cyclization reactions that proceed via η3-allyl intermediates to form the allylic C–O σ-bond.4–6 The development of an alternative palladium(II)-catalyzed construction of enantioenriched 2-vinyl heterocycles 1–3 is the subject of this account.
 |
(1) |
Our investigations in this area are an outgrowth of the recently reported enantioselective synthesis of 3-phenoxy-1-alkenes from the reaction of phenols and allylic trichloroacetimidates using palladium(II) catalysts of the COP family (Figure 1).7–9 Herein we disclose our studies of the intramolecular variant of this chemistry, in which tethered (E)-allylic imidates yield 2-vinylchromanes (1), 2-vinyl-1,4-benzodioxanes (2), and 2,3-dihydro-2-vinyl-2H-1,4-benzoxazines (3) in good yields and high enantiomeric purities (eq 2). We report also that the enantioselective synthesis of chromane 1 can be accomplished with substrates having an acetate rather than a trichloroacetimidate leaving group.
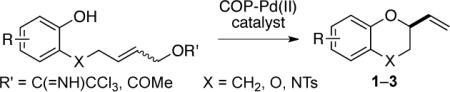 |
(2) |
Figure 1.
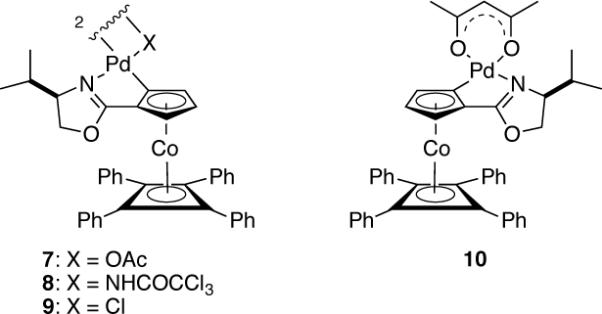
Selected palladium(II) catalysts of the COP family.
Results and Discussion
Enantioselective Synthesis of 2-Vinylchromanes, 2-Vinyl-1,4-benzodioxanes, and 2,3-Dihydro-2-vinyl-2H-1,4-benzoxazines from Allylic Trichloroacetimidate Precursors
Our studies began by examining the cyclization of phenolic (E)-allylic imidate 11a10 in the presence of 2 mol % of various palladium(II) catalysts (Table 1). At 38 °C in CH2Cl2, the COP complexes [(Sp,R)-COP-OAc]2 (7) and (Rp,S)-COP-acac (10) catalyzed the formation of 2-vinylchromane (1a) in high yields and respectively 89 and 80% ee (entries 1 and 2). As expected, allylic imidate rearrangement to form transposed allylic amide 12 was a significant competing process when chloride-bridged complex ent-9 was employed (entry 3).11 Trichloroacetamidate-bridged dimer 8, previously reported by our group to be a kinetically poor catalyst for allylic trichloroacetimidate rearrangements,8 did not suppress completely the competitive formation of allylic amide 12 (entry 4). Reactions were sufficiently rapid that a catalyst loading of 0.5 mol % could be employed in reactions carried out at room temperature where enantioselectivity was improved to 92–94% ee (entries 5 and 6). With [COP-OAc]2 identified as the most effective catalyst, several additional solvents were examined. Enantioselectivity was enhanced to 98% ee in d8-toluene, however competitive formation of amide 12 was observed in this solvent.12 The best combination of catalyst rate and enantioselectivity was obtained in CH2Cl2 at room temperature, conditions that we utilized in most subsequent experiments. The 4-bromo- and 4-methoxyphenol analogues cyclized similarly, although enantioselection was lower with the bromophenol precursor and catalysis rate lower with the methoxy congener (entries 7–9). As observed in related bimolecular reactions,8 enantioselectivity in the cyclization of the 4-bromophenol precursor was increased when CHCl3 was employed as the solvent (entry 8). In marked contrast, the Z allylic imidate stereoisomer cyclized to form 2-vinylchromane (1a) of negligible enantiomeric purity (entry 10).
Table 1.
Catalytic Enantioselective Synthesis of 2-Vinylchromanes 1a–c.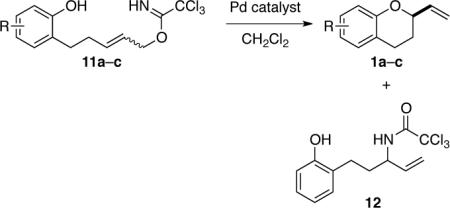
| entrya | substrate | R | catalyst (mol %) | temp (°C) | yield (%)b12 | yield (%)b1 | ee (%)c1 |
|---|---|---|---|---|---|---|---|
| 1 | (E)-11a | H | 7 (2) | 38 | — | 86 | 89 (R)d |
| 2 | (E)-11a | H | 10 (2) | 38 | — | 72 | 80 (S) |
| 3 | (E)-11a | H | ent-9 (2) | 38 | 43 | 41 | 80 (S) |
| 4 | (E)-11a | H | 8 (2) | 38 | 13 | 81 | 87 (R) |
| 5 | (E)-11a | H | 7 (0.5) | 23 | — | 91e | 94 (R)e |
| 6 | (E)-11a | H | ent-7 (0.5) | 23 | — | 97e | 92 (S)e |
| 7 | (E)-11b | 4-Br | 7 (0.5) | 23 | — | 96e | 80e |
| 8f | (E)-11b | 4-Br | 7 (0.5) | 23 | — | 94e | 90e |
| 9g | (E)-11c | 4-OMe | 7 (0.5) | 23 | — | 92e | 91 (R)d,e |
| 10 | (Z)-11a | H | ent-7 (0.5) | 23 | — | 92e | 9 (R)e |
[11] = 0.2 M; reaction time 8–18 h.
Isolated yield after purification on silica gel.
Determined by HPLC analysis using a enantioselective stationary phase.
Absolute configuration determined by comparison of optical rotation data with that reported in the literature.4b
Mean values of duplicate reactions.
The solvent was CHCl3.
Reaction time was 30 h.
2-Vinyl-1,4-benzodioxane (2) and 2,3-dihydro-2-vinyl-2H-1,4-benzoxazine (3) can be prepared in similar fashion in high yield and enantiopurity although catalysis rate is somewhat slower (Table 2). For example, in the presence of 0.5 mol % of COP catalyst 7, 2-vinylbenzodioxane 2 was formed in 92% ee; however, a reaction time of 72 h was required (entry 1). Increasing the catalyst loading to 2 mol % gave 2 and 3 in high yield and respectively 94% and 98% ee after reaction times of 15–18 h (entries 2 and 3). The E trisubstituted allylic precursor 15 provided benzodioxane 17 in 85% ee, albeit in only 30% yield after a reaction time of 72 h (entry 4). Although the yield of 17 was modest, this transformation is notable because it represented the first example of COP-catalyzed formation of a tetrasubstituted stereocenter.13 The reaction could not be extended to the formation of 2,3-dihydro-2-vinyl-2H-1,4-benzothiopyran (18), presumably as a result of coordination of the palladium(II) catalyst to sulfur.12
Table 2.
Catalytic Enantioselective Synthesis of 2-Vinyl-1,4-benzodioxanes (2) and 2-Vrnyl-2H-1,4-benzoxazines (3).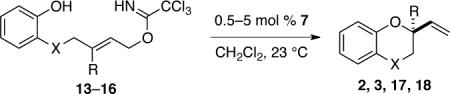
| entry | X | R | imidate | catalyst (mol %) | time (h) | product | yield (%)a | ee (%)b |
|---|---|---|---|---|---|---|---|---|
| 1 | O | H | 13 | 7 (0.5) | 72 | 2 | 92 | 92 (S)c |
| 2 | O | H | 13 | 7 (2) | 28 | 2 | 90 | 94 (S)c |
| 3 | NTs | H | 14 | 7 (2) | 15 | 3 | 98 | 9S (S)c |
| 4 | O | Me | 15 | ent-7 (5) | 72 | 17 | 30 | 85d |
| 5 | S | H | 16 | 7 (5) | 18 | 18 | 0 | — |
Mean yields after purification on silica gel of reactions performed in duplicate.
Determined by HPLC analysis using a enantioselective stationary phase (mean value of duplicate reactions).
Absolute configuration determined by comparison of optical rotation data with those reported in the literature.6c,d
Absolute configuration was not established.
Enantioselective Synthesis of 2-Vinylchromanes from Allylic Acetate Precursors
Trichloacetimidates are widely used leaving groups, largely because they can be prepared in high yields from trichloroacetonitrile and most alcohols—including quite labile ones—under mild conditions (catalytic DBU at room temperature).14,15 The disadvantage in their use is the production of trichloroacetamide (MW = 144) as a byproduct. In a preliminary survey, we found that acetate was an adequate leaving group for the [COP-OAc]2-catalyzed cyclization of phenolic (E)-allylic acetate 19 to form 2-vinylchromane (Table 3).16 In CH2Cl2, cyclohexane or toluene, acetate (E)-19a cyclized in the presence of 2 mol % of [COP-OAc]2 to form ent-1a in 84–85% ee (entries 1–3), whereas significantly lower enantioselectivity was observed in CH3CN, DMF, and THF. Though the use of the alcoholic solvents EtOH, i-PrOH, cyclohexanol, or t-BuOH typically resulted in low conversions (5–20%) and moderate enantioselectivities (72–84% ee), to our surprise, excellent enantioselectivity was obtained in MeOH. Increasing the reaction temperature to 60 °C gave rise to chromane ent-1a in 71% yield and 91% ee after 20 h using 2 mol % of [(Rp,S)-COP-OAc]2 (ent-7) (entry 4). However, enantiomeric purity of the product ent-1a begins to erode at ~15–20 h, which is attributed to protodemetallation of [COP-OAc]2 at 60 °C to generate an achiral palladium(II) catalyst. More success was realized by adding bases. For example, adding K2CO3 (1 equiv) increased conversion in reactions carried out at room temperature in CH2Cl2 or cyclohexane to respectively 73% and >95% (entries 5 and 6). Since enantioselectivity was higher in CH2Cl2, further optimization of the base was carried out in this solvent (entries 7–12). Heterogeneous bases performed better than Et3N (entry 8). As expected in the heterogeneous reactions,17 conversions and enantioselectivities were highly dependent upon the physical state of the solid bases employed. Conversions were generally highest when K2CO3 was used; however, reproducible yields and enantioselectivities were best achieved using powdered, dried KF with rapid stirring. The optimal conditions for the conversion of 19 to ent-1a were found to be: 2 mol % [(Rp,S)-COP-OAc]2 (ent-7), 1 equiv KF, [19] = 1.0 M CH2Cl2, 23 °C (entry 11).
Table 3.
Catalytic Enantioselective Synthesis of 2-Vinylchromane ent-1a from Allylic Acetate Precursor (E)-19a.18
| entrya | solvent | temp (°C) | additiveb | conversion (%)c | ee (%)d |
|---|---|---|---|---|---|
| 1 | CH2Cl2 | 40 | none | 33 | 84 |
| 2 | toluene | 40 | none | 27 | 85 |
| 3 | cyclohexane | 40 | none | 43 | 84 |
| 4e | MeOH | 60 | none | 71 | 91 |
| 5e,f | CH2Cl2 | 23 | K2CO3 | 73 | 91 |
| 6e,f | cyclohexane | 23 | K2CO3 | >95 | 82 |
| 7e | CH2C12 | 23 | K3PO4 | 93 | 73 |
| 8 | CH2Cl2 | 23 | Et3N | 41 | 84 |
| 9e | CH2C12 | 23 | CsF | 26 | 94 |
| 10e,f | CH2C12 | 23 | KF | 55 | 93 |
| 11e,g | CH2Cl2 | 23 | KF | 88 | 92 |
| 12e,h | CH2Cl2 | 23 | KF | 64 | 93 |
[(E)-19a] = 0.2 M, unless otherwise indicated.
1 equiv was used.
Determined by 1H NMR analysis using an internal standard.
Determined by HPLC analysis using a enantioselective stationary phase.
Reaction was heterogeneous.
When the identical reaction was performed in the absence of ent-7, only (E)-19a was observed by 1H NMR analysis.
[(E)-19a] = 1.0M.
1 mol % of [(Rp,S)-COP-OAc]2 (ent-7) was employed.
The results of the synthesis of 2-vinylchromanes ent-1a–c from the corresponding allylic acetate precursors under these optimized conditions are summarized in Table 4. In all three cases, yields (89–95%) and enantioselectivities (88–95%) were excellent. The (Z)-allylic ester (Z)-19a was also transformed to chromane ent-1a under these conditions, although as observed with the corresponding trichloroacetimidate precursor, enantioselectivity was low (entry 5).
Table 4.
Catalytic Enantioselective Synthesis of 2-Vinylchromane ent-1a from Allylic Acetate Precursors 19.a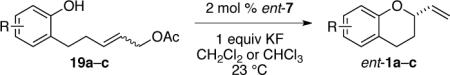
| entryb | R | 19 | solvent | time (h) | yield (%)c | ee (%)d |
|---|---|---|---|---|---|---|
| 1 | H | (E)-19a | CH2Cl2 | 24 | 89 | 94 (S) |
| 2 | 4-Br | (E)-19b | CH2C12 | 6 | 82 | 88 |
| 3 | 4-Br | (E)-19b | CHC13 | 10 | 92 | 90 |
| 4 | 4-OMe | (E)-19c | CH2Cl2 | 36 | 95 | 95 (S) |
| 5 | H | (Z)-19a | CH2Cl2 | 24 | 91 | 18–51 (S) |
All reactions reported in this table were heterogeneous.
[19] = 1.0 M.
Mean yields after purification on silica gel of reactions performed in duplicate.
Determined by HPLC analysis using a enantioselective stationary phase (mean value of duplicate reactions).
