

Abstract
Dosage compensation—equalizing gene expression levels in response to differences in gene dose or copy number—is classically considered to play a critical role in the evolution of heteromorphic sex chromosomes. As the X and Y diverge through degradation and gene loss on the Y (or the W in female-heterogametic ZW taxa), it is expected that dosage compensation will evolve to correct for sex-specific differences in gene dose. Although this is observed in some organisms, recent genome-wide expression studies in other taxa have revealed striking exceptions. In particular, reports that both birds and the silkworm moth (Bombyx mori) lack dosage compensation have spurred speculation that this is the rule for all female-heterogametic taxa. Here, we revisit the issue of dosage compensation in silkworm by replicating and extending the previous analysis. Contrary to previous reports, our efforts reveal a pattern typically associated with dosage compensated taxa: the global male:female expression ratio does not differ between the Z and autosomes. We believe the previous report of unequal male:female ratios on the Z reflects artifacts of microarray normalization in conjunction with not testing a major assumption that the male:female global expression ratio was unbiased for autosomal loci. However, we also find that the global Z chromosome expression is significantly reduced relative to autosomes, a pattern not expected in dosage compensated taxa. This combination of male:female parity with an overall reduction in expression for sex-linked loci is not consistent with the prevailing evolutionary theory of sex chromosome evolution and dosage compensation.
Keywords: sex chromosomes, lepidoptera, microarray, female heterogamety
Introduction
In species with heteromorphic sex chromosomes, the dose of most sex-linked genes differs by a factor of two between the sexes. For instance, consider the evolution of sex chromosomes in male-heterogametic taxa (where males are XY and females are XX). The X and Y chromosomes are initially homologous, but differentiation and degradation of the Y chromosomes results in females retaining two copies of X-linked genes, whereas males have only one (Charlesworth B and Charlesworth D 2000; Charlesworth et al. 2005). Without a mechanism to compensate for this difference in gene dose, global gene expression levels of the sex chromosome relative to the autosomes will be unbalanced between the sexes (Muller 1950; Ohno 1967; Charlesworth 1978; Rice 1987). Having an uncompensated X chromosome in males has classically been considered evolutionarily untenable because of assumed fitness costs associated with functional aneuploidy. It was therefore widely accepted for many years that the evolution of some mechanism for dosage compensating gene expression between males and females was an inescapable consequence of sex chromosome differentiation (Marín et al. 2000; Straub and Becker 2007; Mank 2009; Vicoso and Bachtrog 2009). This scenario applies equally to female-heterogametic taxa where males are ZZ and females are ZW.
The pattern classically associated with sex chromosome dosage compensation is an equal male:female (M:F) global expression ratio for the sex chromosome (Muller 1950; Meyer and Casson 1986; Straub and Becker 2007; Johnston et al. 2008). That an organism would “need” dosage compensation is often justified by the argument that a M:F imbalance in sex chromosome expression cannot be tolerated by finely tuned gene networks involving both sex-linked and autosomal loci (Marín et al. 2000; Straub and Becker 2007). Thus, selection will act to balance M:F global expression between sex chromosomes. However, this explanation obscures a subtle but fundamental distinction between how dosage compensation is diagnosed and the underlying evolutionary process that causes it (Vicoso and Bachtrog 2009). The pattern of equal M:F expression typically used to diagnose dosage compensation is only indirectly related to the causative evolutionary pressures.
To speak of “selection for dosage compensation” is potentially confusing because variation in the M:F ratio of the sex chromosomes per se cannot influence the differential survival and reproduction of an “individual” male or female. Rather, as the sex chromosomes diverge, there will be stabilizing selection to maintain the optimum (and presumably ancestral) global expression ratio between autosomes and the sex chromosome within an individual, either male or female (Ohno 1967; Charlesworth 1978; Rice 1987). Here, it is assumed that sex chromosomes arise from an ancestral state as normal autosomes with equal representation in both sexes and that stabilizing selection will act to maintain as “optimal” the ancestral global expression ratios. Such stabilizing selection should move the sex chromosome to autosome ratio toward unity in the heterogametic sex, though this assumes ancestral global expression levels were on average equal across the chromosomes. Depending on the molecular mechanism by which it is achieved, increased expression of sex-linked loci in the heterogametic sex may also raise expression for the same loci in the homogametic sex, moving females (of an XY species) away from the ancestrally optimum X:autosomal (X:A) ratio as males move toward it (Rice 1987). In such cases, the same stabilizing selective pressures will subsequently act on females to reduce expression. Thus, sexually antagonistic pleiotropy likely plays a key role in the evolutionary dynamics of dosage compensation (Rice 1987; Haig 2006; Engelstädter and Haig 2008; Vicoso and Bachtrog 2009). Ultimately, a sex-limited mechanism for balancing differences in dose between males and females is expected to evolve such that M:F = 1 for the sex chromosome. This theoretical scenario also predicts that the X:A ratio equilibrates at approximately one in both sexes. So while we typically diagnose dosage compensation by comparing the expression levels of sex-linked loci between sexes, the evolution of dosage compensation reflects selective processes concerning the X:A ratio within each sex.
For many years, the assumption was that the evolution of heteromorphic sex chromosomes necessitated the concomitant evolution of dosage compensation. This was supported by pregenomic empirical studies (Muller 1950; Lyon 1961; Meyer and Casson 1986) and reinforced by genome-wide microarray studies conducted in model organisms, such as mouse, Drosophila flies, humans, and Caenorhabditis worms (Gupta et al. 2006; Nguyen and Disteche 2006; Lin et al. 2007; Johnston et al. 2008). Although molecular mechanisms for achieving dosage compensation are known to differ between these species, the consistent expression patterns across diverse lineages clearly point to a consistently potent evolutionary pressure to adjust gene dose imbalances arising from the evolution of heteromorphic sex chromosomes (Marín et al. 2000; Straub and Becker 2007). However, recent transcriptional profiling studies in several more taxa (e.g., bird, moth, schistosome, beetle, platypus, and fish) have reported notable exceptions to this pattern (Ellegren et al. 2007; Itoh et al. 2007, 2010; Deakin et al. 2008; Mank and Ellegren 2009; Zha et al. 2009; Leder et al. 2010; Prince et al. 2010; Vicoso and Bachtrog 2011; Wolf and Bryk 2011). Moreover, the status of dosage compensation in mammals has been recently called into question by a study using RNA-seq to demonstrate substantial technical artifacts in previous microarray studies (Xiong et al. 2010). The RNA-seq results confirmed the parity of M:F expression on the X chromosome but revealed a X:A ratio of ∼0.5. These recent results are currently stimulating a reformulation of theory as well as careful reconsideration of the existing data (Mank 2009; Vicoso and Bachtrog 2009; Naurin et al. 2010; Xiong et al. 2010).