Geometry of the Intramolecular SN2′ Reaction
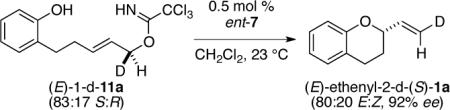
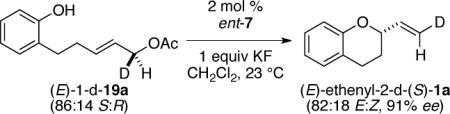
To gain insight into the mechanism of the [COP-OAc]2-catalyzed cyclization reactions in both the imidate and acetate series, the cyclization of two enantioenriched deuterium-labeled substrates was examined (eqs 3 and 4). As in our study of the geometry of related bimolecular SN2′ reactions,7b the allylic alcohol precursors of deuterated enantioenriched imidate and acetate cyclization substrates were prepared by enantioselective Keck reduction of the corresponding enal.12,19 In both series, chirality transfer was complete within experimental error and in accord with an antarafacial SN2′ geometry.
 |
(3) |
 |
(4) |
The antarafacial geometry of the SN2′ cyclization to form 2-vinylchromane (1a) from both the (E)-allylic trichloroacetimidate and (E)-allylic acetate precursors is identical to the geometry of the bimolecular reaction of (E)-allylic imidates and phenol in the presence of COP catalyst 8.7b In the context of an oxypalladation/deoxypalladation mechanism, this result requires that the two steps occur with opposite geometries. In our earlier study of [COP-OAc]2-catalyzed allylic esterification of (Z)-allylic imidates, computational modeling provided evidence for an anti-oxypalladation/syn-deoxypalladation mechanism.7b,20 Such a sequence for the formation of vinylchromane (S)-1a is illustrated in Scheme 1.
Scheme 1.
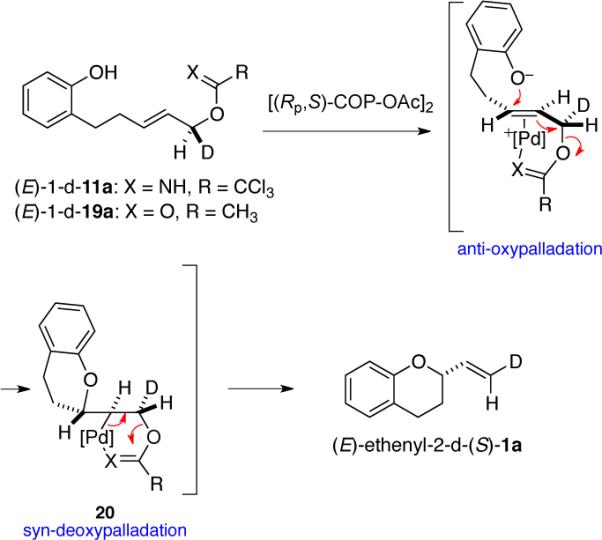
Anti-oxypalladation/Syn-deoxypalladation Sequence for Forming (S)-1a.
Computational Studies
As the oxypalladation step has been determined to be both rate- and enantiodetermining in the [COP-OAc]2-catalyzed bimolecular reaction of (Z)-allylic trichloroacetimidates with carboxylic acid nucleophiles,7b this step of the [COP-OAc]2-catalyzed cyclization of (E)- and (Z)-imidates 11a to form 2-vinylchromane was studied computationally. The objective was to ascertain whether or not an anti-oxypalladation/syn-deoxypalladation mechanism would: (a) explain the observation that stereoinduction was much higher in the [COP-OAc]2-catalyzed cyclization of E allylic imidate (E)-11a than the corresponding Z stereoisomer, and (b) rationalize preferential formation of the S enantiomer of dihydrobenzopyran 1a from the cyclization of (E)-11a catalyzed by [(Rp,S)-COP-OAc]2 (ent-7). The TURBOMOLE v6.221 program at the B3-LYP level of theory22 with a continuum solvation model (COSMO)23 was utilized for this study. All atoms were represented by the def2-TZVP basis set24 except for the carbon and hydrogen atoms of the tetraphenylcyclobutadiene unit of catalyst ent-7, which were represented by the SZ.benzene basis set.25
For each double bond isomer, two pathways were investigated: anti-oxypalladation where the catalyst ent-7 was bound to the imidate nitrogen and the Re (pathways (E)-I or (Z)-I) or Si face (pathways (E)-II or (Z)-II) of the double bond (Figure 2).7b Oxypalladation of Re-bound catalyst complex 24 occurs by transition structure 25 to provide palladated benzopyran complex 26 (Figure 2, pathway (E)-I); this elementary step requires 0.3 kcal/mol in activation energy and is exergonic by 21.0 kcal/mol. Deoxypalladation of 26 would yield (S)-2-vinylchromane. The Si-bound complex 21 undergoes oxypalladation with an activation energy of 1.4 kcal/mol by transition structure 22 to give palladated-chromane complex 23 with the overall release of 20.1 kcal/mol (Figure 2, pathway (E)-II). Further reaction of this intermediate would form (R)-2-vinylchromane. Oxypalladation via pathway (E)-I to form the observed S enantiomer of 1a is calculated to be favored by ΔΔE‡ of 3.7 kcal/mol. In contrast, the transition-state energy differences for oxypalladation of the two (Z)-imidate complexes (pathways (Z)-I and (Z)-II) are calculated to differ in energy by only 0.8 kcal/mol, consistent with the low selectivity observed in the cyclization of the (Z)-imidate precursor.
Figure 2.

Calculated reaction coordinate diagrams for oxypalladation of (E)- and (Z)-allylic trichloroacetimidates 11a with COP catalyst ent-7.
Analysis of the two calculated transition structures, 22 and 25, for cyclization of the (E)-allylic imidate substrate provided some insight into the factors contributing to the high levels of enantioselectivity observed in the [COP-OAc]2-catalyzed cyclization reaction (Figure 3).26 The closest contact (2.36 Å) seen in low energy transition structure 25 is between H4 and the cyclopentadiene fragment (HCp), with the remaining interatomic distances being outside of van der Waals contact. In contrast, oxypalladation transition structure 22 that leads to the minor observed enantiomer, shows several destabilizing steric interactions. In particular, imidate hydrogen H4 is 2.26 Å from the cyclopentadiene fragment (HCp), and H5 is 2.73 Å from the tetraphenylcyclobutadiene fragment (Cfloor). Unlike transition structure 25, structure 22 has van der Waals contacts between the hydrogens of the methylene chain (H4 and H5) of the substrate and the COP ligand (Cfloor and HCp).27
Figure 3.
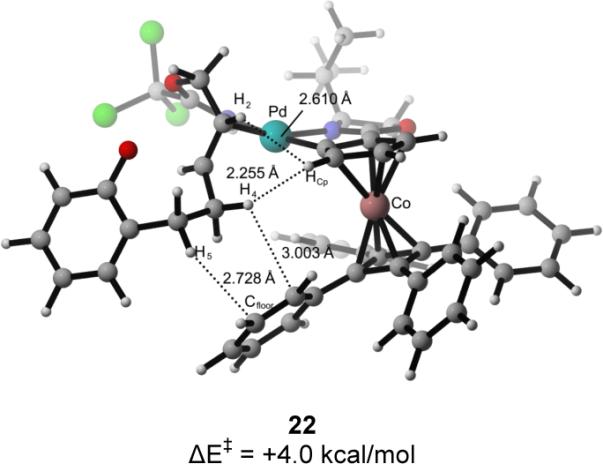
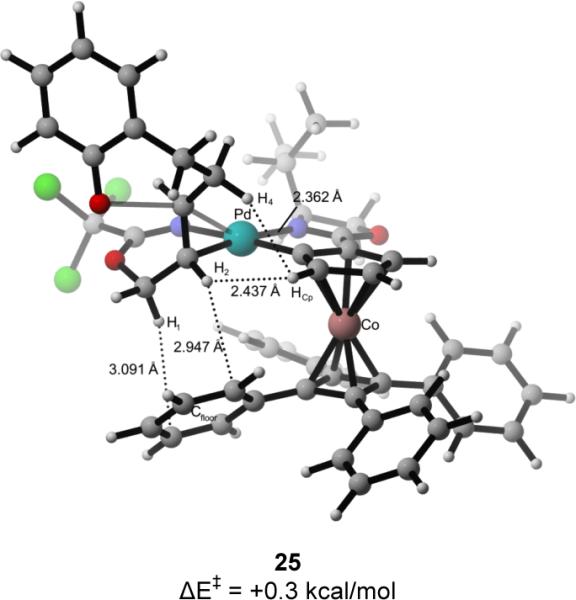
Calculated transition structures for intramolecular oxypalladation of (E)-allylic trichloroacetimidates.
Conclusion
Using phenol-tethered (E)-allylic trichloroacetimidate precursors, the catalytic enantioselective synthesis of 2-vinylchromanes, 2-vinyl-1,4-benzodioxanes, and 2,3-dihydro-2-vinyl-2H-1,4-benzoxazines depicted in eq 2 takes place at room temperature in 90–98% yield and 87–98% ee under neutral conditions using 0.5 mol % of the palladacyclic catalyst [COP-OAc]2. For the preparation of 2-vinylchromane (1a), 2-vinyl-1,4-benzodioxane (2) and 2,3-dihydro-4-(4-toluenesulfonyl)-2-vinyl-2H-1,4-benzoxazine (3) the catalytic enantioselective syntheses reported herein are the first to provide these products in both >90% yield and >90% ee.4–6 We also demonstrate for the first time that acetate is a competent leaving group in COP-catalyzed enantioselective SN2′ substitution reactions. In this way, 2-vinylchromanes 1a–c were prepared in 82–95% yield and 88–95% ee.
Deuterium labeling experiments establish that the catalytic enantioselective cyclizations of both the (E)-allylic trichloroacetimidate and acetate precursors proceed by an antarafacial geometry identical to that of the bimolecular reaction of (E)-allylic trichloroacetimidates with phenols.7b DFT-modeling studies of the synthesis of 2-vinylchromane from the allylic imidate precursor are consistent with an anti-oxypalladation/syn-deoxypalladation identical to that of the corresponding bimolecular reaction. We speculate that cyclization of the allylic acetate precursor takes place in a similar fashion (Scheme 1), although additional experimentation will be required to establish this point. The computational studies rationalize the observation that enantioselection is significantly higher with the E than the Z allylic trichloroacetimidate substrate as well as the overall sense of stereoinduction. This model for stereoinduction supports earlier findings that the positioning of the tetraphenylcyclobutadiene group beneath the palladium square-plane is a key factor in achieving high levels of enantioselection in reactions catalyzed by COP complexes.7b
Experimental Section
General Procedure for Synthesis of 2-Vinylheterocycles from Allylic Trichloroacetimidate Precursors
A sealable ½-dram glass vial equipped with a Teflon® cap was charged with the trichloroacetimidate precursor (0.077 mmol). A solution of [(Sp,R)-COP-OAc]2 (7) or its enantiomer (ent-7) (0.001 M in CH2Cl2 or CHCl3, 0.4 mL, 0.0004 mmol) was added via syringe. The reaction vial was sealed under an Ar atmosphere and the orange solution was maintained at room temperature. After the reported reaction time, the solution was concentrated in vacuo and the residue was purified by flash chromatography on silica gel to afford the 2-vinylheterocycle product.
(R)-2,3-Dihydro-2-vinyl-2H-1-benzopyran (1a)
Following the general procedure for imidate cyclization with catalyst 7 (0.001 M in CH2Cl2, 0.4 mL, 0.0004 mmol), imidate (E)-11a (25 mg, 0.077 mmol) was converted to 1a (11 mg, 0.070 mmol, 91%), a clear colorless oil: 94% ee by enantioselective HPLC analysis [OJ column + OJ guard; flow: 0.5 mL/min; 100% n-hexane; λ = 280 nm; major enantiomer tR = 22.56 min, minor enantiomer tR = 25.41 min]; [α]D25 −76.5, [α]57725 −85.0, [α]54625 −95.4, [α]43525 −168.8, [α]40525 −202.3, (c 0.27, CHCl3). 1H NMR and optical rotation data were consistent with previously reported values.4b
(R)-6-Bromo-2,3-dihydro-2-vinyl-2H-1-benzopyran (1b).28
Following the general procedure for imidate cyclization with catalyst 7 (0.001 M in CHCl3, 0.4 mL, 0.0004 mmol), imidate (E)-11b (30 mg, 0.075 mmol) was converted to 1b (17 mg, 0.070 mmol, 94%), a clear colorless oil: 90% ee by enantioselective HPLC analysis [OJ column; flow: 0.5 mL/min; 100% n-hexane; λ = 230 nm; major enantiomer tR = 28.85 min, minor enantiomer tR = 32.63 min]; [α]D25 −63.4, [α]57725 −67.7, [α]54625 −78.5, [α]43525 −133.1, (c 0.40, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.20–7.18 (m, 2H), 6.74 (d, J = 8.3, 1H), 5.96 (ddd, J = 16.3, 10.6, 5.6, 1H), 5.37 (d, J = 17.3, 1H), 5.25 (d, J = 10.6, 1H), 4.57–4.53 (m, 1H), 2.87–2.80 (m, 1H), 2.77–2.72 (m, 1H), 2.09–2.04 (m, 1H), 1.87–1.79 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 153.7 (C), 137.2 (CH), 132.1 (CH), 130.3 (CH), 124.1 (C), 118.7 (CH), 116.7 (CH), 112.3 (C), 76.3 (CH), 27.1 (CH2), 24.1 (CH2); IR (thin film) 3083, 2927, 1648, 1236, 1040, 992 cm−1; HRMS (GC-MS) m/z calcd for C11H11BrO, (M)+ 237.9993, found 237.9985.
(R)-2,3-Dihydro-6-methoxy-2-vinyl-2H-1-benzopyran (1c)
Following the general procedure for imidate cyclization with catalyst 7 (0.001 M in CH2Cl2, 0.6 mL, 0.0006 mmol), imidate (E)-11c (40 mg, 0.11 mmol) was converted to 1c (19 mg, 0.10 mmol, 92%), a clear colorless oil: 91% ee by enantioselective HPLC analysis [OJ column + OJ guard; flow: 0.5 mL/min; 99:1 n-hexane:isopropanol; λ = 210 nm; major enantiomer tR = 24.82 min, minor enantiomer tR = 27.55 min]; [α]D25 −83.7, [α]57725 −86.6, [α]54625 −99.3, [α]43525 −161.5, [α]40525 −188.8, (c 0.54, CHCl3). 1H NMR and optical rotation data were consistent with previously reported values.4b
(S)-2,3-Dihydro-2-vinyl-2H-1,4-benzodioxane (2).29
Following the general procedure for imidate cyclization with catalyst 7 (0.004 M in CH2Cl2, 0.6 mL, 0.0023 mmol), imidate 13 (38 mg, 0.12 mmol) was converted to 2 (17 mg, 0.11 mmol, 90%), a clear colorless oil: 94% ee by enantioselective HPLC analysis [OJ column + OJ guard; flow: 0.5 mL/min; 99:1 n-hexane:isopropanol; λ = 280 nm; major enantiomer tR = 18.90 min, minor enantiomer tR = 20.19 min]; [α]D25 −6.5, [α]57725 −3.7, [α]54625 −4.1, (c 0.16, CHCl3). 1H NMR and optical rotation data were consistent with previously reported values.6c
(S)-2,3-Dihydro-4-(4-toluenesulfonyl)-2-vinyl-2H-1,4-benzoxazine (3).29
Following the general procedure for imidate cyclization with catalyst 7 (0.004 M in CH2Cl2, 0.5 mL, 0.00021 mmol), imidate 14 (50 mg, 0.10 mmol) was converted to 3 (32 mg, 0.10 mmol, 98%), a pale yellow oil: 98% ee by enantioselective HPLC analysis [AD column; flow: 1.0 mL/min; 80:20 n-hexane:isopropanol; λ = 280 nm; minor enantiomer tR = 6.09 min, major enantiomer tR = 6.49 min]; [α]D25 +72.8, [α]57725 +70.9, [α]54625 +75.0, [α]43525 +195.3, (c 0.25, CHCl3). 1H NMR and optical rotation data were consistent with previously reported values.6d
(R)-2,3-Dihydro-2-methyl-2-vinyl-1,4-benzodioxane (17).28,29
Following the general procedure for imidate cyclization with catalyst ent-7 (0.01 M in CH2Cl2, 0.3 mL, 0.0030 mmol), imidate 15 (20 mg, 0.059 mmol) was converted to 17 (3.2 mg, 0.018 mmol, 30%), a pale yellow oil: 85% ee by enantioselective HPLC analysis [OJ column; flow: 0.5 mL/min; 99.5:0.5 n-hexane:isopropanol; λ = 280 nm; major enantiomer tR = 13.59 min, minor enantiomer tR = 14.95 min]; [α]D25 −3.6, [α]57725 −3.0, [α]54625 −8.1, [α]43525 −6.0, (c 0.05, CHCl3). 1H NMR data were consistent with previously reported values.30
(S,E)-2-(2-Deuterovinyl)-1-benzopyran ((E)-ethenyl-2-d-(S)-1a).31,32
Following the general procedure for imidate cyclization with catalyst ent-7 (0.001 M in CH2Cl2, 0.4 mL, 0.0004 mmol), imidate (E)-1-d-11a (25 mg, 0.077 mmol) was converted to (E)-ethenyl-2-d-(S)-1a (9 mg, 0.058 mmol, 75%), a clear colorless oil. Analysis by electrospray mass spectrometry indicated a >96:4 ratio of d1-2-d-(S)-1a:d0-2-d-(S)-1a. The E:Z-ratio was calculated to be 80:20 using 1H NMR spectroscopy: 92% ee by enantioselective HPLC analysis [OJ column + OJ guard; flow: 0.5 mL/min; 100% n-hexane; λ = 280 nm; minor enantiomer tR = 22.23 min, major enantiomer tR = 24.51 min]; [α]D25 2.5, (c 0.33, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 7.12–7.09 (m, 1H), 7.06 (d, J = 7.4, 1H), 6.87–6.84 (m, 2H), 5.38 (d, J = 17.2, 1H), 4.58–4.56 (m, 1H), 2.91–2.84 (m, 1H), 2.81–2.76 (m, 1H), 2.11–2.06 (m, 1H), 1.90–1.83 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 154.6 (C), 137.6 (CH), 129.6 (CH), 127.4 (CH), 121.9 (C), 120.3(CH), 117.0 (CH), 116.1 (t, JCD = 23.8, CDH), 76.2 (CH), 27.6 (CH2), 24.3 (CH2); IR (thin film) 3081, 2919, 1231, 1048, 975 cm−1; HRMS (CI) m/z calcd for C11H12DO, (M + H)+ 162.1029, found 162.1023.
General Procedure for Synthesis of 2-Vinylchromanes from Allylic Acetate Precursors
A sealable, oven-dried ½-dram glass vial equipped with a stir bar and a Teflon® cap was charged with [(Rp,S)-COP-OAc]2 (ent-7) or its enantiomer (7) (2.7 mg, 0.0018 mmol) and KF (5.3 mg, 0.091 mmol). The vial was sealed with a Teflon® cap, then flame-dried and back-filled with Ar twice. Once the vial cooled to room temperature, acetate (0.091 mmol) in CH2Cl2 or CHCl3 (0.09 mL, 1.0 M) was added via syringe. The reaction vial was sealed under an Ar atmosphere, placed in an aluminum block, and the reaction was stirred vigorously at room temperature. After the reported reaction time, the heterogeneous brown reaction mixture was filtered, the filtrate was concentrated in vacuo, then the residue was purified by flash chromatography on silica gel to afford the 2-vinylchromane product.
(S)-2,3-Dihydro-2-vinyl-2H-1-benzopyran (ent-1a)
Following the general procedure for acetate cyclization with catalyst ent-7 (2.7 mg, 0.0018 mmol), acetate (E)-19a (1.0 M in CH2Cl2, 0.09 mL, 0.091 mmol) was converted to ent-1a (13 mg, 0.081 mmol, 89%), a clear colorless oil: 94% ee by enantioselective HPLC analysis [OJ column + OJ guard; flow: 0.5 mL/min; 100% n-hexane; λ = 280 nm; minor enantiomer tR = 20.94 min, major enantiomer tR = 22.92 min]. 1H NMR data were consistent with previously reported values.4b
(S)-6-Bromo-2,3-dihydro-2-vinyl-2H-1-benzopyran (ent-1b)
28 Following the general procedure for acetate cyclization with catalyst ent-7 (2.0 mg, 0.0013 mmol), acetate (E)-19b (1.0 M in CHCl3, 0.07 mL, 0.067 mmol) was converted to ent-1b (15 mg, 0.061 mmol, 92%), a clear colorless oil: 90% ee by enantioselective HPLC analysis [OJ column; flow: 0.5 mL/min; 100% n-hexane; λ = 230 nm; minor enantiomer tR = 30.55 min, major enantiomer tR = 32.78 min]. See characterization data for 1b for spectral details.
(S)-2,3-Dihydro-6-methoxy-2-vinyl-2H-1-benzopyran (ent-1c)
Following the general procedure for acetate cyclization with catalyst ent-7 (1.8 mg, 0.0012 mmol), acetate (E)-19c (1.0 M in CH2Cl2, 0.06 mL, 0.060 mmol) was converted to ent-1c (10.8 mg, 0.057 mmol, 95%), a clear colorless oil: 95% ee by enantioselective HPLC analysis [OJ column + OJ guard; flow: 0.5 mL/min; 99:1 n-hexane:isopropanol; λ = 210 nm; minor enantiomer tR = 22.64 min, major enantiomer tR = 24.19 min]. 1H NMR data were consistent with previously reported values.4b
(S,E)-2-(2-Deuterovinyl)-1-benzopyran ((E)-ethenyl-2-d-(S)-1a)
31,32 Following the general procedure for acetate cyclization with catalyst ent-7 (2.7 mg, 0.0018 mmol), acetate (E)-1-d-19a (20 mg, 0.090 mmol) was converted to (E)-ethenyl-2-d-(S)-1a (14 mg, 0.083 mmol, 92%), a clear colorless oil. Analysis by electrospray mass spectrometry indicated a >96:4 ratio of d1-2-d-(S)-1a:d0-2-d-(S)-1a. The E:Z-ratio was calculated to be 82:18 using 1H NMR spectroscopy: 91% ee by enantioselective HPLC analysis [OJ column + OJ guard; flow: 0.5 mL/min; 100% n-hexane; λ = 280 nm; minor enantiomer tR = 20.82 min, major enantiomer tR = 22.48 min]. See characterization data above for spectral details.
Synthesis of Cyclization Precursors
2-(4,4-Dibromobut-3-enyl)phenol (34)
Using a procedure by Kinoshita,33 chroman-2-ol (33) (2.00 g, 13.3 mmol) was treated with CBr4 (8.83 g, 26.6 mmol) and PPh3 (13.9 g, 53.3 mmol). The crude light yellow residue was purified by flash chromatography (99.5:0.5 CH2Cl2-acetone) to provide dibromide 34 (3.65 g, 11.9 mmol, 90%) as a pale yellow oil: 1H NMR (500 MHz, CDCl3) δ 7.13–7.11 (m, 2H), 6.90 (t, J = 7.4, 1H), 6.75 (d, J = 7.9, 1H), 6.47 (t, J = 7.2, 1H), 4.74–4.70 (m, 1H), 2.75 (t, J = 7.7, 2H), 2.44 (app q, J = 7.5, 2H); 13C NMR (125 MHz, CDCl3) δ 153.6 (C), 138.1 (CH), 130.5 (CH), 127.7 (CH), 126.9 (C), 121.0 (CH), 115.4 (CH), 89.3 (C), 33.2 (CH2), 28.3 (CH2); IR (thin film) 3541, 3032, 2927, 1591, 753 cm−1; HRMS (CI) m/z calcd for C10H11Br2O, (M + H)+ 305.9078, found 305.9074.
2-(5-Hydroxypent-3-ynyl)phenol (35)
Using a modification of the procedure by Kinoshita,33 a solution of 34 (3.55 g, 11.6 mmol) in THF (39 mL) was cooled to −78 °C in a 100-mL round-bottomed flask. A solution of n-BuLi (2.0 M in hexane, 21 mL, 42.0 mmol) was added via syringe over 20 min. The reaction mixture was stirred at −78 °C for 40 min then paraformaldehyde (1.04 g, 34.8 mmol) was added in a single portion. The reaction mixture was allowed to warm to 0 °C and stirred for 1 h, then allowed to warm to ambient temperature and stirred for 1 h. Saturated aqueous NH4Cl (60 mL) was added and the resulting mixture was extracted with EtOAc (3 × 50 mL), dried over Na2SO4, filtered, and concentrated in vacuo to give a pale yellow residue that was purified by flash chromatography (97.5:2.5 to 92:8 CH2Cl2-acetone) to provide propargyl alcohol 35 (1.33 g, 7.6 mmol, 65%) as a clear colorless oil: 1H NMR (500 MHz, CDCl3) δ 7.16–7.11 (m, 2H), 6.90 (t, J = 7.4, 1H), 6.79 (d, J = 7.9, 1H), 4.96 (s, 1H), 4.25 (s, 2H), 2.87 (t, J = 7.3, 2H), 2.55 (t, J = 7.3, 2H); 13C NMR (125 MHz, CDCl3) δ 153.8 (C), 130.7 (CH), 128.0 (CH), 127.0 (C), 121.0 (CH), 115.8 (CH), 86.5 (C), 79.3 (C), 51.5 (CH2), 29.8 (CH2), 19.6 (CH2); IR (thin film) 3364, 2929, 2223, 1181, 1006 cm−1; HRMS (ESI) m/z calcd for C11H12O2Na, (M + Na)+ 199.0735, found 199.0730.
(E)-2-(5-Hydroxypent-3-enyl)phenol ((E)-36)
Using a modification of a procedure by Denmark,34 Red-Al (65% in toluene, 0.66 mL, 2.1 mmol) and Et2O (0.6 mL) were cooled to 0 °C in a 10-mL round-bottomed flask. A solution of alkyne 35 (150 mg, 0.85 mmol) in Et2O (0.6 mL) was added via syringe over 5 min, then the solution was allowed to warm to room temperature. After 16 h, the solution was cooled to 0 °C and saturated aqueous Rochelle's salt (5 mL) was added slowly. The mixture was stirred for 1 h, extracted with EtOAc (3 × 5 mL), dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography (94:6 CH2Cl2-acetone) to yield alkene (E)-36 (107 mg, 0.600 mmol, 71%) as a clear colorless oil: 1H NMR (500 MHz, CDCl3) δ 7.12–7.07 (m, 2H), 6.87 (t, J = 7.4, 1H), 6.76 (d, J = 8.0, 1H), 5.78 (dt, J = 15.4, 6.6, 1H), 5.68 (dt, J = 15.4, 5.8, 1H), 5.48 (br s, 1H), 4.10 (d, J = 5.4, 2H), 2.72 (t, J = 7.4, 2H), 2.38 (app q, J = 7.3, 2H); 13C NMR (125 MHz, CDCl3) δ 153.8 (C), 133.1 (CH), 130.4 (CH), 129.4 (CH), 128.0 (C), 127.4 (CH), 120.8 (CH), 115.5 (CH), 63.9 (CH2), 32.5 (CH2), 29.9 (CH2); IR (thin film) 3307, 3035, 2850, 1668, 966 cm−1; HRMS (ESI) m/z calcd for C11H14O2Na, (M + Na)+ 201.0892, found 201.0892.
(E)-5-(2-Hydroxyphenyl)pent-2-enyl 2′,2′,2′-trichloroacetimidate ((E)-11a)
In a 10-mL round-bottomed flask, alcohol (E)-36 (25 mg, 0.14 mmol) was dissolved in CH2Cl2 (0.56 mL), then DBU (24 μL, 0.17 mmol) was added via syringe. Trichloroacetonitrile (17 μL, 0.17 mmol) was added via syringe and the solution was maintained at room temperature. After 1 h, the reaction mixture was concentrated in vacuo and the residue was purified by flash chromatography (98.5:0.5:1 CH2Cl2-acetone-Et3N) to provide imidate (E)-11a (43 mg, 0.13 mmol, 96%) as a pale yellow oil: 1H NMR (500 MHz, CDCl3) δ 8.28 (br s, 1H), 7.12–7.08 (m, 2H), 6.88 (t, J = 7.4, 1H), 6.76 (d, J = 7.9, 1H), 5.95 (dt, J = 15.4, 6.6, 1H), 5.73 (dt, J = 15.3, 6.2, 1H), 4.82 (br s, 1H), 4.76 (d, J = 5.9, 2H), 2.74 (t, J = 7.7, 2H), 2.45–2.40 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 163.0 (C), 153.7 (C), 136.2 (CH), 130.5 (CH), 127.8 (C), 127.4 (CH), 123.7 (CH), 120.9 (CH), 115.5 (CH), 91.6 (C), 70.1 (CH2), 32.5 (CH2), 29.7 (CH2); IR (thin film) 3328, 3067, 2927, 1658, 1607, 1592 cm−1; HRMS (CI) m/z calcd for C13H18Cl3N2O2, (M + NH4)+ 339.0434, found 339.0436.