The most striking and well-researched exception to the expected pattern is found in birds, which are female heterogametic. Both chicken and zebra finch show pervasive male-biased expression for most Z-linked genes. Also, the global Z:A ratio in males is distinctly greater in males (Z:A ≈ 1) than in females (Z:A ≈ 0.8). These patterns demonstrate global dosage compensation is incomplete in birds (Ellegren et al. 2007; Itoh et al. 2007, 2010; Melamed and Arnold 2007; Mank and Ellegren 2009). This raised the question whether the same pattern might be found in all female-heterogametic taxa. In 2009, Zha et al. published a microarray-based genome-wide analysis in the silkworm, Bombyx mori, which appeared to answer this question affirmatively. Like birds, Lepidoptera (moths and butterflies) are female-heterogametic. Zha et al. (2009) reported a chromosome-wide pattern of male-biased expression on the Z and concluded that B. mori, also like birds, lacked complete sex chromosome dosage compensation. This result furthered the notion that dosage compensation was indeed primarily limited to male-heterogametic taxa and inspired the hypothesis that the effects of unbalanced gene dose may be different—perhaps worse—for males relative to females (Mank 2009; Vicoso and Bachtrog 2009; Leder et al. 2010; Naurin et al. 2010). The Z:A ratio was not previously examined in B. mori.
Unfortunately, the results of Zha et al. (2009) rest on an unverified analytical assumption. In asserting that a male-biased global expression ratio on the Z chromosome indicates a lack of dosage compensation, they assumed—but did not examine—that the M:F global expression ratio is unbiased for autosomal loci. In this article, we replicate and extend this analysis to examine this assumption as well as the previously unexamined Z:autosome (Z:A) expression ratios for B. mori. Our analysis indicates that the male-biased expression previously reported for the Z chromosome in B. mori primarily reflects an artifact of microarray data normalization and that the global M:F expression ratio of Z-linked genes is comparable to that of autosomal loci. However, we also find that the Z:A expression ratios are substantially less than 1, indicating that M:F parity on the sex chromosome may have evolved without conservation of the ancestral Z:A expression levels.
Materials and Methods
Raw microarray genepix image files as well as data normalized by Xia et al. (2007) were downloaded from the B. mori microarray database hosted by SilkDB.org (Duan et al. 2010). Xia et al. (2007) initially generated microarray data from nine tissues, whereas Zha et al. (2009) considered only five of these tissues in their analyses of dosage compensation. Here, we consider all nine tissues for which microarray data are available. All statistical analyses were completed using the R statistical computing software (R Development Core Team 2008), with microarray data analyzed using the LIMMA package in the BioConductor software suite (Smyth 2004). These were two-color microarrays so we initially normalized the raw data “within array” only using a print-tip loess (PTL) method with the “normexp” background correction and an offset value of 10 (Smyth and Speed 2003). Values from this single within-array normalization were used to examine the M:F expression ratios between chromosomes.
We applied an additional between-array quantile normalization to the PTL-normalized data before estimating sex-specific (single-color) expression levels. Sex-specific expression values were used to examine the Z:A expression ratio and also to evaluate whether a given probe was expressed in each tissue. To confirm that our results were robust to different methods of normalization, the data were also analyzed using the “variance stabilizing normalization” (VSN) method which concurrently provides both within- and between-array normalization. The normalized data from Xia et al. (2007) were provided as single-channel intensity values. Our analysis includes 5 tissues previously analyzed by Zha et al. (2009) as well as 4 more not previously analyzed (table 1). Arrays for gonads were normalized separately from somatic tissues because of major differences in the variance of expression ratios between the two groups of tissues.
Table 1.
Summary of Microarray Analysis of Bombyx mori Male:Female Expression Ratios on the Z and Autosomes from Nine Different Tissues
| Gonad | Head | Integument | Malpighian Tubules | Ant/Med Silk Gland | Posterior Silk Gland | Fat Body | Mid Gut | Hemocyte | |
| Number of biological replicates performed | 3 | 1 | 1 | 2 | 3 | 2 | 1 | 1 | 1 |
| PTL normalization | |||||||||
| Number of autosomal probes included | 11,650 | 11,134 | 11,448 | 10,803 | 10,660 | 9,814 | 10,659 | 9,446 | 9,991 |
| Mean autosomal M:F expression ratio | 1.318 | 0.985 | 1.016 | 1.002 | 1.032 | 0.993 | 1.018 | 0.996 | 0.996 |
| Median autosomal M:F expression ratio | 1.061 | 1.006 | 1.003 | 1.005 | 1.018 | 0.996 | 0.990 | 0.994 | 1.005 |
| Number of Z-linked probes Z included | 383 | 319 | 322 | 284 | 261 | 244 | 289 | 258 | 262 |
| Mean Z-linked M:F ratio | 1.749 | 1.046 | 1.049 | 1.031 | 1.053 | 1.014 | 1.017 | 1.006 | 0.991 |
| Median Z-linked M:F ratio | 1.286 | 1.040 | 1.052 | 1.015 | 1.031 | 1.019 | 0.995 | 1.004 | 0.999 |
| MWU P value: autosomal ≠ Z inked | 2.21 × 10−04 | 7.64 × 10−6 | 2.26 × 10−04 | 0.309 | 0.227 | 0.071 | 0.883 | 0.488 | 0.533 |
| Data normalized by Xia et al (2007) | |||||||||
| Number of autosomal probes includeda | 11,648 | 11,129 | 11,443 | 10,803 | 10,660 | 9,814 | 10,654 | 9,431 | 9,979 |
| Mean autosomal M:F expression ratio | 1.940 | 1.218 | 1.355 | 1.241 | 1.070 | 0.864 | 1.128 | 0.955 | 0.980 |
| Median autosomal M:F expression ratio | 1.485 | 1.241 | 1.319 | 1.256 | 1.051 | 0.870 | 1.074 | 0.957 | 0.989 |
| Number of Z-linked probes Z includeda | 383 | 319 | 322 | 284 | 261 | 244 | 289 | 258 | 262 |
| Mean Z-linked M:F ratio | 2.778 | 1.323 | 1.427 | 1.273 | 1.106 | 0.876 | 1.153 | 0.970 | 0.977 |
| Median Z-linked M:F ratio | 1.944 | 1.301 | 1.425 | 1.255 | 1.054 | 0.874 | 1.098 | 0.964 | 0.967 |
| MWU P value: autosomal ≠ Z linked | 2.70 × 10−05 | 3.51 × 10−07 | 5.39 × 10−06 | 0.394 | 0.118 | 0.493 | 0.485 | 0.467 | 0.369 |
NOTE.—Data reflect the probe-wise analysis using the ML expression threshold. Results for other normalizations, the uniform expression thresholds, and gene-wise analyses are in supplementary tables 1 and 2 (Supplementary Material online).
In several cases, the background correction and normalization used by Xia et al. (2007) left negative or zero intensity values in the red or green channels, which precluded calculating an M value (which involves a logarithmic conversion of the intensities). When this occurred in all arrays for a given tissue at the same probe, it was impossible to calculate the M:F ratio, and the spot was excluded.
Expression Thresholds for Active Genes
We used two different approaches for determining whether a probe’s signal indicated an actively expressed gene. First, we followed the precedent of Xia et al. (2007), who used a uniform threshold at a signal intensity of 400. We calculated the median sex-specific intensity for each probe using the data normalized by Xia et al. (2007). Any probe with an intensity >400 in either sex we considered to be actively expressed. We refer to this as the “uniform” expression threshold. However, we felt this approach was overly conservative and did not account for differences in hybridization efficiencies and background intensities between probes which can produce dramatically different signal:noise ratios (Li and Wong 2001). We therefore attempted to fit a model to infer whether the signal intensity of a given probe was significantly greater than background noise.
For each combination of tissue type and sex, we discriminated between those probes which showed genuine expression and those that did not using an expectation-maximization algorithm. We extracted the normalized log data from each channel from the appropriate microarray for each probe associated with a gene. We can assume that, as biological replicates, the data should be identically distributed across channels, and so we performed quantile normalization on these data.
We assumed that the observed (log) data for each probe consisted of either noise or both noise and signal, plus an array-specific noise. We assumed that the noise was normally distributed and that the signal was gamma distributed. For a given probe i from sample j, the observed (log) data pij
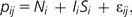 |
where Ni ∼ N(μ, σ) is probe-specific noise, Si ∼ Γ(α, β) is probe-specific signal, εij ∼ N(0, ζ) is experimental noise, and Ii ∈ {0, 1} is an indicator function indicating whether or not this gene is expressed. We began with an assumption that Ii = 0 if the mean expression of probe i is less than the mean of the mean expression of all other probes and Ii = 1 otherwise.
We next deduced by maximum likelihood (ML) the parameters μ, σ, α, β, and ζ. Given these parameters, we evaluated the likelihood of each pij under the alternative assumptions that Ii = 0 or Ii = 1, choosing the value of Ii that maximizes the likelihood of pij. We then iterated the estimation of the initial parameters μ, σ, α, β, and ζ and the Ii values until no further change in the Ii occurs. We thus acquired for each probe an indicator function identifying whether or not the probe contained some genuine signal. For subsequent analyses of M:F ratios, a probe was considered actively expressed if a genuine signal was detected in either sex. We refer to this as the ML expression threshold.
Male:Female Expression Ratios
For each tissue’s set of arrays, the M:F expression ratio was estimated by fitting a linear model for each spot. We compared the M:F expression ratios between Z-linked and autosomal loci using both probe-wise and gene-wise analyses separately for each of the nine tissues. For the probe-wise analysis, we first considered both the uniform and ML expression thresholds. We used the same list of 691 Z–linked probes reported by Zha et al. (2009). (Note: Zha et al. [2009] nominally report 697, but six probes are duplicated in the published list.) We replicated the analysis three times, successively using the data normalized via PTL, VSN, or by Xia et al. (2007). For several probes, the background corrected and normalized data reported by Xia et al. (2007) included negative or zero intensity values in the red or green channels, which precluded calculating an M value [the red vs. green log-ratio; log2(red/green)]. When this occurred in all arrays for a given tissue at the same probe, it was not possible to calculate the M:F ratio and the spot was excluded from analysis in that tissue.
For the gene-wise analysis, we averaged (median) the M:F ratio for all active probes corresponding to a given gene. Unfortunately, there is not a systematic correspondence between probes and genes because the microarrays considered here were designed several years ago using an early draft of the B. mori genome along with an extensive collection of expressed sequence tags (Xia et al. 2004, 2007; Zha et al. 2009). Recently, an improved draft genome and annotation was published (The International Silkworm Genome 2008), but a genic analysis of the present microarray data requires mapping the probe set onto the current set of predicted genes (Zha et al. 2009). To accomplish this, we used BLAT (Kent 2002) to align the 69mer microarray probe sequences to the consensus gene set downloaded from SilkDB (Duan et al. 2010). Probes aligning for less than 60 contiguous bp or with less than 97% identity were excluded from the gene-wise analysis. For each gene’s associated active probes, we calculated the median log2(M:F) obtained from the linear model. Probes mapping to scaffolds not yet assigned to a chromosome were excluded from the analysis. We performed this gene-wise analysis using the ML threshold and the PTL and VSN normalizations.
We used a nonparametric Mann–Whitney U (MWU) test for significant differences in the distribution of the M:F expression ratios for Z-linked versus autosomal loci. Because we examined nine different tissues, we applied a Bonferroni correction for repeated tests, which in this case requires a P value less than 0.0055 for significance. We visualized localized patterns of gene expression by plotting genic values for log2(M:F) expression against gene location along the concatenated Z-chromosome scaffolds. This expression map also included a sliding window analysis in which we plotted the mean log-ratio of a 1-Mb window shifted by 100-Kb steps.
Simulation Power Analyses
An absence of significant differences in the distribution of M:F expression ratios might be attributed to a lack of statistical power. In order to explore this possibility, we performed a set of simulation studies to discover what level of difference in distributions is detectable in these data. For each simulation, we randomly selected (with replacement) two samples of M:F ratios from active autosomal loci from a particular tissue. The first larger sample was intended to reflect “autosomal” loci, and the sampled values were not modified. The second, smaller sample reflected “sex-linked” loci, assumed to show some level of fold change across the sample. To reflect this, we added some (log2) fold change to the original values sampled. For each tissue, we iterated over a series of fold-change values ranging from 0.7 to 1.3 at steps of 0.02 holding other factors constant. We ran 500 simulations for each set of parameters we explored, performing an MWU test of means on each Z versus autosome comparison and reporting “power” as the proportion of times the null hypothesis of “no difference” was rejected using a P value of 0.0055.
We performed simulations reflecting both probe-wise and gene-wise data sets, focusing on the PTL normalization and using the ML expression threshold. For the probe-wise data set, we used sample sizes of 10,000 and 250 for autosomal and “Z-linked” loci, respectively. For the gene-wise data set, we used 5,000 and 200, respectively. These sample sizes were chosen to approximate the actual sample sizes observed the data (table 1 and supplementary table 1, Supplementary Material online).
Z:Autosome Expression Ratios
The microarray design employed here requires both within- and between-array normalization for estimating the sex-specific Z:A ratios. Because the experimental design employed here required directly, competitively hybridizing male and female samples to the same array, within-array normalization alone is sufficient when direct comparison of male versus female expression is made. In this case, it is the intensity “ratio” at a given probe that is the operative datum. Between-array normalization becomes necessary when seeking indirect sex-specific comparisons between tissues or loci. Such analyses must combine single-color intensity data across arrays to estimate expression at each probe for one sex independently of expression in the other sex for the same probe. VSN provides both within- and between- normalization in one step but PTL normalization does not (Smyth et al. 2009). We therefore applied an additional between-array quantile normalization to the signal intensities from the PTL normalization before estimating sex-specific expression levels. We were thus able to use single-color intensities from both VSN and PTL normalizations to compare the average expression level for Z-linked loci relative to autosomal loci separately for each sex. Mean single-value intensities (log2 transformed) were obtained for each tissue and sex by fitting a linear model for each probe (Smyth and Speed 2003; Smyth 2004; Smyth et al. 2009).
As with the M:F ratios, we conducted both probe-wise and gene-wise analyses. Gene-wise expression values were calculated as above, using the ML expression thresholds as determined for each sex independently. For each tissue in each sex, we applied an MWU test for a difference in mean expression levels between Z-linked and autosomal probes. All tests were conducted on log2-transformed intensities, but we report the Z:A mean expression ratios calculated from absolute (untransformed) signal intensities.