(Z)-2-(5-Hydroxypent-3-enyl)phenol ((Z)-36)
Using a modification of a procedure by Taber,35 a 10-mL round-bottomed flask was evacuated and refilled with nitrogen three times. The flask was charged with Ni(OAc)2·4H2O (106 mg, 0.426 mmol) and evacuated and refilled with nitrogen three additional times before degassed EtOH (1.5 mL) and NaBH4 (20 mg, 0.54 mmol) were added to form a black suspension. Ethylene diamine (0.11 mL, 1.7 mmol) was added via syringe, followed by a degassed aqueous solution of NaOH (2.0 M, 7 μL, 0.01 mmol). Alkyne 35 (500 mg, 2.84 mmol) was dissolved in degassed EtOH (1.5 mL) and added via syringe to the black suspension. Hydrogen gas was bubbled vigorously through the reaction mixture for 5 min, then a balloon of H2 was placed over the reaction mixture, which was allowed to stir for 18 h then filtered through a short pad of silica gel using EtOH as the eluent. The organic layer was concentrated in vacuo to yield alkene (Z)-36 (506 mg, 2.84 mmol, 100%) as a pale yellow oil: 1H NMR (500 MHz, CDCl3) δ 7.10–7.07 (m, 2H), 6.86 (t, J = 7.4, 1H), 6.78 (d, J = 7.9, 1H), 5.70–5.66 (m, 2H), 4.07 (d, J = 6.2, 2H), 2.71 (t, J = 7.4, 2H), 2.43 (app q, J = 7.3, 2H); 13C NMR (125 MHz, CDCl3) δ 154.1 (C), 133.2 (CH), 130.8 (CH), 128.8 (CH), 127.8 (C), 127.6 (CH), 120.5 (CH), 115.6 (CH), 58.3 (CH2), 30.6 (CH2), 27.8 (CH2); IR (thin film) 3367, 2932, 1592, 1180, 1017, 721 cm−1; HRMS (CI) m/z calcd for C11H16NO, (M + NH4 − H2O)+ 178.1232, found 178.1240.
(Z)-5-(2-Hydroxyphenyl)pent-2-enyl 2′,2′,2′-trichloroacetimidate ((Z)-11a)
Following the procedure described for the preparation of (E)-11a, alcohol (Z)-36 (100 mg, 0.561 mmol) was treated with DBU and Cl3CCN. After 2 h, the reaction mixture was concentrated in vacuo and the residue was purified by flash chromatography (98.5:0.5:1 CH2Cl2-acetone-Et3N) to provide imidate (Z)-11a (136 mg, 0.424 mmol, 76%) as a pale yellow oil: 1H NMR (500 MHz, CDCl3) δ 8.29 (br s, 1H), 7.12–7.09 (m, 2H), 6.87 (t, J = 7.4, 1H), 6.78 (d, J = 8.1, 1H), 5.91 (br s, 1H), 5.83 (dt, J = 10.6, 7.9, 1H), 5.67 (dt, J = 10.8, 6.8, 1H), 4.87 (d, J = 6.8, 2H), 2.73 (t, J = 7.8, 2H), 2.48 (app q, J = 7.7, 2H); 13C NMR (125 MHz, CDCl3) δ 163.2 (C), 154.0 (C), 135.2 (CH), 130.5 (CH), 128.6 (CH), 127.5 (C), 123.3 (CH), 120.8 (CH), 115.6 (CH), 91.6 (C), 65.4 (CH2), 30.5 (CH2), 28.0 (CH2); IR (thin film) 3333, 3029, 2944, 1650, 1608, 1593 cm−1; HRMS (ESI) m/z calcd for C13H14Cl3NO2Na, (M + Na)+ 343.9988, found 343.9981.
6-Bromochroman-2-ol (38)
A 250-mL round-bottomed flask was charged with 6-bromochroman-2-one (37)36 (9.9 g, 44 mmol) and toluene (110 mL). The solution was cooled to −78 °C and DIBAL (25% in toluene, 30 mL, 48 mmol) was added over 10 min via syringe. The reaction mixture was stirred at −78 °C for 4 h, then allowed to warm to 0 °C. Water (50 mL) and saturated aqueous Rochelle's salt (75 mL) were added and the resultant mixture was allowed to warm to room temperature and stirred for 1 h. The biphasic mixture was extracted with Et2O (3 × 70 mL) and the organic layer was dried over Na2SO4, filtered, then concentrated in vacuo to yield a residue that was purified by flash chromatography (98.5:1.5 CH2Cl2-acetone) to provide lactol 38 (8.2 g, 36 mmol, 82%) as a pale yellow oil. Product was of sufficient purity to be carried on to the next step. 1H NMR data were consistent with previously reported values.37
(E)-Ethyl 5-(5-bromo-2-hydroxyphenyl)pent-2-enoate (39)
In a 250-mL round-bottomed flask, lactol 38 (8.19 g, 35.6 mmol) was dissolved in benzene (100 mL), then treated with carboethoxymethylenetriphenylphosphorane (13.1 g, 37.5 mmol) in benzene (100 mL) and the solution was maintained at room temperature for 15 h. Evaporation of solvent using a rotary evaporator left a viscous oil that was dissolved in CH2Cl2 (70 mL). Celite (2 g) was added to the suspension and the organic solvent was removed in vacuo to yield a powder that was loaded on silica gel and purified by flash chromatography (92:8 to 0:100 hexanes-EtOAc) to provide ester 39 (6.95 g, 24.5 mmol, 67%) as a white solid: mp 87–89 °C; 1H NMR (500 MHz, CDCl3) δ 7.22 (s, 1H), 7.18 (dd, J = 8.5, 2.4, 1H), 7.06–7.00 (m, 1H), 6.65 (d, J = 8.5, 1H), 5.87 (dd, J = 15.6, 1.4, 1H), 5.75 (br s, 1H), 4.21 (q, J = 3.4, 2H), 2.74 (t, J = 7.3, 2H), 2.54–2.49 (m, 2H), 1.30 (t, J = 4.4, 3H); 13C NMR (125 MHz, CDCl3) δ 167.3 (C), 153.0 (C), 148.6 (CH), 132.9 (CH), 130.3 (CH), 129.7 (C), 121.8 (CH), 117.1 (CH), 112.7 (C), 60.6 (CH2), 32.2 (CH2), 28.7 (CH2), 14.4 (CH3); IR (thin film) 3381, 2983, 1693, 1272, 1039 cm−1; HRMS (ESI) m/z calcd for C13H15BrO3Na, (M + Na)+ 321.0102, found 321.0098.
(E)-4-Bromo-2-(5-hydroxypent-3-enyl)phenol ((E)-40)
A 25-mL round-bottomed flask was charged with ester 39 (500 mg, 1.76 mmol) and toluene (4.4 mL). The solution was cooled to −78 °C and DIBAL (25% in toluene, 2.2 mL, 3.5 mmol) was added over 5 min via syringe. The reaction mixture was stirred at −78 °C for 3.5 h, then allowed to warm to 0 °C. Water (5 mL) and saturated aqueous Rochelle's salt (5 mL) were added and the resultant mixture was allowed to warm to room temperature and stirred for 1 h. The biphasic mixture was extracted with CH2Cl2 (3 × 10 mL) and the organic layer was dried over Na2SO4, filtered, then concentrated in vacuo to yield a residue that was purified by flash chromatography (98:2 to 96:4 CH2Cl2-MeOH) to provide alcohol (E)-40 (243 mg, 0.945 mmol, 54%): 1H NMR (500 MHz, CDCl3) δ 7.22 (s, 1H), 7.17 (dd, J = 8.5, 2.4, 1H), 6.64 (d, J = 8.5, 1H), 5.75 (dt, J = 15.3, 6.5, 1H), 5.67 (dt, J = 18.0, 6.4, 1H), 4.11 (d, J = 5.8, 2H), 2.67 (t, J = 7.7, 2H), 2.37–2.33 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 153.0 (C), 133.0 (CH), 132.6 (CH), 130.4 (C), 130.0 (CH), 129.6 (CH), 117.2 (CH), 112.7 (C), 63.8 (CH2), 32.3 (CH2), 29.7 (CH2); IR (thin film) 3325, 3029, 2927, 1669, 812 cm−1; HRMS (ESI) m/z calcd for C11H13BrO2Na, (M + Na)+ 278.9997, found 278.9995.
(E)-5-(5-Bromo-2-hydroxyphenyl)pent-2-enyl 2′,2′,2′-trichloroacetimidate ((E)-11b)
Following the procedure described for the preparation of (E)-11a, alcohol (E)-40 (243 mg, 0.945 mmol) was treated with DBU and Cl3CCN. After 1 h, the reaction mixture was concentrated in vacuo and the residue was purified by flash chromatography (98.5:0.5:1 CH2Cl2-acetone-Et3N) to provide imidate (E)-11b (272 mg, 0.677 mmol, 72%) as a pale yellow powder: mp 108–111 °C; 1H NMR (500 MHz, CDCl3) δ 8.29 (br s, 1H), 7.23 (s, 1H), 7.18 (dd, J = 8.4, 2.4, 1H), 6.64 (d, J = 8.5, 1H), 5.91 (dt, J = 15.5, 6.7, 1H), 5.70 (dt, J = 15.4, 6.1, 1H), 4.94 (br s, 1H), 4.76 (d, J = 6.0, 2H), 2.70 (t, J = 7.2, 2H), 2.42–2.38 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 163.0 (C), 152.9 (C), 135.5 (CH), 133.1 (CH), 130.3 (C), 130.1 (CH), 124.0 (CH), 117.2 (CH), 112.8 (C), 91.6 (C), 70.0 (CH2), 32.3 (CH2), 29.5 (CH2); IR (thin film) 3326, 3033, 2922, 1657, 971 cm−1; HRMS (ESI) m/z calcd for C13H13BrCl3NO2Na, (M + Na)+ 421.9093, found 421.9083.
(E)-2-(5-Hydroxypent-3-enyl)-4-methoxyphenol ((E)-42)
A 50-mL round-bottomed flask was charged with (E)-5-(2-((tert-butyldimethylsilyl)oxy)-5-methoxyphenyl)pent-2-en-1-ol (41)4a (1.63 g, 5.05 mmol) and THF (25 mL), then TBAF (1.0 M in THF, 7.6 mL, 7.6 mmol) was added. The reaction mixture was stirred at room temperature for 2 h, then the reaction mixture was concentrated in vacuo and the residue was purified by flash chromatography (70:30 to 50:50 to 40:60 hexanes-EtOAc) to provide diol (E)-42 (714 mg, 3.43 mmol, 85%) as a clear colorless oil: 1H NMR (500 MHz, CDCl3) δ 6.70–6.69 (m, 2H), 6.64–6.62 (m, 1H), 5.78 (dt, J = 15.4, 6.5, 1H), 5.69 (dt, J = 15.4, 5.8, 1H), 4.10 (d, J = 5.1, 2H), 3.76 (s, 3H), 2.69 (t, J = 7.4, 2H), 2.40–2.35 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 153.3 (C), 148.0 (C), 133.0 (CH), 129.5 (C), 129.1 (CH), 116.2 (CH), 116.0 (CH), 111.9 (CH), 63.6 (CH2), 55.9 (CH3) 32.4 (CH2), 30.0 (CH2); IR (thin film) 3366, 2935, 1611, 1509, 971 cm−1; HRMS (ESI) m/z calcd for C12H16O3Na, (M + Na)+ 231.0997, found 231.0994.
(E)-5-(2-Hydroxy-5-methoxyphenyl)pent-2-enyl 2′,2′,2′-trichloroacetimidate ((E)-11c)
A ½-dram glass vial equipped with a Teflon® cap was charged with NaH (60% dispersion in mineral oil, 11 mg, 0.26 mmol) and Et2O (0.2 mL). The suspension was cooled to 0 °C and stirred for 15 min. Diol (E)-42 (50 mg, 0.24 mmol) was added in 0.28 mL Et2O via syringe. Trichloroacetonitrile (25 μL, 0.25 mmol) was added via syringe and the reaction mixture was stirred at 0 °C for 1 h. The suspension was allowed to warm to room temperature and stirred for 15 min. The reaction mixture was filtered through silica gel using 50:50:1 CH2Cl2-acetone-Et3N as the eluent. The filtrate was concentrated in vacuo and the residue was purified by flash chromatography (98.5:0.5:1 CH2Cl2-acetone-Et3N) to yield imidate (E)-11c (60 mg, 0.17 mmol, 71%) as a clear colorless oil. Imidate (E)-11c was used directly in the [COP-OAc]2 (7)-catalyzed cyclization reactions: 1H NMR (500 MHz, CDCl3) δ 8.29 (br s, 1H), 6.70–6.69 (m, 2H), 6.64–6.62 (m, 1H), 5.94 (dt, J = 15.4, 6.7, 1H), 5.72 (dt, J = 15.4, 6.2, 1H), 4.76 (d, J = 6.2, 2H), 4.61 (br s, 1H), 3.76 (s, 3H), 2.71 (t, J = 7.3, 2H), 2.44–2.39 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 162.9 (C), 153.9 (C), 147.5 (C), 135.9 (CH), 129.1 (C), 123.9 (CH), 116.3 (CH), 116.1 (CH), 112.0 (CH), 91.6 (C), 70.0 (CH2), 55.8 (CH3), 32.6 (CH2), 30.0 (CH2); IR (thin film) 3372, 3334, 2924, 1660, 1080, 971 cm−1; HRMS (ESI) m/z calcd for C14H16Cl3NO3Na, (M + Na)+ 374.0093, found 374.0082.
(E)-2-(4-Tetrahydro-2H-pyran-2-yloxy)but-2-enyloxy)phenol (48)
In a 5-mL round-bottomed flask, catechol (45) (0.400 g, 3.63 mmol) in DMF (11 mL) was cooled to 0 °C. In a single portion, K2CO3 (0.100 g, 0.726 mmol) was added and the suspension was stirred for 5 min. (E)-2-((4-Bromobut-2-en-1-yl)oxy)tetrahydro-2H-pyran (47)38 (0.170 g, 0.726 mmol) in DMF (1.1 mL) was added via syringe and the reaction mixture was allowed to warm to room temperature. After 20 h, water (20 mL) and brine (10 mL) were added and the mixture was extracted with CH2Cl2 (3 × 10 mL), dried over Na2SO4, filtered, and concentrated in vacuo to yield a residue that was purified by flash chromatography (75:25 hexanes-Et2O) to provide ether 48 (147 mg, 0.557 mmol, 77%) as a clear colorless oil: 1H NMR (500 MHz, CDCl3) δ 6.95–6.81 (m, 4H), 6.04–5.95 (m, 2H), 5.67 (s, 1H), 4.66 (t, J = 3.5, 1H), 4.62 (d, J = 4.1, 2H), 4.31 (dd, J = 13.4, 4.2, 1H), 4.05 (dd, J = 13.6, 4.9, 1H), 3.88 (td, J = 11.0, 2.8, 1H), 3.54–3.51 (m, 1H), 1.89–1.83 (m, 1H), 1.78–1.72 (m, 1H), 1.64–1.54 (m, 4H); 13C NMR (125 MHz, CDCl3) δ 146.0 (C), 145.7 (C), 131.1 (CH), 127.0 (CH), 121.9 (CH), 120.2 (CH), 114.8 (CH), 112.3 (CH), 98.3 (CH), 69.1 (CH2), 66.9 (CH2), 62.4 (CH2), 30.7 (CH2), 25.5 (CH2), 19.6 (CH2); IR (thin film) 3393, 2942, 1609, 1115, 972 cm−1; HRMS (ESI) m/z calcd for C15H20O4Na, (M + Na)+ 287.1259, found 287.1261.
(E)-4-(2-Hydroxyphenoxy)but-2-enyl 2′,2′,2′-trichloroacetimidate (13)
In a 5-mL round-bottomed flask, protected alcohol 48 (30 mg, 0.11 mmol) and p-TsOH·H2O (4 mg, 0.02 mmol) were dissolved in MeOH (3.0 mL). The solution was refluxed for 3 h then allowed to cool to room temperature. The solvent was removed in vacuo to yield a viscous oil that was dissolved in CH2Cl2 (0.57 mL), then DBU (23 μL, 0.16 mmol) was added via syringe. Trichloroacetonitrile (14 μL, 0.14 mmol) was added via syringe and the solution was maintained at room temperature. After 10 min, the reaction mixture was concentrated in vacuo and the residue was purified by flash chromatography (60:40:1 pentane-Et2O-Et3N) to provide imidate 13 (28 mg, 0.086 mmol, 76%) as a pale yellow oil: 1H NMR (500 MHz, CDCl3) δ 8.37 (br s, 1H), 6.96 (d, J = 7.8, 1H), 6.91–6.82 (m, 3H), 6.13 (dt, J = 15.8, 5.1, 1H), 6.06 (dt, J = 16.1, 4.9, 1H), 5.65 (s, 1H), 4.87 (d, J = 5.1, 2H), 4.66 (d, J = 5.0, 2H); 13C NMR (125 MHz, CDCl3) δ 162.6 (C), 146.0 (C), 145.5 (C), 129.1 (CH), 127.3 (CH), 122.1 (CH), 120.3 (CH), 115.0 (CH), 112.3 (CH), 91.4 (C), 68.8 (CH2), 68.5 (CH2); IR (thin film) 3411, 3338, 2926, 1663, 1473, 982 cm−1; HRMS (ESI) m/z calcd for C12H12Cl3 NO3Na, (M + Na)+ 345.9781, found 345.9787.
(E)-N-(4-Hydroxybut-2-enyl)-N-(2-hydroxyphenyl)-4-methylbenzenesulfonamide (50)
In a 250-mL round-bottomed flask, PPh3 (889 mg, 3.39 mmol) was added to a mixture of N-(2-hydroxyphenyl)-4-methylbenzenesulfonamide (43) (0.800 g, 3.39 mmol), (E)-4-((tetrahydro-2H-pyran-2-yl)oxy)but-2-en-1-ol (44)38 (583 mg, 3.39 mmol), and diethyl azodicarboxylate (40% in toluene, 1.5 mL, 3.4 mmol) in THF (39 mL) at 0 °C. The reaction mixture was allowed to warm to room temperature. After 18 h, the reaction mixture was concentrated in vacuo to give a residue that was filtered through silica gel using 75:25 petroleum ether-EtOAc as eluent. The pale yellow oil and p-TsOH·H2O (77 mg, 0.41 mmol) were dissolved in MeOH (12 mL) and refluxed for 18 h. The solution was allowed to cool to room temperature and the solvent was removed in vacuo. The residue was purified by flash chromatography (70:30 to 60:40 petroleum ether-EtOAc) to provide diol 50 (437 mg, 1.31 mmol, 64%) as a pale yellow oil: 1H NMR (500 MHz, DMSO) δ 9.53 (s, 1H), 7.56 (d, J = 8.2, 2H), 7.35 (d, J = 8.1, 2H), 7.11 (td, J = 8.0, 1.5, 1H), 6.93 (dd, J = 7.8, 1.5, 1H), 6.79 (dd, J = 8.1, 1.1, 1H), 6.72 (td, J = 7.6, 1.1, 1H), 5.57 (dt, J = 15.5, 4.6, 1H), 5.50 (dt, J = 15.5, 5.9, 1H), 4.64 (t, J = 5.4, 1H), 4.13 (d, J = 5.5, 2H), 3.78 (t, J = 4.2, 2H), 2.39 (s, 3H); 13C NMR (125 MHz, DMSO) δ 154.9 (C), 142.8 (CH), 137.2 (CH), 133.9 (CH), 132.0 (C), 129.4 (2C), 127.3 (CH), 124.8 (CH), 124.2 (CH), 118.6 (CH), 116.4 (CH), 60.6 (CH2), 50.9 (CH2), 21.0 (CH3); IR (thin film) 3416, 3049, 2923, 1598, 1337, 1090 cm−1; HRMS (ESI) m/z calcd for C17H19NO4SNa, (M + Na)+ 356.0933, found 356.0931.
(E)-4-(N-(2-Hydroxyphenyl)-4-methylphenylsulfonamido)but-2-enyl 2′,2′,2′-trichloroacetimidate (14)
Following the procedure described for the preparation of (E)-11a, alcohol 50 (193 mg, 0.579 mmol) was treated with DBU and Cl3CCN. After 1 h, the reaction mixture was concentrated in vacuo and the residue was purified by flash chromatography (99.5:0.5:1 CH2Cl2-acetone-Et3N) to provide imidate 14 (249 mg, 0.521 mmol, 90%) as a pale yellow oil: 1H NMR (500 MHz, DMSO) δ 9.57 (br s, 1H), 9.25 (br s, 1H), 7.57 (d, J = 8.2, 2H), 7.35 (d, J = 8.2, 2H), 7.10 (td, J = 8.1, 1.3, 1H), 6.92 (d, J = 7.8, 1H), 6.79 (d, J = 8.1, 1H), 6.70 (t, J = 7.7, 1H), 5.78–5.68 (m, 2H), 4.62 (d, J = 4.5, 2H), 4.19 (d, J = 4.0, 2H), 2.38 (s, 3H); 13C NMR (125 MHz, DMSO) δ 159.7 (C), 154.9 (C), 142.8 (C), 137.2 (C), 132.0 (CH), 129.6 (CH), 129.4 (CH), 129.3 (CH), 127.3 (CH), 126.6 (CH), 124.8 (C), 118.7 (CH), 116.5 (CH), 90.7 (C), 67.6 (CH2), 50.6 (CH2), 21.0 (CH3); IR (thin film) 3436, 3336, 2924, 1664, 1344, 1090 cm−1; HRMS (ESI) m/z calcd for C19H19Cl3N2O4SNa, (M + Na)+ 499.0029, found 499.0024.
(E)-tert-Butyldimethyl(3-methyl-4-(2-triisopropylsilyloxy)phenoxy)but-2-enyloxy)silane (54)
In a 200-mL round-bottomed flask, PPh3 (1.34 g, 5.11 mmol) was added to a mixture of 2-((triisopropylsilyl)oxy)phenol (52) (1.30 g, 4.87 mmol), (E)-4-((tert-butyldimethylsilyl)oxy)-2-methylbut-2-en-1-ol (53)39 (1.12 mg, 5.17 mmol), and diethyl azodicarboxylate (40% in toluene, 2.3 mL, 5.2 mmol) in THF (54 mL) at 0 °C. The reaction mixture was allowed to warm to room temperature. After 3 h, the reaction mixture was concentrated in vacuo to give a residue that was purified by flash chromatography (99.5:0.5 to 98.5:1.5 pentane-Et2O) to provide ether 54 (1.61 g, 3.17 mmol, 65%) as a clear colorless oil: 1H NMR (500 MHz, CDCl3) δ 6.90–6.86 (m, 3H), 6.83–6.81 (m, 1H), 5.72 (m, 1H), 4.41 (s, 2H), 4.28 (d, J = 6.2, 2H), 1.77 (s, 3H), 1.32–1.26 (m, 3H), 1.13 (d, J = 6.1, 18H), 0.93 (s, 9H), 0.09 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 150.3 (C), 145.9 (C), 132.6 (C), 128.3 (CH), 121.4 (CH), 121.1 (CH), 120.5 (CH), 114.2 (CH), 74.3 (CH2), 60.1 (CH2), 26.1 (CH3), 18.1 (C, CH3), 14.3 (CH3), 13.0 (CH), −5.0 (CH3); IR (thin film) 3064, 2947, 1267, 1116, 1073, 921 cm−1; HRMS (ESI) m/z calcd for C26H48O3Si2Na, (M + Na)+ 487.3040, found 487.3038.
(E)-2-(4-Hydroxy-2-methylbut-2-enyloxy)phenol (55)
A 50-mL round-bottomed flask was charged with bis-silyl ether 54 (1.61 g, 3.17 mmol) and THF (16 mL), then TBAF (1.0 M in THF, 7.0 mL, 7.0 mmol) was added. The reaction mixture was stirred at room temperature for 3 h, then the reaction mixture was concentrated in vacuo and the residue was purified by flash chromatography (94:6 CH2Cl2-acetone) to provide diol 55 (616 mg, 3.17 mmol, 100%) as a clear colorless oil: 1H NMR (500 MHz, CDCl3) δ 6.95 (d, J = 7.7, 1H), 6.94–6.80 (m, 3H), 6.23 (br s, 1H), 5.77 (t, J = 6.6, 1H), 4.44 (s, 2H), 4.23 (d, J = 6.9, 2H), 2.60 (br s, 1H), 1.76 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 145.9 (C), 145.8 (C), 133.8 (C), 127.2 (CH), 121.8 (CH), 120.1 (CH), 115.1 (CH), 112.5 (CH), 73.8 (CH2), 58.8 (CH2), 14.0 (CH3); IR (thin film) 3418, 2924, 1679, 1596, 845 cm−1; HRMS (ESI) m/z calcd for C11H14O3Na, (M + Na)+ 217.0841, found 217.0841.
(E)-4-(2-Hydroxyphenoxy)-3-methylbut-2-enyl 2′,2′,2′-trichloroacetimidate (15)
Following the procedure described for the preparation of (E)-11a, alcohol 55 (26 mg, 0.13 mmol) was treated with DBU and Cl3CCN. After 5 min, the reaction mixture was concentrated in vacuo and the residue was purified by flash chromatography (97.5:1.5:1 CH2Cl2-acetone-Et3N) to provide imidate 15 (41 mg, 0.12 mmol, 91%) as a pale yellow oil: 1H NMR (500 MHz, CDCl3) δ 8.33 (br s, 1H), 6.95 (dd, J = 7.8, 1.5, 1H), 6.91–6.80 (m, 3H), 5.88–5.85 (m, 1H), 5.64 (s, 1H), 4.91 (d, J = 6.7, 2H), 4.54 (s, 2H), 1.88 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 162.6 (C), 145.9 (C), 145.6 (C), 137.3 (C), 121.9 (CH), 121.4 (CH), 120.11 (CH), 115.0 (CH), 112.4 (CH), 91.4 (C), 73.6 (CH2), 65.4 (CH2), 14.3 (CH3); IR (thin film) 3414, 3337, 2923, 1662, 1501, 988 cm−1; HRMS (ESI) m/z calcd for C13H14Cl3NO3Na, (M + Na)+ 359.9937, found 359.9928.
(E)-2-(4-(Tetrahydro-2H-pyran-2-yloxy)but-2-enylthio)phenol (49)
Following the procedure described for the preparation of 48, 2-mercaptophenol (46) (902 mg, 7.15 mmol) was alkylated using (E)-2-((4-bromobut-2-en-1-yl)oxy)tetrahydro-2H-pyran (47).38 After 24 h, water (20 mL) and brine (10 mL) were added and the mixture was extracted with CH2Cl2 (3 × 20 mL), dried over Na2SO4, filtered, and concentrated in vacuo to yield a residue that was purified by flash chromatography (70:30 hexanes-Et2O) to provide thioether 49 (319 mg, 1.14 mmol, 80%) as a pale yellow oil: 1H NMR (500 MHz, CDCl3) δ 7.42 (dd, J = 7.7, 1.6, 1H), 7.28–7.23 (m, 1H), 6.98 (dd, J = 8.1, 1.1, 1H), 6.88–6.84 (m, 1H), 6.68 (s, 1H), 5.73 (dt, J = 15.2, 7.5, 1H), 5.41 (dt, J = 15.2, 5.5, 1H), 4.56 (t, J = 2.9, 1H), 4.11 (dd, J = 12.9, 4.8, 1H), 3.89 (dd, J = 13.0, 6.3, 1H), 3.83 (td, J = 11.0, 3.0, 1H), 3.52–3.46 (m, 1H), 3.32 (d, J = 7.5, 2H), 1.85–1.79 (m, 1H), 1.73–1.68 (m, 1H), 1.62–1.51 (m, 4H); 13C NMR (125 MHz, CDCl3) δ 157.3 (C), 136.7 (CH), 136.4 (C), 131.5 (CH), 130.7 (CH), 127.6 (CH), 120.8 (CH), 114.9 (CH), 97.8 (CH), 66.7 (CH2), 62.2 (CH2), 38.8 (CH2), 30.7 (CH2), 25.6 (CH2), 19.5 (CH2); IR (thin film) 3404, 2940, 1470, 1119, 966 cm−1; HRMS (ESI) m/z calcd for C15H20O3SNa, (M + Na)+ 303.1031, found 303.1029.