RNA-Seq Analysis of Z:Autosome Expression Ratio
To corroborate our inferences of the Z:A ratio based on microarray data, we compared average gene expression between chromosomes using a previously published RNA-seq data set (Zemach et al. 2010). A single lane of Illumina single-end 36-bp sequencing RNA-seq expression data was available from the GenBank GEO database (GPL9151), generated from an entire B. mori larva of unknown sex. Expression was estimated as counts of reads mapped per kilobase of coding sequence. A measure of mapping “uniqueness” was associated with each gene indicating how many other gene models were hit on average by each read (Zilberman D, personal communication). All statistical tests were performed on the complete data set (12,021 expressed loci) as well as on the subset of genes with perfectly unique mapping (11,031 loci where uniqueness = 0). We applied an MWU test for a difference in mean expression levels between Z-linked and autosomal probes. We also iteratively tested each of the 27 autosomes for a global difference in expression from the remaining autosomes, applying a Bonferroni correction such that statistical significance occurred at P < 0.00179 = 0.05/28. In all tests, we excluded loci on scaffolds not assigned to chromosomes.
To provide further context for these RNA-seq expression data, we endeavored to infer the sex of the sequenced silkworm caterpillar by assaying single nucleotide polymorphisms (SNPs) in the data, making the assumption that only a male would show heterozygosity on the Z chromosome. Sequencing reads were mapped to coding sequences using Bowtie followed by SNP calling via SAMtools pileup (Langmead et al. 2009; Li et al. 2009). Analyses were performed with software implemented in the Galaxy analysis pipeline using the default parameters (Goecks et al. 2010). We considered only sites with SNP quality ≥20 and where at least ten reads mapped with basecall quality ≥20.
Results and Discussion
We have reanalyzed previously published microarray data comparing male versus female expression for nine tissues in the silkworm B. mori with the explicit goal of reexamining the issue of M:F expression ratios on the Z chromosome as well as assessing the Z:A expression ratios (Xia et al. 2007). Our results are contrary to a previous analysis of these data reporting a global Z chromosome M:F ratio > 1 but do suggest a lack of dosage compensation based on Z:A ratios (Zha et al. 2009). We begin our discussion by addressing the root of the differences between the previous interpretation and our current conclusions. We then evaluate the global M:F expression ratio of the Z chromosome and the sex-specific Z:A ratios. Finally, we consider these results in the broader context of our current understanding of dosage compensation and the methods available to assess it.
Reconsidering Previous Analyses
Zha et al. (2009) previously addressed the issue of sex chromosome dosage compensation in B. mori using these same microarray data as normalized by Xia et al. (2007). They found that the distribution of M:F expression log ratios was significantly greater than zero for Z-linked loci and interpreted this result as a lack of dosage compensation. The discrepancy between our results and this previous finding can be explained by two interacting factors. First, Zha et al. (2009) tested for male-biased expression of Z-linked loci using a t-test against a null hypothesis of a mean of zero. This approach implicitly assumes that the M:F expression ratios of autosomes is on average equal and that the associated distribution of log ratios are centered around zero. However, this assumption was never examined. Second, the normalization performed by Xia et al. (2007) and utilized by Zha et al. (2009) left a substantial male bias in the microarray data set. Xia et al. (2004) eschewed the standard loess normalization, choosing instead to normalize their data relative to four highly expressed “house-keeping” genes. This approach generated a substantial overall bias toward higher male expression in the data that is greatly mitigated by more standard treatments of the data we have applied here (fig. 1; supplementary fig. 1, Supplementary Material online). Using the original male-biased normalization, Zha et al. (2009) detected a significant male bias in expression of Z-linked genes but failed to test whether M:F expression ratios among autosomes were unbiased and did not directly compare the Z chromosome to the autosomes.
FIG. 1.—

The effect of different normalization methods on the Bombyx mori microarray data sets. The log ratio of M:F expression is plotted on the y axis against the log of average spot intensity. This is analogous to a standard MA plot, but here, spot values are averaged across a pair of dye-swap replicates for each tissue (no biological replicates were performed for these tissues, for details, see Xia et al. 2007). Only active probes (ML threshold) are plotted here, with Z-linked probes reported by Zha et al. (2009) highlighted in red. The original normalization by Xia are in the left panels with the more conventional normalization methods of PTL and VSN plotted center and right, respectively. Plots for the remaining seven tissues sampled by Xia et al. (2007) are in supplementary materials.
Male:Female Expression Ratios of the Z and Autosomes
A direct comparison reveals that the M:F expression ratios are highly consistent between the Z chromosome and autosomes (fig. 2 and table 1; see also supplementary figs. 2–4 and tables 1 and 2, Supplementary Material online). Mean and median M:F expression ratios from the two groups of chromosomes generally differ by only a few percentage points in somatic tissues. Even for head and integument, which show significant differences across all the different analyses, the average discrepancy is typically less than 10%. A biological explanation for the significant difference arising in these two tissues is not readily apparent. However, a biological explanation for this observation may be unnecessary since these two tissues are among those with only a single biological replicate (Xia et al. 2007). This limited sampling substantially constrains the confidence of the inference. The dye effects in this data set are quite variable across arrays (data not shown) and the significant difference in these two tissues may simply be an artifact of uneven dye bias occurring between dye replicates of a single biological replicate. Qualitatively speaking, the overlap in distributions of M:F expression ratios for Z-linked and autosomal loci appear no more different for head and integument than the other somatic tissues (fig. 2 and supplementary figs. 2–4, Supplementary Material online).
FIG. 2.—

Comparison of male and female gene expression for genes located on autosomes (black) versus the Z chromosome (red). The distribution densities of the log ratio male:female expression values are plotted for active probes (ML threshold). Individual data points are plotted unidimensionally along the x axis. The data as normalized by Xia et al. (2004) are plotted in the top row, whereas the bottom row are the same data normalized using a PTL method. The dotted vertical line delineates where the male:female ratio equals 1 [log2(M:F) = 0]. Comparable plots are included in the supplementary materials for additional tissues, the VSN normalization, and gene-wise analyses, and with the uniform threshold applied.
Overall, these results are not consistent with the hypothesis that B. mori has incomplete dosage compensation. For contrast, consider the case in birds. It is well established that both chicken and zebra-finch lack a mechanism for global dosage compensation, and in both species, Z-linked loci have approximately 30% greater expression in males (Itoh et al. 2007, 2010; Melamed and Arnold 2007; Mank and Ellegren 2009). A similar pattern was also reported in crows (Wolf and Bryk 2011). The pattern observed in B. mori of essentially equal M:F expression on the Z chromosome is far more consistent with observations from taxa classically considered to be dosage compensated, such as mammals and Drosophila than it is with birds.
Gonad is the one tissue examined here showing a substantial difference in average M:F expression between the Z and autosomes (table 1, supplementary table 1, Supplementary Material online). This pattern is likely explained by the fact that the Z chromosome is enriched for testis-specific genes, previously reported by Arunkumar et al. (2009) and borne out by our results (see “sex-specific” analyses below). Relative to other tissues, an excess of testis-specific loci should increase average M:F expression ratio of the Z. This hypothesis was supported by reanalyzing the data while excluding sex-specific genes. Requiring genes to be expressed in both sexes removed the significant difference between the Z and autosomes for gonadal M:F ratios and reduced the difference in mean absolute M:F ratio by 25% (supplemental table 1, Supplementary Material online).