(E)-2-(4-Hydroxybut-2-enylthio)phenol (51)
In a 25-mL round-bottomed flask, protected alcohol 49 (319 mg, 1.14 mmol) and p-TsOH·H2O (43 mg, 0.23 mmol) were dissolved in MeOH (9.5 mL) and the solution was refluxed for 6 h, then allowed to cool to room temperature. After 18 h, the solvent was removed in vacuo and the residue was purified by flash chromatography to yield alcohol 51 (142 mg, 0.723 mmol, 64%) as a clear colorless oil. 1H NMR data were consistent with those previously reported.40
(E)-4-(2-Hydroxyphenylthio)but-2-enyl 2′,2′,2′-trichloroacetimidate (16)
Following the procedure described for the preparation of (E)-11a, alcohol 51 (132 mg, 0.673 mmol) was treated with DBU and Cl3CCN. After 5 min, the reaction mixture was concentrated in vacuo and the residue was purified by flash chromatography (98.5:0.5:1 CH2Cl2-acetone-Et3N) to provide imidate 16 (191 mg, 0.563 mmol, 84%) as a pale yellow oil: 1H NMR (500 MHz, CDCl3) δ 8.30 (br s, 1H), 7.42 (dd, J = 7.7, 1.4, 1H), 7.29–7.25 (m, 1H), 6.99 (d, J = 8.2, 1H), 6.86 (t, J = 7.6, 1H), 6.66 (s, 1H), 5.87 (dt, J = 15.3, 7.5, 1H), 5.48 (dt, J = 15.3, 5.8, 1H), 4.69 (d, J = 5.8, 2H), 3.33 (d, J = 7.5, 2H); 13C NMR (125 MHz, CDCl3) δ 162.4 (C), 157.3 (C), 136.7 (CH), 131.5 (CH), 129.8 (CH), 127.0 (CH), 120.7 (CH), 117.7 (C), 114.9 (CH), 91.4 (C), 68.6 (CH2), 38.4 (CH2); IR (thin film) 3408, 3337, 2916, 1662, 1470, 984 cm−1; HRMS (GC-MS) m/z calcd for C12H16Cl3N2O2S, (M + NH4)+ 356.9998, found 357.0004.
(E)-5-(2-Hydroxyphenyl)pent-2-enyl acetate ((E)-19a)
In a 25-mL round-bottomed flask, alcohol (E)-36 (700 mg, 3.93 mmol) in THF (5.9 mL) was cooled to 0 °C, then acetic anhydride (1.1 mL, 12 mmol) was added via syringe. BF3·OEt2 (0.12 mL, 0.98 mmol) was added via syringe and the solution was maintained at 0 °C. After 1.5 h, cold aqueous NaHCO3 (5% solution, 10 mL) was added, the mixture was stirred for 2 min, extracted with CH2Cl2 (3 × 10 mL), dried over Na2SO4, filtered, and concentrated in vacuo to yield a residue that was purified by flash chromatography (80:20 hexanes-Et2O) to give acetate (E)-19a (691 mg, 3.14 mmol, 80%) as a clear colorless oil: 1H NMR (500 MHz, CDCl3) δ 7.12–7.08 (m, 2H), 6.87 (t, J = 7.4, 1H), 6.76 (d, J = 7.9, 1H), 5.86 (dt, J = 15.4, 7.7, 1H), 5.61 (dt, J = 15.4, 6.6, 1H), 5.16 (br s, 1H), 4.53 (d, J = 6.5, 2H), 2.72 (t, J = 7.4, 2H), 2.41–2.37 (m, 2H), 2.08 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 171.4 (C), 153.7 (C), 135.9 (CH), 130.4 (CH), 127.8 (C), 127.4 (CH), 124.4 (CH), 120.9 (CH), 115.5 (CH), 65.4 (CH2), 32.5 (CH2), 30.0 (CH2), 21.2 (CH3); IR (thin film) 3411, 2925, 1739, 1672, 1235, 965 cm−1; HRMS (ESI) m/z calcd for C13H16O3Na, (M + Na)+ 243.0997, found 243.0992.
(Z)-5-(2-Hydroxyphenyl)pent-2-enyl acetate ((Z)-19a)
Following the procedure described for the preparation of (E)-19a, alcohol (Z)-36 (100 mg, 0.561 mmol) was acetylated using acetic anhydride (0.16 mL, 1.7 mmol) to give a residue that was purified by flash chromatography (80:20 hexanes-Et2O) to provide acetate (Z)-19a (93 mg, 0.42 mmol, 75%) as a clear colorless oil: 1H NMR (500 MHz, CDCl3) δ 7.11–7.08 (m, 2H), 6.88 (t, J = 7.4, 1H), 6.80 (d, J = 7.7, 1H), 5.78–5.68 (br m, 2H), 5.58–5.53 (m, 1H), 4.59 (d, J = 7.2, 2H), 2.69 (t, J = 7.4, 2H), 2.45 (app q, J = 8.0, 2H), 2.07 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 171.6 (C), 154.0 (C), 134.8 (CH), 130.5 (CH), 127.6 (CH), 127.4 (C), 123.9 (CH), 120.7 (CH), 115.7 (CH), 60.5 (CH2), 30.4 (CH2), 28.1 (CH2), 21.2 (CH3); IR (thin film) 3306, 3026, 2929, 1731, 1609, 754 cm−1; HRMS (ESI) m/z calcd for C13H16O3Na, (M + Na)+ 243.0997, found 243.0997.
(E)-5-(5-Bromo-2-hydroxyphenyl)pent-2-enyl acetate ((E)-19b)
Following the procedure described for the preparation of (E)-19a, allylic alcohol (E)-40 (100 mg, 0.389 mmol) was acetylated using acetic anhydride (0.11 mL, 1.2 mmol) to give a residue that was purified by flash chromatography (80:20 to 75:25 pentane-Et2O) to provide acetate (E)-19b (89 mg, 0.30 mmol, 77%) as a pale yellow oil: 1H NMR (500 MHz, CDCl3) δ 7.21 (s, 1H), 7.18–7.16 (m, 1H), 6.65 (d, J = 8.4, 1H), 5.82 (dt, J = 15.4, 6.8, 1H), 5.59 (dt, J = 15.4, 6.5, 1H), 5.28 (br s, 1H), 4.52 (d, J = 6.4, 2H), 2.68 (t, J = 7.4, 2H), 2.36 (app q, J = 7.3, 2H), 2.08 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 171.5 (C), 152.9 (C), 135.3 (CH), 133.0 (CH), 130.4 (C), 130.0 (CH), 124.8 (CH), 117.2 (CH), 112.8 (C), 65.4 (CH2), 32.3 (CH2), 29.5 (CH2), 21.2 (CH3); IR (thin film) 3459, 3021, 2924, 1710, 1269, 966 cm−1; HRMS (ESI) m/z calcd for C13H15BrO3Na, (M + Na)+ 321.0102, found 321.0105.
(E)-5-(2-Hydroxy-5-methoxyphenyl)pent-2-enyl acetate ((E)-19c)
Following the procedure described for the preparation of (E)-19a, allylic alcohol (E)-42 (60 mg, 0.29 mmol) was acetylated using acetic anhydride (54 μL, 0.58 mmol) to give a residue that was purified by flash chromatography (90:10 hexanes-EtOAc) to provide acetate (E)-19c (52 mg, 0.21 mmol, 72%) as a clear colorless oil: 1H NMR (500 MHz, CDCl3) δ 6.71–6.62 (m, 3H), 5.85 (dt, J = 15.3, 6.7, 1H), 5.60 (dt, J = 15.3, 6.4, 1H), 4.60 (br s, 1H), 4.52 (d, J = 6.4, 2H), 3.76 (s, 3H), 2.69 (t, J = 7.5, 2H), 2.38 (app q, J = 7.2, 2H), 2.07 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 171.2 (C), 153.8 (C), 147.6 (C), 135.7 (CH), 129.1 (C), 124.6 (CH), 116.2 (CH), 116.0 (CH), 112.0 (CH), 65.4 (CH2), 55.8 (CH3), 32.6 (CH2), 30.0 (CH2), 21.2 (CH3); IR (thin film) 3397, 3028, 2917, 1708, 1611, 801 cm−1; HRMS (ESI) m/z calcd for C14H18O4Na, (M + Na)+ 273.1103, found 273.1109.
Synthesis of Deuterium-Labeled Cyclization Precursors (E)-1-d-11a and (E)-1-d-19a
tert-Butyl-(2-(4,4-dibromobut-3-enyl)phenoxy)dimethylsilane (56)
A 250-mL round-bottomed flask was charged with alcohol 34 (12.0 g, 39.2 mmol). Dimethylformamide (26 mL) was added by syringe, followed by imidazole (5.34 g, 78.4 mmol), then TBSCl (6.80 g, 45.1 mmol). The reaction mixture was stirred for 2 h, then water (100 mL) was added. The resulting mixture was extracted with Et2O (4 × 75 mL). The combined organic extracts were dried over Na2SO4, filtered, then concentrated in vacuo to give a light yellow residue that was purified by flash chromatography (99.75:0.25 hexanes-Et2O) to provide silyl ether 56 (14.2 g, 33.7 mmol, 86%) as a clear colorless oil: 1H NMR (500 MHz, CDCl3) δ 7.16–7.11 (m, 2H), 6.94 (t, J = 6.6, 1H), 6.83 (d, J = 8.0, 1H), 6.44 (t, J = 7.1, 1H), 2.75 (t, J = 7.5, 2H), 2.42 (app q, J = 7.5, 2H), 1.06 (s, 9H), 0.29 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 153.8 (C), 138.3 (CH), 131.2 (C), 130.4 (CH), 127.5 (CH), 121.3 (CH), 118.6 (CH), 89.1 (C), 33.6 (CH2), 28.8 (CH2), 26.0 (CH3), 18.4 (C), −4.0 (CH3); IR (thin film) 3020, 2955, 1600, 922, 810 cm−1; HRMS (CI) m/z calcd for C16H28Br2NOSi, (M + NH4)+ 436.0307, found 436.0291.
Methyl 5-(2-(tert-butyldimethylsilyloxy)phenyl)pent-2-ynoate (57)
Using a modification of a procedure by Kinoshita,33 a solution of bromide 56 (14.2 g, 33.7 mmol) in THF (110 mL) was cooled to −78 °C in a 1-L round-bottomed flask. A solution of n-BuLi in hexane (1.6 M, 52 mL, 84 mmol) was added to the clear solution via syringe over 20 min. The pale yellow reaction mixture was stirred at −78 °C for 30 min then methyl chloroformate (7.80 mL, 101 mmol) was added over via syringe over 5 min. The reaction mixture was allowed to warm to 0 °C and stirred for 10 min, then allowed to warm to ambient temperature and stirred for 15 min. Saturated aqueous NH4Cl (120 mL) was added and the resulting mixture was extracted with Et2O (3 × 75 mL), dried over Na2SO4, filtered, and concentrated in vacuo to give a pale yellow residue that was purified by flash chromatography (99:1 hexanes-Et2O) to provide ynoate 57 (10.6 g, 33.3 mmol, 99%) as a pale yellow oil: 1H NMR (500 MHz, CDCl3) δ 7.17 (d, J = 7.5, 1H), 7.14–7.11 (m, 1H), 6.91 (t, J = 7.5, 1H), 6.80 (d, J = 8.1, 1H), 3.76 (s, 3H), 2.88 (t, J = 7.7, 2H), 2.61 (t, J = 7.7, 2H), 1.03 (s, 9H), 0.26 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 154.4 (C), 153.7 (C), 130.5 (CH), 130.2 (C), 127.9 (CH), 121.2 (CH), 118.5 (CH), 89.5 (C), 73.2 (C), 52.7 (CH3), 29.4 (CH2), 25.9 (CH3), 19.3 (CH2), 18.3 (C), −4.0 (CH3); IR (thin film) 3011, 2931, 2239, 1718, 1435, 1108 cm−1; HRMS (ESI) m/z calcd for C18H26O3SiNa, (M + Na)+ 341.1549, found 341.1544.
5-(2-(tert-Butyldimethylsilyloxy)phenyl)-1,1-bis-deuteropent-2-yn-1-ol (58)
In a 1-L round-bottomed flask, a solution of ynoate 57 (9.0 g, 28 mmol) in Et2O (140 mL) was cooled to 0 °C. Lithium aluminum deuteride (1.76 g, 42.0 mmol) was added portion-wise over 30 min and gas evolution was observed. After the reaction mixture was stirred at 0 °C for 45 min, water (50 mL) was added slowly, followed by aqueous NaOH solution (0.1 N, 20 mL), followed by more water (50 mL). The resulting mixture was allowed to warm to ambient temperature, extracted with Et2O (4 × 75 mL), dried over Na2SO4, filtered, and concentrated in vacuo to give a residue that was purified by flash chromatography (90:10 hexanes-EtOAc) to provide propargyl alcohol 58 (6.23 g, 21.1 mmol, 75%) as a clear colorless oil. Analysis by electrospray mass spectrometry indicated a 327:5:1 ratio of d2-58:d1-58:d0-58: 1H NMR (500 MHz, CDCl3) δ 7.19 (d, J = 7.5, 1H), 7.13–7.10 (m, 1H), 6.91 (t, J = 7.4, 1H), 6.81 (d, J = 8.1, 1H), 2.83 (t, J = 7.7, 2H), 2.50 (t, J = 7.6, 2H), 1.05 (s, 9H), 0.26 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 153.7 (C), 131.2 (C), 130.5 (CH), 127.5 (C), 121.1 (CH), 118.5 (CH), 86.2 (C), 78.8 (C), 50.9 (quintet, JCD = 22.4, CD2), 30.3 (CH2), 26.0 (CH2), 19.4 (CH3), 18.3 (C), −4.0 (CH3); IR (thin film) 3243, 3030, 2929, 2248, 1453, 1175 cm−1; HRMS (ESI) m/z calcd for C17H24D2O2SiNa, (M + Na)+ 315.1725, found 315.1725.
(Z)-5-(2-(tert-Butyldimethylsilyloxy)phenyl)-1,1-bisdeuteropent-2-en-1-ol (59)
Following the procedure described for the preparation of (Z)-36,35 propargyl alcohol 58 (3.65 g, 12.5 mmol) was reduced using catalytic Ni(OAc)2·4H2O (466 mg, 1.87 mmol) to give a residue that was purified by filtering the reaction mixture through a short pad of silica gel using EtOH as the eluent. The organic layer was concentrated in vacuo to yield alkene 59 (3.68 g, 12.5 mmol, 100%) as a pale yellow oil. Analysis by electrospray mass spectrometry indicated a 226:4:1 ratio of d2-59:d1-59:d0-59: 1H NMR (500 MHz, CDCl3) δ 7.13–7.08 (m, 2H), 6.90 (t, J = 7.4, 1H), 6.82 (d, J = 7.9, 1H), 5.61–5.59 (m, 2H), 2.69 (t, J = 7.5, 2H), 2.39 (app q, J = 7.2, 2H), 1.05 (s, 9H), 0.26 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 153.7 (C), 132.1 (CH), 132.1 (C), 130.7 (CH), 129.1 (C), 127.2 (CH), 120.9 (CH), 118.5 (CH), 57.8 (quintet, JCD = 21.5, CD2), 30.5 (CH2), 27.8 (CH2), 25.9 (CH3), 18.3 (C), −4.0 (CH3); IR (thin film) 3338, 3017, 2930, 1107, 755, 701 cm−1; HRMS (ESI) m/z calcd for C17H26D2O2SiNa, (M + Na)+ 317.1882, found 317.1884.