Because we are basing an argument for the parity of M:F expression for sex-linked genes on the general lack of a statistical difference in the global M:F ratio between the Z and autosomes, we conducted simulation power analyses on our data. Our goal was to explore what fold change in global expression between the Z and autosomes would consistently produce a statistically significant result given the particularities of our data. Our simulations indicate that there is substantial power to detect global differences in the M:F ratio between the Z and autosomes, particularly in somatic tissues (fig. 3). At fold changes greater than 1.1 or less than 0.9, there appears to be nearly perfect power in somatic tissues to detect differences with our significance threshold of α = 0.0055. Previous empirical examples from birds and schistosomes as well as experimental manipulations in other organisms indicate that uncompensated differences in gene dose should produce expression fold changes that fall in this range of “perfect” detection (Mank and Ellegren 2009; Itoh et al. 2010; McAnally and Yampolsky 2010; Zhang et al. 2010; Vicoso and Bachtrog 2011). Therefore, if a global expression difference between the sexes truly exists due to partial or completely uncompensated Z-linked gene dose, it is very likely we would have discovered it.
FIG. 3.—

Simulation statistical power analysis of M:F global expression ratio. The mean (±standard deviation) statistical power across eight somatic tissues and gonads is plotted for a series of simulated M:F global fold-change differences. Here, power is calculated from 500 simulations at each point and reflects the proportion of times a MWU test rejects the null hypothesis of equal means for the two samples with a significance threshold of α = 0.0055. For each tissue, two samples corresponding to autosomal (A) and Z-linked (Z) loci were randomly generated from autosomal loci (PTL normalization; ML threshold), with the Z sample subsequently modified to reflect the fold change being examined. Blue points reflect sample sizes of A = 10,000 and Z = 250 generated from the probe-wise analysis. Green points reflect sample sizes of A = 5,000 and Z = 200 generated from the gene-wise analysis. Red and black points reflect gonads only, probe-wise and gene-wise analyses, respectively. Points have been slightly offset to facilitate plotting.
Normalizations, Thresholds, and Probe-Gene Mapping
Our results concerning direct comparisons of M:F expression ratios are consistent across several different analytical treatments of the data. The general observation that M:F expression ratios are highly concordant between autosomes and Z-chromosomes is not altered by various approaches to background correction and data normalization, expression thresholds, nor the grouping of probes by gene. Judging from comparisons of array-specific MA-plots (results not shown) as well as array-averaged plots of expression ratio versus signal intensity (supplementary fig. 1, Supplementary Material online), we believe that the PTL normalization and normexp background correction is a better treatment of these data than VSN for removing dye bias and other artifacts present in the data. For this reason, we have concentrated our discussion primarily on results from the PTL normalization. Both PTL and VSN appear to provide substantial improvement over the normalization utilized by Xia et al. (2007) (fig. 1 and supplementary fig. 1, Supplementary Material online).
The ML method for discerning expressed probes—those with hybridization signal above background noise—was a less restrictive filter than the uniform threshold applied by Xia et al. (2007). The ML threshold indicated informative expression signal existed for many more probes than relative to the uniform application of a single intensity threshold of 400 and yielded as much as twice as many informative probes in several tissues. This is equally true whether the uniform threshold is applied either to the PTL-normalized data or the data as normalized by Xia et al. (2007) (results not shown). Nonetheless, the ML method very effectively separates probes with strong signal intensities from those with weaker ones (supplemental fig. 5, Supplementary Material online).
There were 13,183 microarray probes successfully mapped to the updated B. mori gene set yielding 10,644 genes represented by at least one probe and up to a maximum of seven. Six hundred and three probes mapped to 495 coding sequences predicted on the Z chromosome. Six hundred and one of these Z-gene probes overlapped with the 691 Z-linked probes identified by Zha et al. (2009). Plotting M:F fold change of genes along the Z chromosome does not reveal any obvious “neighborhood” effects with localized deviations from the overall pattern of equal M:F expression (fig. 4, supplementary figs. 6 and 7, Supplementary Material online).
FIG. 4.—

Expression map of Bombyx mori Z chromosome genes. Log2(M:F) ratios for each gene (points) are plotted along with the mean of a 1-Mbp sliding window average (red line) shifted every 100 Kb. The data reflect the PTL normalization and include all Z-linked coding sequences for which corresponding active (ML expression threshold) probes were identified in the gene-wise analysis. A unidimensional plot of gene position is projected along the x axis; the green bar (lower left) represents the sliding window size. Expression maps for the other remaining tissues and also after applying the signal-intensity threshold are in supplementary materials.
Complete listings of the ML expression indicator for each probe and tissue along with mapping of probes to genes were deposited in the DRYAD data repository (doi: 10.5061/dryad.8716).
Sex-Specific Patterns of Z Versus Autosomes
Theory concerning the evolution of dosage compensation predicts that stabilizing selection to conserve the ancestral expression ratios between autosomes and the nascent sex chromosomes should produce a global pattern of equal M:F sex ratios on the sex chromosomes. This selective pressure should also generate a pattern of Z:A close to unity in both sexes, assuming that the ancestral global expression levels were approximately equal across the chromosomes (Charlesworth 1978, 1996; Rice 1987; Engelstädter and Haig 2008). Our analysis suggests that B. mori does not meet this second prediction. In somatic tissues for both males and females, average Z expression is consistently significantly lower than average autosomal expression, with Z:A ratios falling in the range of 0.6–0.8 (fig. 5, table 2, supplementary table 3 and fig. 8, Supplementary Material online). This result holds under both probe-wise and gene-wise analyses and both VSN and PTL normalizations. We do not present the analyses using the uniform cutoff because this method strongly biases the data; removing the majority of weakly expressed loci compresses the mean expression of the two groups of genes.
FIG. 5.—

Expression of Z-linked and autosomal loci in males and females. Plotted data reflect the probe-wise analysis with PTL normalization and the ML expression threshold. Plots for all tissues are in supplementary figure 8 (Supplementary Material online).
Table 2.
Summary of Expression Levels Compared between Z and Autosomes
| Number of Male Z Probes Above Threshold | Number of Female Z Probes Above Threshold | Number of Male Autosomal Probes Above Threshold | Number of Female Autosomal Probes Above Threshold | Male Z:A Ratio (Intensities Not Log Transformed) | Female Z:A Ratio (Intensities Not Log Transformed) | Male MWU P Value, Mean Expression of Z Versus A | Female MWU P Value, Mean Expression of Z Versus A | |
| Gonad | 341 | 303 | 10,309 | 10,042 | 1.141 | 0.736 | 0.512 | 1.76 × 10−03 |
| Head | 306 | 285 | 10,800 | 10,410 | 0.766 | 0.761 | 7.63 × 10−04 | 1.78 × 10−04 |
| Integument | 309 | 291 | 10,990 | 10,667 | 0.818 | 0.828 | 0.035 | 0.072 |
| Malphigian tubules | 255 | 268 | 9,842 | 10,258 | 0.702 | 0.694 | 1.60 × 10−03 | 7.17 × 1004 |
| Ant/Med silk gland | 233 | 249 | 9,800 | 10,277 | 0.703 | 0.672 | 4.81 × 10−03 | 9.89 × 10−04 |
| Posterior silk gland | 227 | 229 | 9,163 | 9,534 | 0.692 | 0.678 | 4.92 × 10−03 | 0.036 |
| Fat body | 267 | 271 | 10,042 | 9,912 | 0.821 | 0.799 | 0.185 | 0.044 |
| Mid gut | 243 | 238 | 9,078 | 8,942 | 0.722 | 0.698 | 1.21 × 10−03 | 1.21 × 10−03 |
| Hemocyte | 251 | 240 | 9,577 | 9,257 | 0.672 | 0.680 | 1.60 × 10−04 | 4.19 × 10−04 |
NOTE. —Data reflect probe-wise analysis with PTL normalization and ML expression threshold. Additional tables for VSN and gene-wise analyses are in supplementary table 3 (Supplementary Material online).
This difference in global expression ratios appears to be a phenomenon unique to the Z chromosome. We tested for a difference in global expression between each individual autosome and the remaining autosomes just as we tested for a difference between Z and autosomes. Although a few autosomes occasionally showed a significantly different average expression level relative to the other autosomes, no consistent pattern was observed across tissues as was observed for the sex chromosome (supplementary table 4, Supplementary Material online).
Comparing global expression of Z versus autosomes using RNA-seq data produces results consistent with those obtained from the single-channel microarray analysis. The RNA-seq estimates of gene expression also indicate a significantly reduced global expression on the Z, with mean and median expression of Z-linked genes both approximately one-half the autosomal averages (table 3). As with the microarray data, this pattern appears to be unique to the Z chromosome. In both the full and “unique” RNA-seq data sets, the Z has the lowest median expression of all chromosomes and no other chromosome showed a significantly lower average expression relative to the other autosomes (fig. 6, supplementary table 5, Supplementary Material online).
Table 3.
Summary of RNA-Seq Expression Levels Compared between Z and Autosomes
| All Loci |
Loci Where All Reads Map Uniquely |
|||
| Z (N = 500) | Autosomes (N = 10,881) | Z (N = 485) | Autosomes (N = 10,003) | |
| Mean expression | 237.7 | 611.7 | 242.2 | 500.6 |
| Median expression | 24 | 43.4 | 24.2 | 43.7 |
| MWU P value: autosomal ≠ Z linked | 1.14 × 10−12 | 6.45 × 10−12 | ||
FIG. 6.—

Comparison of global expression levels of across chromosomes. Plotted data reflect absolute RNA-seq estimates of gene expression (reads per kilobase [RPK]). Box widths are proportional to the square root of the count of expressed genes on each chromosome (i.e., sample size). Outliers are not plotted.
The RNA-seq data represent a single lane of Illumina sequencing generated as part of a study unrelated to dosage compensation from an individual of unknown sex (Zemach et al. 2010). Because discerning the sex of the sampled individual would make these data more meaningful in the context of dosage compensation, we assayed SNPs in the data, assuming that only a male would show heterozygosity on the Z. This is a reasonable assumption given that there is ample allozyme evidence for diploid, heterozygous expression of Z-linked loci in male Lepidoptera (Mallet et al. 1993; Raijmann et al. 1997); there is also little evidence for any Z-W homology in Bombyx so it is unlikely that apparent Z-linked heterozygosity could arise from pseudoautosomal regions such as occurs between X and Y chromosomes in humans (Fujii and Shimada 2007). We identified 182 heterozygous Z-linked SNPs, a count which falls squarely in the range detected among autosomes using the same criteria (132–569 SNPs per chromosome). Thus, we believe these RNA-seq expression data were obtained from a male silkworm. Accordingly, the reduced global expression of the Z occurs despite a diploid complement of sex chromosomes.
It is perplexing to observe equal M:F expression on the Z, whereas global expression levels are substantially reduced relative to the autosomes. It suggests that B. mori may achieve parity of M:F expression on the Z by male silencing of one Z chromosome such that both sexes are effectively haploid for the sex chromosome. This scenario is inconsistent with leading theories regarding the evolution of sex chromosomes and dosage compensation which predict that parity of M:F ratios should be accompanied by equal Z:A ratios (Charlesworth 1978, 1996; Rice 1987; Engelstädter and Haig 2008; Vicoso and Bachtrog 2009). All previous microarray assays of taxa showing balanced sex chromosome M:F ratios (Drosophila, Caenorhabditis, Mammals) have shown X:A ratios ∼ 1 despite divergent dosage compensating mechanisms, providing support for both major theoretical predictions (Gupta et al. 2006; Nguyen and Disteche 2006; Lin et al. 2007; Johnston et al. 2008). However, microarray estimates of absolute levels of gene expression are often unreliable because absolute signal strength depends substantially on the hybridization efficiency of probes. Hybridization efficiency is largely determined by probe sequence and therefore varies substantially between probes (Li and Wong 2001; Gentleman and Carey 2005; Draghici et al. 2006). Thus, these microarray results should be interpreted with some caution.
Although not without its own shortcomings, RNA-seq is generally considered to give more accurate estimates of absolute expression levels than microarrays (Fu et al. 2009). It was nonetheless a surprise when a recent study using RNA-seq reported that the X:A ratio in both male and female mammals is actually ∼0.5 (Xiong et al. 2010). Our observations in B. mori are consistent with this revised X:A estimate for mammals, where one copy of the female X chromosome is inactivated (Payer and Lee 2008). In contrast to the discrepant results from mammals, the RNA-seq expression estimates in silkworm support the microarray-based observation of a distinctly reduced Z:A ratio. This consistency bolsters the argument that this is a real biological phenomenon and not an artifact of the technology. However, the RNA-seq data considered here are limited in that they reflect data from only a single individual. It will be very important to use RNA-seq or other similarly accurate methods to verify this preliminary indication of reduced expression of Z-linked loci in B. mori across multiple individuals of both sexes as well as in other lepidopteran species. Widespread observations of equal M:F ratios with reduced Z:A ratios cannot be easily accommodated by the current theories of sex chromosome dosage compensation.
Male gonads (testes) stand out in this sex-specific analysis as having a global expression profile distinctly different from all the other tissue/sex combinations examined. Testes show a Z:A ratio ∼ 1 and an average expression not significantly different from the autosomes (table 1, supplementary fig. 8 and table 3, Supplementary Material online). Based on ML expression threshold, there is a disproportionately large number of Z-linked genes uniquely expressed in the testes relative to autosomes (Z: 27/495 = 5.5% vs. A: 259/9653 = 2.7%; Fisher’s Exact test P < 0.002). This is consistent with a previous report that the Z is enriched for testis-specific genes (Arunkumar et al. 2009). Our data show that mean expression of testis-specific genes is 20% greater than the mean testes expression of genes expressed in testes and at least one other tissue (MWU, P < 0.001). Thus, the distinct expression profile of testes may be largely explained by an enriched portion of highly expressed genes on the Z chromosome.
Conclusions
Our work here demonstrates an important exception to the emerging consensus that ZW taxa are not dosage compensated or at least that they do not have equal M:F expression of the sex chromosome. Genome-wide patterns of sex chromosome dosage compensation have been assayed in two other taxa where female heterogamety evolved independently: birds and schistosomes (Mank and Ellegren 2009; Itoh et al. 2010; Vicoso and Bachtrog 2011). Both of these taxa show a reduced Z:A ratio in females but not in males. Consistent with this pattern, birds also show an elevated M:F expression ratio on the Z chromosome. The M:F ratio was not directly examined in schistosome, but an equal M:F ratio would be surprising given the discrepancy in Z:A ratios between sexes. Strikingly, our revised appraisal of these patterns indicating an equal M:F expression ratio in B. mori provides a distinct departure from these other ZW taxa. Without consideration of the Z:A ratio, this result alone would suggest that B. mori is dosage compensated.