(Z)-5-(2-(tert-Butyldimethylsilyloxy)phenyl)-1-deuteropent-2-enal ((Z)-60)
In a 100-mL round-bottomed flask, a solution of 59 (1.0 g, 3.4 mmol) in CH2Cl2 (34 mL) was cooled to 0 °C. N,N-Diisopropylethylamine (4.1 mL, 24 mmol) was added via syringe and the solution was maintained at 0 °C for 10 min. DMSO (2.4 mL, 34 mmol) was added via syringe and the solution was maintained at 0 °C for 10 min, then SO3·pyridine (2.16 g, 13.6 mmol) was added in a single portion. The reaction mixture was stirred at 0 °C for 45 min, then saturated aqueous NaHCO3 (40 mL) was added and the resulting mixture was extracted with CH2Cl2 (3 × 30 mL). The organic layer was washed with water (2 × 30 mL), dried over Na2SO4, filtered, and concentrated in vacuo to give a pale yellow residue. After azeotropic removal of pyridine with toluene (2 × 100 mL), the residue (988 mg, 3.37 mmol, 99%, 90:10 Z:E by 1H NMR analysis) was carried directly to the next synthetic step. Analysis by electrospray mass spectrometry indicated a >96:4 ratio of d1-(Z)-60:d0-(Z)-60: 1H NMR (500 MHz, CDCl3) δ 7.13–7.05 (m, 2H), 6.89 (t, J = 7.3, 1H), 6.82 (d, J = 8.3, 1H), 6.69–6.63 (m, 1H), 5.94 (d, J = 11.2, 1H), 2.89 (app q, J = 7.5, 2H), 2.81 (t, J = 7.3, 2H), 1.03 (s, 9H), 0.25 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 190.6 (t, JCD = 25.7, CDO), 153.7 (C), 152.3 (CH), 130.6 (CH), 130.6 (C), 130.5 (CH), 127.7 (CH), 121.2 (CH), 118.6 (CH), 30.3 (CH2), 28.4 (CH2), 25.9 (CH3), 18.3 (C), −4.0 (CH3); IR (thin film) 3023, 2955, 2857, 1668, 1252, 927 cm−1; HRMS (ESI) m/z calcd for C17H25DO2SiNa, (M + Na)+ 314.1663, found 314.1659.
(E)-5-(2-(tert-Butyldimethylsilyloxy)phenyl)-1-deuteropent-2-enal ((E)-60)
In a 25-mL round-bottomed flask, (Z)-60 (938 mg, 3.22 mmol) was dissolved in hexanes (10 mL). Silica gel (500 mg) and pyridine (0.20 mL, 2.5 mmol) were added in a single portion and the slurry was stirred at ambient temperature for 15 h. The reaction mixture was filtered and concentrated in vacuo, then pyridine was azeotropically removed with toluene (2 × 25 mL) to give aldehyde (E)-60 (938 mg, 3.22 mmol, 100%, >20:1 E:Z by 1H NMR analysis) as a clear colorless oil. Analysis by electrospray mass spectrometry indicated a >96:4 ratio of d1-(E)-60:d0-(E)-60: 1H NMR (500 MHz, CDCl3) δ 7.13–7.10 (m, 2H), 6.91–6.82 (m, 2H), 6.81 (d, J = 7.8, 1H), 6.14 (d, J = 15.5, 1H), 2.80 (t, J = 7.3, 2H), 2.63 (app q, J = 6.8, 2H), 1.02 (s, 9H), 0.25 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 193.9 (t, JCD = 26.3, CDO), 158.2 (CH), 153.7 (C), 133.1 (t, JCD = 3.8, CH), 131.0 (C), 130.2 (CH), 127.6 (CH), 121.3 (CH), 118.6 (CH), 33.1 (CH2), 29.2 (CH2), 25.9 (CH3), 18.3 (C), –4.0 (CH3); IR (thin film) 3033, 2930, 2857, 1711, 1491, 1146 cm−1; HRMS (ESI) m/z calcd for C17H25DO2SiNa, (M + Na)+ 314.1663, found 314.1655.
(S,E)-5-(2-(tert-Butyldimethylsilyloxy)phenyl)-1-deuteropent-2-en-1-ol ((E)-61).31
Using a modification of a procedure by Keck,19 titanium isopropoxide (89 μL, 0.32 mmol) was added to a stirring mixture of (R)-BINOL (184 mg, 0.644 mmol), trifluoroacetic acid (0.5 M in CH2Cl2, 0.13 mL, 0.064 mmol), molecular sieves (4 Å, 1.7 g) and diethyl ether (12 mL) in a 100-mL round-bottomed flask. The red-orange mixture was heated to reflux for 1 h, allowed to cool to room temperature, and α,β-unsaturated aldehyde (E)-60 (938 mg, 3.22 mmol) in Et2O (10 mL) was added. The resulting mixture was stirred at room temperature for 10 min, then cooled to −78 °C and Bu3SnH (1.0 mL, 3.9 mmol) was added dropwise. The reaction mixture was then transferred to a −20 °C freezer and left for 18 h, without stirring. The mixture was removed from the freezer and saturated aqueous NaHCO3 (10 mL) was added. The mixture was stirred for 1 h at room temperature, then filtered through a pad of celite. The layers were separated, and the aqueous layer was extracted with Et2O (3 × 5 mL). The combined organic layers were washed with water (2 × 5 mL), dried over Na2SO4, filtered, and concentrated in vacuo to give a residue that was purified by flash chromatography (88:12 hexanes-Et2O) to provide allylic alcohol (E)-61 (488 mg, 1.67 mmol, 62% brsm) as a clear colorless oil. A modified Mosher's ester analysis12 of the product showed it to be of 65% enantiomeric excess and predominantly the (S) enantiomer. Analysis by electrospray mass spectrometry indicated a >96:4 ratio of d1-(E)-61:d0-(E)-61: [α]D25 −2.7, [α]57725 −3.6, [α]54625 −3.8, [α]43525 −3.3, (c = 0.33, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 7.12 (d, J = 7.4, 1H), 7.08 (t, J = 7.9, 1H), 6.88 (t, J = 7.3, 1H), 6.79 (d, J = 7.9, 1H), 5.75 (dt, J = 15.5, 6.5, 1H), 5.66 (dd, J = 15.4, 5.8, 1H), 4.07 (br s, 1H), 2.68 (t, J = 7.6, 2H), 2.34 (app q, J = 7.8, 2H), 1.03 (s, 9H), 0.25 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 153.7 (C), 133.1 (CH), 132.4 (C), 130.4 (CH), 129.4 (CH), 127.1 (CH), 121.1 (CH), 118.5 (CH), 63.6 (t, JCD = 22.5, CDH), 32.8 (CH2), 30.5 (CH2), 26.0 (CH3), 18.4 (C), −4.0 (CH3); IR (thin film) 3338, 3022, 2929, 1105, 755, 701 cm−1; HRMS (ESI) m/z calcd for C17H27DO2SiNa, (M + Na)+ 316.1819, found 316.1818.
(S,E)-2-(5-Deutero-5-hydroxypent-3-enyl)phenol ((E)-62)
In a 25-mL round-bottomed flask, silyl ether (E)-61 (468 mg, 1.59 mmol) was dissolved in THF (8.0 mL). A solution of TBAF (1.0 M in THF, 1.9 mL, 1.9 mmol) was added via syringe. After 18 h, the reaction mixture was concentrated in vacuo to give a pale yellow residue that was purified by flash chromatography (90:10 to 85:15 CH2Cl2-acetone) to provide diol (E)-62 (285 mg, 1.59 mmol, 100%) as a clear colorless oil. Analysis by electrospray mass spectrometry indicated a >96:4 ratio of d1-(E)-62:d0-(E)-62: [α]D25 −8.3, [α]57725 −8.5, [α]54625 −10.7, [α]43525 −3.9, (c = 0.11, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 7.12–7.06 (m, 2H), 6.86 (t, J = 7.9, 1H), 6.78 (d, J = 7.7, 1H), 6.62 (br s, 1H), 5.77 (dt, J = 15.4, 6.5, 1H), 5.65 (dd, J = 15.5, 5.8, 1H), 4.07 (d, J = 5.4, 1H), 2.72 (t, J = 7.8, 2H), 2.51 (br s, 1H), 2.38 (app q, J = 7.2, 2H); 13C NMR (125 MHz, CDCl3) δ 154.0 (C), 133.4 (CH), 130.3 (CH), 128.8 (CH), 128.2 (C), 127.3 (CH), 120.5 (CH), 115.5 (CH), 63.4 (t, JCD = 21.3, CDH), 32.5 (CH2), 29.8 (CH2); IR (thin film) 3336, 3070, 2922, 1180, 1042, 973 cm−1; HRMS (ESI) m/z calcd for C11H13DO2Na, (M + Na)+ 202.0954, found 202.0951.
(S,E)-1-Deutero-5-(2-hydroxyphenyl)pent-2-enyl 2′,2′,2′-trichloroacetimidate ((E)-1-d-11a)
Following the procedure described for the preparation of (E)-11a, alcohol (E)-62 (100 mg, 0.558 mmol) was treated with DBU and Cl3CCN. After 30 min, the reaction mixture was concentrated in vacuo and the residue was purified by flash chromatography (97:2:1 toluene-acetone-Et3N) to provide imidate (E)-1-d-11a (101 mg, 0.312 mmol, 56%) as a pale yellow oil. Analysis by electrospray mass spectrometry indicated a >96:4 ratio of d1-(E)-1-d-11a:d0-(E)-1-d-11a: [α]D25 −43.8, [α]57725 −40.2, [α]54625 −27.2, [α]43525 −24.6, (c = 0.17, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 8.30 (br s, 1H), 7.13–7.08 (m, 2H), 6.88 (t, J = 7.5, 1H), 6.76 (d, J = 8.0, 1H), 5.96 (dt, J = 15.4, 6.7, 1H), 5.86 (br s, 1H), 5.73 (dd, J = 15.5, 6.1, 1H), 4.76 (d, J = 5.4, 1H), 2.75 (t, J = 7.8, 2H), 2.43 (app q, J = 7.3, 2H); 13C NMR (125 MHz, CDCl3) δ 163.1 (C), 153.8 (C), 136.4 (CH), 130.4 (CH), 127.9 (C), 127.3 (CH), 123.4 (CH), 120.7 (CH), 115.4 (CH), 91.6 (C), 69.9 (t, JCD = 21.3, CDH), 32.5 (CH2), 29.6 (CH2); IR (thin film) 3332, 3036, 2928, 1660, 1593, 972 cm−1; HRMS (ESI) m/z calcd for C13H13DCl3NO2Na, (M + Na)+ 345.0051, found 345.0056.
(S,E)-1-Deutero-5-(2-hydroxyphenyl)pent-2-enyl acetate ((E)-1-d-19a)
Following the procedure described for the preparation of acetate (E)-19a, allylic alcohol (E)-62 (50 mg, 0.28 mmol) was acetylated using acetic anhydride (80 μL, 0.84 mmol) to give a residue that was purified by flash chromatography (85:15 to 80:20 pentane-Et2O) to provide acetate (E)-1-d-19a (55 mg, 0.25 mmol, 89%) as a clear colorless oil. Analysis by electrospray mass spectrometry indicated a >96:4 ratio of d1-(E)-1-d-19a:d0-(E)-1-d-19a: [α]D25 −16.8, [α]57725 −18.4, [α]54625 −15.3, (c = 0.16, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 7.12–7.06 (m, 2H), 6.88 (t, J = 7.4, 1H), 6.76 (d, J = 7.9, 1H), 5.86 (dt, J = 15.6, 6.5, 1H), 5.60 (dd, J = 15.6, 6.4, 1H), 4.74 (br s, 1H), 4.50 (d, J = 5.8, 1H), 2.72 (t, J = 7.5, 2H), 2.39 (app q, J = 7.0, 2H), 2.07 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 171.2 (C), 153.6 (C), 135.9 (CH), 130.4 (CH), 127.8 (C), 127.4 (CH), 124.5 (CH), 121.0 (CH), 115.5 (CH), 65.1 (t, JCD = 21.3, CDH), 32.6 (CH2), 29.7 (CH2), 21.2 (CH3); IR (thin film) 3408, 3035, 2918, 1708, 1242, 969 cm−1; HRMS (ESI) m/z calcd for C13H15DO3Na, (M + Na)+ 244.1060, found 244.1054.
(S,E)-5-(2-(tert-Butyldimethylsilyloxy)phenyl)-1-deuteropent-2-enyl (R)-3′,3′,3′-trifluoro-2′-methoxy-2′-phenylpropanoate ((1S,2′R)-(E)-63)
In a 5-mL round-bottomed flask, allylic alcohol (E)-61 (5 mg, 0.02 mmol), DMAP (0.2 mg, 0.002 mmol), and (R)-2-trifluoromethyl-2-methoxy-2-phenylacetic acid (5 mg, 0.02 mmol) were dissolved in CH2Cl2 (0.34 mL). N,N′-Dicyclohexylcarbodiimide (5 mg, 0.02 mmol) was added at room temperature and the reaction mixture was stirred for 18 h. Saturated aqueous NaHCO3 (1 mL) was added and the resulting mixture was extracted with Et2O (3 × 0.5 mL), dried over Na2SO4, filtered, and concentrated in vacuo to give a residue that was purified by flash chromatography (95:5 pentane-Et2O). The ester product (7 mg, 0.01 mmol, 80%) was obtained as a 82.5:17.5 mixture of (1S,2′R)-(E)-63:(1R,2′R)-(E)-63 as a clear colorless oil. Analysis by electrospray mass spectrometry indicated a >96:4 ratio of d1-(1S,2′R)-(E)-63:d0-(1S,2′R)-(E)-63: Major diastereomer (1S,2′R)-(E)-63: [α]D25 28.1, [α]57725 28.7, [α]54625 31.9, (c = 1.10, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 7.53–7.52 (m, 2H), 7.41–7.40 (m, 3H), 7.10–7.06 (m, 2H), 6.87 (d, J = 6.7, 1H), 6.78 (d, J = 7.9, 1H), 5.90 (dt, J = 15.3, 6.7, 1H), 5.62 (dd, J = 15.4, 6.6, 1H), 4.71 (d, J = 6.5, 1H), 3.55 (s, 3H), 2.67 (t, J = 7.5, 2H), 2.35 (app q, J = 7.1, 2H), 1.01 (s, 9H), 0.24 (s, 6H); 2H NMR (61 MHz, CDCl3) δ 4.81 (s, 12H); 13C NMR (125 MHz, CDCl3) δ 166.5 (C), 153.7 (C), 138.0 (CH), 132.5 (C), 132.0 (C), 130.3 (CH), 129.7 (CH), 128.5 (CH), 127.5 (CH), 127.2 (CH), 124.6 (C), 122.8 (CH), 122.3 (C), 121.1 (CH), 118.5 (CH), 66.8 (t, JCD = 23.5, CDH), 55.6 (CH3), 32.7 (CH2), 30.2 (CH2), 25.9 (CH3), 18.3 (C), −4.0 (CH3); IR (thin film) 3034, 2930, 1748, 1453, 1253, 1169 cm−1; HRMS (ESI) m/z calcd for C27H34DF3O4SiNa, (M + Na)+ 532.2217, found 532.2202.