However, the M:F ratio is only an indirect measure of dosage compensation. Evolutionary theory emphasizes that dosage compensation reflects the process of stabilizing selection on expression levels of the proto-sex chromosomes during sex chromosome divergence and degradation. Equal M:F expression is a by-product of this primary evolutionary phenomenon (Charlesworth 1978, 1996; Rice 1987; Engelstädter and Haig 2008). Thus, our observation of a consistently low Z:A ratio arguably overturns the claim of dosage compensation based on M:F ratios and presents a paradox: how to explain the evolution of equal M:F expression ratios on the Z while rejecting the existence of dosage compensation?
We can see a few possible solutions for this paradox. To begin with, it may not be a paradox at all but rather an artifact of microarray analysis. Unfortunately, microarrays —until recently our primary tool for assessing genome-wide expression patterns—are poorly suited for estimating absolute expression differences between loci, as is required for assessing X:A or Z:A ratios (Li and Wong 2001; Gentleman and Carey 2005; Xiong et al. 2010). However, microarray estimates of relative expression, as used for estimating M:F ratios, have not been similarly called into question. Thus, although the “definitive” criterion for dosage compensation rests on the X:A or Z:A ratios and requires estimating absolute expression levels, microarray data are far better suited to examining the relative M:F expression ratios. Further thorough assays of the Z:A ratios using more accurate methods of estimating absolute expression such as RNA-seq are needed.
Nonetheless, the limited RNA-seq data that are currently available do support the microarray results and are consistent with the recent RNA-seq results in mammals showing X:A is substantially less than 1 (Xiong et al. 2010). There is not currently a strong evolutionary hypothesis which might explain reduced X:A or Z:A with equal M:F ratios, though one possible answer might come from considering evolution of sex chromosome inactivation via sexual antagonism (Rice 1987; Haig 2006; Engelstädter and Haig 2008). If inactivation arose or equilibrated at a point before all sex-linked loci recovered their ancestral expression in the haploid state, then global sex-linked M:F ratios would be equal but X:A or Z:A ratios would be less than 1.
Another rather straightforward resolution to this paradox may be that the proto-sex chromosome just happened to have an unusually low average expression and that a low Z:A ratio does not actually indicate a lack of dosage compensation. In the case of B. mori, this seems unlikely because none of the other 27 chromosomes show such a consistently different average expression. Nonetheless, it is difficult to completely exclude this scenario without much broader phylogenetic sampling.
Ultimately, the existing data and analyses currently present a complex and changing picture of dosage compensation that is not entirely consistent with any single evolutionary theory. With less than a dozen taxa yet surveyed comprehensively, the evolutionary genomics community is still in the early stages of documenting genome-wide patterns of male versus female expression. New theory and new data will certainly help resolve our understanding of dosage compensation and the evolution of sex chromosomes, but it should not be overlooked that critical reevaluation of existing data sets can also be a valuable contribution toward this effort (Xiong et al. 2010).
Supplementary Material
Supplementary figures S1–S8 and tables S1–S5 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
We thank Chris Jiggins, Gos Micklem, and members of their laboratory groups for support and assistance with this project. Daujun Cheng helpfully clarified several details of the original microarray analysis. Judith Mank generously provided valuable discussions, guidance, and feedback on analyses and the manuscript. Three anonymous reviewers offered several helpful suggestions and critiques. This work was supported by the National Science Foundation (NSF-0905698 to J.R.W.) and the European Commission Seventh Framework Programme (233325 to T.J.H.).
References
- Arunkumar KP, Mita K, Nagaraju J. The silkworm Z chromosome is enriched in testis-specific genes. Genetics. 2009;182:493–501. doi: 10.1534/genetics.108.099994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B. Model for evolution of Y chromosomes and dosage compensation. Proc Natl Acad Sci U S A. 1978;75:5618–5622. doi: 10.1073/pnas.75.11.5618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B. The evolution of chromosomal sex determination and dosage compensation. Curr Biol. 1996;6:149–162. doi: 10.1016/s0960-9822(02)00448-7. [DOI] [PubMed] [Google Scholar]
- Charlesworth B, Charlesworth D. The degeneration of Y chromosomes. Philos Trans R Soc Lond B Biol Sci. 2000;355:1563–1572. doi: 10.1098/rstb.2000.0717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth D, Charlesworth B, Marais G. Steps in the evolution of heteromorphic sex chromosomes. Heredity. 2005;95:118–128. doi: 10.1038/sj.hdy.6800697. [DOI] [PubMed] [Google Scholar]
- Deakin JE, Hore TA, Koina E, Marshall Graves JA. The status of dosage compensation in the multiple X chromosomes of the platypus. PLoS Genet. 2008;4:e1000140. doi: 10.1371/journal.pgen.1000140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draghici S, Khatri P, Eklund AC, Szallasi Z. Reliability and reproducibility issues in DNA microarray measurements. Trends Genet. 2006;22:101–109. doi: 10.1016/j.tig.2005.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan J, et al. SilkDB v2.0: a platform for silkworm (Bombyx mori) genome biology. Nucleic Acids Res. 2010;38:D453–D456. doi: 10.1093/nar/gkp801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegren H, et al. Faced with inequality: chicken do not have a general dosage compensation of sex-linked genes. BMC Biol. 2007;5:40. doi: 10.1186/1741-7007-5-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelstädter J, Haig D. Sexual antagonism and the evolution of X chromosome inactivation. Evolution. 2008;62:2097–2104. doi: 10.1111/j.1558-5646.2008.00431.x. [DOI] [PubMed] [Google Scholar]
- Fu X, et al. Estimating accuracy of RNA-Seq and microarrays with proteomics. BMC Genomics. 2009;10:161. doi: 10.1186/1471-2164-10-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii T, Shimada T. Sex determination in the silkworm, Bombyx mori: a female determinant on the W chromosome and the sex-determining gene cascade. Semin Cell Dev Biol. 2007;18:379–388. doi: 10.1016/j.semcdb.2007.02.008. [DOI] [PubMed] [Google Scholar]
- Gentleman R, Carey V, Huber W, Irizarry R, Dudoit S. Bioinformatics and computational biology solutions using R and bioconductor. New York: Springer; 2005. Analysis overview; pp. 183–185. [Google Scholar]
- Goecks J, Nekrutenko A, Taylor J. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11:R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta V, et al. Global analysis of X-chromosome dosage compensation. J Biol. 2006;5:3. doi: 10.1186/jbiol30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haig D. Self-imposed silence: parental antagonism and the evolution of X-chromosome inactivation. Evolution. 2006;60:440–447. [PubMed] [Google Scholar]