(S,E)-5-(2-(tert-Butyldimethylsilyloxy)phenyl)-1-deuteropent-2-enyl (S)-3′,3′,3′-trifluoro-2′-methoxy-2′-phenylpropanoate ((1S,2′S)-(E)-63)
Following the procedure described for the preparation of (1S,2′R)-(E)-63, allylic alcohol (E)-61 (5 mg, 0.02 mmol) was acylated using (S)-2-trifluoromethyl-2-methoxy-2-phenylacetic acid (5 mg, 0.02 mmol) to give a residue that was purified by flash chromatography (95:5 pentane-Et2O). The ester product (6 mg, 0.01 mmol, 69%) was obtained as a 82.5:17.5 mixture of (1S,2′S)-(E)-63:(1R,2′S)-(E)-63 as a clear colorless oil. Analysis by electrospray mass spectrometry indicated a >96:4 ratio of d1-(1S,2′S)-(E)-63:d0-(1S,2′S)-(E)-63: Major diastereomer (1S,2′S)-(E)-63: [α]D25 −28.6, [α]57725 −30.8, [α]54625 −35.7, (c = 1.60, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 7.54–7.52 (m, 2H), 7.41–7.40 (m, 3H), 7.10–7.06 (m, 2H), 6.87 (d, J = 7.3, 1H), 6.79 (d, J = 7.9, 1H), 5.90 (dt, J = 15.3, 6.6, 1H), 5.62 (dd, J = 15.3, 6.5, 1H), 4.77 (d, J = 6.2, 1H), 3.55 (s, 3H), 2.67 (t, J = 7.5, 2H), 2.35 (app q, J = 7.0, 2H), 1.01 (s, 9H), 0.24 (s, 6H); 2H NMR (61 MHz, CDCl3) δ 4.74 (s, 12H); 13C NMR (125 MHz, CDCl3) δ 166.5 (C), 153.7 (C), 138.0 (CH), 132.5 (C), 132.0 (C), 130.3 (CH), 129.7 (CH), 128.5 (CH), 127.5 (CH), 127.2 (CH), 124.6 (C), 122.8 (CH), 122.3 (C), 121.1 (CH), 118.5 (CH), 66.8 (t, JCD = 21.9, CDH), 55.6 (CH3), 32.7 (CH2), 30.2 (CH2), 25.9 (CH3), 18.4 (C), −4.0 (CH3); IR (thin film) 3032, 2930, 1748, 1453, 1254, 1169 cm−1; HRMS (ESI) m/z calcd for C27H34DF3SiNa, (M + Na)+ 532.2217, found 532.2198.
Supplementary Material
Acknowledgement
We thank the NSF (CHE-9726471) for financial support. Additional unrestricted support from Amgen, Merck and Pfizer is also gratefully acknowledged. The NIH Predoctoral Fellowship Program (1F31GM089137) and the UC Irvine Faculty Mentor Program are acknowledged for financial support of ACO, and BMS for fellowship support for JSC. Computational studies were performed on hardware purchased with funding from CRIF (CHE-0840513), and NMR and mass spectra were obtained at UC Irvine using instrumentation acquired with the assistance of NSF and NIH shared instrumentation programs. Three-dimensional renderings of computed structures were generated using the CYLview program.41
Footnotes
Supporting Information Available. General experimental methods, tables of additional optimization experiments and reaction of imidate 16 with Pd(OAc)2, synthetic schemes for the synthesis of cyclization precursors, 1H and 13C NMR data for new compounds, and copies of chromatography traces used to determine enantiomeric purity. General computational details, calculated close contacts in transition structures 22, 25, 28, and 31, three-dimensional models of 28 and 31, and XYZ coordinates. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.The systematic name for chromane is 3,4-dihydro-2H-1-benzopyran.
- 2.A substructure search in SciFinder of chromane structure 4 (R1 = an alkyl side chain and the oxacyclic ring otherwise unsubstituted) and substance role = biological study finds >2800 compounds.
- 3.For two illustrative examples, see:Carreño MC, Hernándes-Torres G, Urbano A, Colobert F. Eur. J. Org. Chem. 2008;12:2035–2038.. Liu Y-C, Sperry J, Rathwell DCK, Brimble MA. Synlett. 2009;5:793–797..
- 4.Pd(0)-catalyzed synthesis of 2-vinyl-2H-1-benzopyrans: Trost BM, Shen HC, Dong L, Surivet J-P, Sylvain C. J. Am. Chem. Soc. 2004;126:11966–11983. doi: 10.1021/ja048078t.. Trost BM, Shen HC, Dong L, Surivet J-P. J. Am. Chem. Soc. 2003;125:9276–9277. doi: 10.1021/ja036052g.. Labrosse J-R, Poncet C, Lhoste P, Sinou D. Tetrahedron: Asymmetry. 1999:1069–1078.
- 5.Ir(I)-catalyzed synthesis of 2-vinyl-2H-1-benzopyrans: Welter C, Dahnz A, Brunner B, Streiff S, Dübon P, Helmchen G. Org. Lett. 2005;7:1239–1242. doi: 10.1021/ol047351t.
- 6.(a) Ito K, Imahayashi Y, Kuroda T, Eno S, Saito B, Katsuki T. Tetrahedron Lett. 2004;45:7277–7281. [Google Scholar]; (b) Massacret M, Lakhmiri R, Lhoste P, Nguefack C, Abdelouahab FBB, Fadel R, Sinou D. Tetrahedron: Asymmetry. 2000;11:3561–3568. [Google Scholar]; (c) Massacret M, Lhoste P, Lakhmiri R, Parella T, Sinou D. Eur. J. Org. Chem. 1999:2665–2673. [Google Scholar]; (d) Lhoste P, Massacret M, Sinou D. Bull. Soc. Chim. Fr. 1997;134:343–347. [Google Scholar]; (e) Yamazaki A, Achiwa I, Achiwa K. Tetrahedron: Asymmetry. 1996;7:403–406. [Google Scholar]; (f) Massacret M, Goux C, Lhoste P, Sinou D. Tetrahedron Lett. 1994;35:6093–6096. [Google Scholar]; Pd(0)-catalyzed synthesis of 2-vinylbenzodioxanes and 2-vinyl-2H-1,4-benzoxazines
- 7.(a) Kirsch SF, Overman LE, White NS. Org. Lett. 2007;9:911–913. doi: 10.1021/ol070110b. [DOI] [PubMed] [Google Scholar]; (b) Cannon JS, Kirsch SF, Overman LE, Sneddon HF. J. Am. Chem. Soc. 2010;132:15192–15203. doi: 10.1021/ja106688j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olson AC, Overman LE, Sneddon HF, Ziller JW. Adv. Synth. Catal. 2009;351:3186–3192. doi: 10.1002/adsc.200900678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Stevens AM, Richards CJ. Organometallics. 1999;18:1346–1348. [Google Scholar]; (b) Anderson CE, Kirsch SF, Overman LE, Richards CJ, Watson MP. Org. Synth. 2007;84:148–155. [Google Scholar]; (c) Anderson CE, Overman LE, Richards CJ, Watson MP, White N. Org. Synth. 2007;84:139–147. [Google Scholar]
- 10.Prepared using conventional chemistry, see the Experimental Section
- 11.Anderson CE, Overman LE. J. Am. Chem. Soc. 2003;125:12412–12413. doi: 10.1021/ja037086r. [DOI] [PubMed] [Google Scholar]
- 12.See Supporting Information for more details
- 13.When imidate 15 was subjected to reaction conditions in the absence of catalyst, 2-vinylbenzodioxane 17 was not observed in the 1H NMR of the crude reaction mixture, indicating that trichloroacetamidate ionization followed by nucleophilic attack of phenol is not likely
- 14.Numata M, Sugimoto M, Koike K, Ogawa T. Carbohydr. Res. 1987;163:209–225. doi: 10.1016/0008-6215(87)80182-9. [DOI] [PubMed] [Google Scholar]
- 15.Carpenter NE, Overman LE. In: Organic Reactions. Overman LE, editor. Vol. 66. Wiley; Hoboken, NJ: 2005. pp. 1–107. [Google Scholar]
- 16.Other COP catalysts were found to be less effective.12
- 17.For example, see: Meyers C, Maes BUW, Loones KTJ, Bal G, Lemière GLF, Dommisse RA. J. Org. Chem. 2004;69:6010–6017. doi: 10.1021/jo049774e..
- 18.These entries represent a small selection of reaction optimization data. A complete report of solvents and additives examined can be found in the Supporting Information
- 19.Keck GE, Krishnamurthy D. J. Org. Chem. 1996;61:7638–7639. doi: 10.1021/jo961593s. [DOI] [PubMed] [Google Scholar]
- 20.For a recent comprehensive review of Pd(II)-catalyzed nucleopalladation of alkenes, see McDonald RI, Liu G, Stahl SS. Chem. Rev. 2011;111:2981–3019. doi: 10.1021/cr100371y..
- 21.(a) TURBOMOLE. V6.2. Turbomole GmbH; Karlsruhe, Germany: 2009. http://www.turbomole.com. [Google Scholar]; (b) Treutler O, Ahlrichs R. J. Chem. Phys. 1995;102:346–354. [Google Scholar]
- 22.(a) Slater JC. The Self-Consistent Field for Molecules and Solids. Vol. 4. McGraw-Hill; New York: 1974. Quantum Theory of Molecules and Solids. [Google Scholar]; (b) Vosko SH, Wilk L, Nusair M. Can. J. Phys. 1980;58:1200–1211. [Google Scholar]; (c) Becke AD. Phys. Rev. A. 1988;38:3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]; (d) Lee C, Yang W, Parr RG. Phys. Rev. B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]; (e) Miehlich B, Savin A, Stoll H, Preuss H. Chem. Phys. Lett. 1989;157:200–206. [Google Scholar]; (f) Becke AD. J. Chem. Phys. 1993;98:5648–5652. [Google Scholar]; (g) Stephens PJ, Devlin FJ, Chabalowski CF, Frisch MJ. J. Phys. Chem. 1994;98:11623–11627. [Google Scholar]
- 23.Klamt A, Schüürmann G. J. Chem. Soc., Perkin Trans. 1993;2:799–805. [Google Scholar]
- 24.(a) Eichkorn K, Weigend F, Treutler O, Ahlrichs R. Theor. Chem. Acc. 1997;97:119–124. [Google Scholar]; (b) Schäfer A, Huber C, Ahlrichs R. J. Chem. Phys. 1994;100:5829–5835. [Google Scholar]; (c) Weigend F, Furche F, Ahlrichs R. J. Chem. Phys. 2003;119:12753–12762. [Google Scholar]
- 25.For further computational details, see the Supporting Information
- 26.A table of calculated close contacts can be found in the Supporting Information
- 27.The corresponding models for transition structures 28 and 31 for cyclization of (Z)-11a can be found in the Supporting Information
- 28.The absolute configuration of this compound has been tentatively assigned based on the stereochemical assignment of products 1a and 1c, for which the absolute configuration has been established.4b
- 29.The change in absolute configuration is a consequence of different CIP priorities of substituents at the stereocenter
- 30.Birch AJ, Maung M, Pelter A. Aust. J. Chem. 1969;22:1923–1932. [Google Scholar]
- 31.Different batches of (E)-61, the precursor to (E)-1-d-11a and (E)-1-d-19a, were formed with different enantiomeric excesses. The enhancement of all batches was sufficient to determine the stereospecificity of the reactions depicted in eqs 3 and 4. The same level of stereospecificity was observed when starting imidates ((E)-1-d-11a) or acetates ((E)-1-d-19a) of different enantiomeric purity were exposed to the reaction conditions
- 32.The NMR data provided for this compound12 was obtained using a synthetic sequence that originated from (E)-61 with 72% ee. Therefore, the NMR spectra indicate that the major:minor isomer ratio of the product is approximately 86:14
- 33.Mori M, Tonogaki K, Kinoshita A. Org. Synth. 2005;81:1–13. [Google Scholar]
- 34.Jones TK, Denmark SE. Org. Synth. 1986;64:182–185. [Google Scholar]
- 35.(a) Taber DF, Herr RJ, Gleave DM. J. Org. Chem. 1997;62:194–198. doi: 10.1021/jo9616365. [DOI] [PubMed] [Google Scholar]; (b) Brown CA, Ahuja VK. J. Org. Chem. 1973;38:2226–2230. [Google Scholar]
- 36.Davies SG, Pyatt D. J. Organomet. Chem. 1990;387:381–390. [Google Scholar]
- 37.Verdaguer X, Berk SC, Buchwald SL. J. Am. Chem. Soc. 1995;117:12641–12642. [Google Scholar]
- 38.Caldwell JJ, Cameron ID, Christie SDR, Hay AM, Johnstone C, Kerr WJ, Murray A. Synthesis. 2005;19:3293–3296. [Google Scholar]
- 39.Marecak DM, Horiuchi Y, Arai H, Shimonaga M, Maki Y, Koyama T, Ogura K, Prestwich GD. Bioorg. Med. Chem. Lett. 1997;7:1973–1978. [Google Scholar]
- 40.Crich D, Krishnamurthy V, Brebion F, Karatholuvhu M, Subramanian V, Hutton TK. J. Am. Chem. Soc. 2007;129:10282–10294. doi: 10.1021/ja072969u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.CYLview, 1.0b. Legault, C. Y., Université de Sherbrooke, 2009; http://www.cylview.org.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.