- Itoh Y, et al. Dosage compensation is less effective in birds than in mammals. J Biol. 2007;6:2. doi: 10.1186/jbiol53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh Y, et al. Sex bias and dosage compensation in the zebra finch versus chicken genomes: general and specialized patterns among birds. Genome Res. 2010;20:512–518. doi: 10.1101/gr.102343.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston CM, et al. Large-scale population study of human cell lines indicates that dosage compensation is virtually complete. PLoS Genet. 2008;4:e9. doi: 10.1371/journal.pgen.0040009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ. BLAT—the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leder EH, et al. Female-biased expression on the X chromosome as a key step in sex chromosome evolution in threespine sticklebacks. Mol Biol Evol. 2010;27:1495–1503. doi: 10.1093/molbev/msq031. [DOI] [PubMed] [Google Scholar]
- Li C, Wong WH. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc Natl Acad Sci U S A. 2001;98:31–36. doi: 10.1073/pnas.011404098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, et al. Dosage compensation in the mouse balances up-regulation and silencing of X-linked genes. PLoS Biol. 2007;5:e326. doi: 10.1371/journal.pbio.0050326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon MF. Gene action in the X-chromosome of the mouse (Mus musculus L.) Nature. 1961;190:372–373. doi: 10.1038/190372a0. [DOI] [PubMed] [Google Scholar]
- Mallet J, Korman A, Heckel DG, King P. Biochemical genetics of Heliothis and Helicoverpa (Lepidoptera: Noctuidae) and evidence for a founder event in Helicoverpa zea. Ann Entomol Soc Am. 1993;86:189–197. [Google Scholar]
- Mank JE. The W, X, Y and Z of sex-chromosome dosage compensation. Trends Genet. 2009;25:226–233. doi: 10.1016/j.tig.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mank JE, Ellegren H. All dosage compensation is local: gene-by-gene regulation of sex-biased expression on the chicken Z chromosome. Heredity. 2009;102:312–320. doi: 10.1038/hdy.2008.116. [DOI] [PubMed] [Google Scholar]
- Marín I, Siegal ML, Baker BS. The evolution of dosage-compensation mechanisms. Bioessays. 2000;22:1106–1114. doi: 10.1002/1521-1878(200012)22:12<1106::AID-BIES8>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- McAnally AA, Yampolsky LY. Widespread transcriptional autosomal dosage compensation in Drosophila correlates with gene expression level. Genome Biol Evol. 2010;2:44–52. doi: 10.1093/gbe/evp054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melamed E, Arnold AP. Regional differences in dosage compensation on the chicken Z chromosome. Genome Biol. 2007;8:R202. doi: 10.1186/gb-2007-8-9-r202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer BJ, Casson LP. Caenorhabditis elegans compensates for the difference in X chromosome dosage between the sexes by regulating transcript levels. Cell. 1986;47:871–881. doi: 10.1016/0092-8674(86)90802-0. [DOI] [PubMed] [Google Scholar]
- Muller HJ. Evidence of the precision of genetic adaptation. Harvey Lect. 1950;43:165–229. [Google Scholar]
- Naurin S, Hansson B, Bensch S, Hasselquist D. Why does dosage compensation differ between XY and ZW taxa? Trends Genet. 2010;26:15–20. doi: 10.1016/j.tig.2009.11.006. [DOI] [PubMed] [Google Scholar]
- Nguyen DK, Disteche CM. Dosage compensation of the active X chromosome in mammals. Nat Genet. 2006;38:47–53. doi: 10.1038/ng1705. [DOI] [PubMed] [Google Scholar]
- Ohno S. Sex chromosomes and sex-linked genes. Berlin (Germany): Springer; 1967. [Google Scholar]
- Payer B, Lee JT. X chromosome dosage compensation: how mammals keep the balance. Annu Rev Genet. 2008;42:733–772. doi: 10.1146/annurev.genet.42.110807.091711. [DOI] [PubMed] [Google Scholar]
- Prince EG, Kirkland D, Demuth JP. Hyperexpression of the X chromosome in both sexes results in extensive female bias of X-linked genes in the flour beetle. Genome Biol Evol. 2010;2:336–346. doi: 10.1093/gbe/evq024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team. R: a language and environment for statistical computing. Vienna (Austria): R Foundation for Statistical Computing; 2008. [Google Scholar]
- Raijmann LEL, van Ginkel WE, Heckel DG, Menken SBJ. Inheritance and linkage of isozymes in Yponomeuta padellus (Lepidoptera, Yponomeutidae) Heredity. 1997;78:645–654. [Google Scholar]
- Rice WR. Genetic hitchhiking and the evolution of reduced genetic activity of the Y sex chromosome. Genetics. 1987;116:161–167. doi: 10.1093/genetics/116.1.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth Gordon K. Linear models and Empirical Bayes Methods for Assessing Differential Expression in Microarray Experiments. Statistical Applications in Genetics and Molecular Biology. 2004;3(1) doi: 10.2202/1544-6115.1027. doi:10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- Smyth GK, Ritchie M, Thorne N, Wettenhall J, Wei S. limma: Linear Models for Microarray Data User’s Guide [Internet]. [cited 2010 Sep 14] 2009. Available from: http://www.bioconductor.org/packages/bioc/1.8/vignettes/limma/inst/doc/usersguide.pdf. [Google Scholar]
- Smyth GK, Speed T. Normalization of cDNA microarray data. Methods. 2003;31:265–273. doi: 10.1016/s1046-2023(03)00155-5. [DOI] [PubMed] [Google Scholar]
- Straub T, Becker PB. Dosage compensation: the beginning and end of generalization. Nat Rev Genet. 2007;8:47–57. doi: 10.1038/nrg2013. [DOI] [PubMed] [Google Scholar]
- The International Silkworm Genome. The genome of a lepidopteran model insect, the silkworm Bombyx mori. Insect Biochem Mol Biol. 2008;38:1036–1045. doi: 10.1016/j.ibmb.2008.11.004. [DOI] [PubMed] [Google Scholar]
- Vicoso B, Bachtrog D. Progress and prospects toward our understanding of the evolution of dosage compensation. Chromosome Res. 2009;17:585–602. doi: 10.1007/s10577-009-9053-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicoso, Bachtrog Lack of Global Dosage Compensation in Schistosoma mansoni, a Female-Heterogametic Parasite. Genome Biology and Evolution. 2011;3:230–235. doi: 10.1093/gbe/evr010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf JB, Bryk J. General lack of global dosage compensation in ZZ/ZW systems? Broadening the perspective with RNA-seq. BMC Genomics. 2011;12:91. doi: 10.1186/1471-2164-12-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Q, et al. A draft sequence for the genome of the domesticated silkworm (Bombyx mori) Science. 2004;306:1937–1940. doi: 10.1126/science.1102210. [DOI] [PubMed] [Google Scholar]
- Xia Q, et al. Microarray-based gene expression profiles in multiple tissues of the domesticated silkworm, Bombyx mori. Genome Biol. 2007;8:R162. doi: 10.1186/gb-2007-8-8-r162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong, et al. RNA sequencing shows no dosage compensation of the active X-chromosome. Nat Genet. 2010;42(12):1043–7. doi: 10.1038/ng.711. doi:10.1038/ng.711. [DOI] [PubMed] [Google Scholar]
- Zemach A, McDaniel IE, Silva P, Zilberman D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science. 2010;328:916–919. doi: 10.1126/science.1186366. [DOI] [PubMed] [Google Scholar]
- Zha X, et al. Dosage analysis of Z chromosome genes using microarray in silkworm, Bombyx mori. Insect Biochem Mol Biol. 2009;39:315–321. doi: 10.1016/j.ibmb.2008.12.003. [DOI] [PubMed] [Google Scholar]
- Zhang Y, et al. Expression in aneuploid Drosophila S2 cells. PLoS Biol. 2010;8:e1000320. doi: 10.1371/journal.pbio.1000320. [DOI] [PMC free article] [PubMed] [Google Scholar]