

Abstract
Multicolour single molecule fluorescence imaging enables the study of multiple proteins in the membranes of living cells. We describe the use of a supercontinuum laser as the excitation source, show its comparability with multiplexed single-wavelength lasers and demonstrate that it can be used to study membrane proteins such as the ErbB receptor family. We discuss the benefits of white-light sources for single molecule fluorescence, in particular their ease of use and the freedom to use the most appropriate dye without being constrained by available laser wavelengths.
OCIS codes: (170.2520) Fluorescence microscopy, (110.4234) Multispectral and hyperspectral imaging
1. Introduction
Protein networks in cells are highly complex, involving large numbers of molecules. The ErbB signalling pathways [1], for example, involve hundreds of interacting molecules. Our understanding of their dependence, for example on drug application, would benefit from methods which allow multiple proteins to be imaged simultaneously.
Single molecule fluorescence methods [2] are well suited to studying signalling events because they allow molecules to be studied at physiological concentrations. Furthermore, rare or unsynchronised signalling events are not masked by the ensemble average. By using multiple fluorophore tags, multiple proteins in a pathway can be studied using multicolour fluorescence imaging. The properties of the fluorescence from the individual molecules, including intensity, position and wavelength, permit, for example, the diffusion rates and oligomerisation of signalling proteins to be determined through time [3].
The mercury or Xenon arc lamp is the standard light source for conventional widefield fluorescence microscopy. At its simplest, multicolour fluorescence imaging of a single sample can be achieved by using separate bandpass filters for each fluorophore in turn. To image the different fluorophores simultaneously, the microscope can be equipped with multiband filters and image splitting optics, in which dichroic beamsplitters direct the fluorescence to different regions of the detector according to their wavelength.
However, these light sources are rarely used for objective-type total internal reflection fluorescence microscopy (TIRFM) [4], which has become the method of choice for imaging single molecules in the plasma membranes of live cells [3,5–7]. In a TIRF microscope, the excitation beam must exit the objective lens at an angle greater than the critical angle for the sample being imaged and therefore be restricted to passing through points within a thin annulus in the back focal plane. This would involve blocking most of the output of the lamp, which is designed to fill the back aperture of the objective, and the resultant power throughput is insufficient for single molecule TIRFM. Laser excitation, which is easily focused to a point at the edge of the back focal plane, is therefore usually used in TIRFM. Lasers have an additional advantage in that their output is polarised, which is useful for many applications [8].
Multicolour laser excitation can be achieved either using a laser with multiple lines or by combining the beams from multiple lasers. For reasons of cost and size, compact lasers are usually employed, but these are not tuneable and hence it is necessary to realign different lasers into the microscope each time the excitation wavelength needs to be changed. Furthermore, lasers are not available for all wavelengths. Highly complex setups are required when the number of lasers is increased to three or four [9]. Although devices to combine laser outputs are available commercially, there is not day-to-day freedom to change the lasers within them.
Choosing a fluorophore for a particular experiment involves balancing its photophysical properties (excitation and emission spectra, brightness, photostability), the excitation sources available and the ease of tagging the target molecule with the dye. Cost considerations mean that most laboratories do not possess lasers that excite optimally all of the fluorophores that are used.
The ideal light source for single molecule TIRFM would combine the spectral properties of an arc lamp with the power and control of a laser. We present here, therefore, the application of white-light supercontinuum laser sources to single-molecule TIRFM. This convenience of having multiple wavelengths from a single laser has previously been exploited by other fluorescence microscopies, such as confocal microscopy [10], fluorescence lifetime imaging [11] and STED [12], but this is the first time that a supercontinuum source has been used in wide-field imaging of single molecules. We demonstrate a relatively low cost and compact instrument for multicolour imaging of single molecules in live cell membranes with the flexibility to image a wide range of different fluorophores.
2. Instrumentation
The supercontinuum (SC-450-8, Fianium, Southampton, UK) output nominally covers the range 470-2000 nm. An acousto-optical tuneable filter (AOTF) is used to select up to 8 wavelengths from this range. Because the width of the AOTF passbands increases with wavelength and because the spectral profile of the supercontinuum is not flat, it is necessary to select multiple wavelengths to obtain sufficient power at the blue end of the spectrum. (However, it is not possible to select wavelengths that are very close together without reducing the power throughput.) To excite the three fluorophores Alexa 488, Alexa 546 and Atto 647N, we used the spectrum shown in Fig. 1a . We used 4 of the channels to excite Alexa 488 (476, 481, 487 and 491 nm) and 2 channels to excite Alexa 546 (557 and 564 nm) to achieve a similar power level to the 638 nm channel used to excite Atto 647N.
Fig. 1.
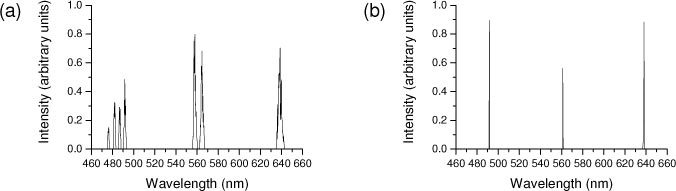
Spectra of laser sources used for multicolour imaging of Alexa 488, Alexa 546 and Atto 647N. (a) Supercontinuum with AOTF, (b) Single-wavelength lasers.
To demonstrate that the supercontinuum laser provides comparable performance to fixed wavelength lasers, we also used three such lasers (Calypso 491 nm, Cobolt, Solna, Sweden; SLIM 561 nm, Oxxius, Lannion, France; IQ1C30 639 nm, Power Technology, Alexander, USA) suited to exciting the same three fluorophores. The narrow spectra of these lasers are shown in Fig. 1b, for comparison.
The output from the AOTF was coupled into the rear port of the microscope (Axio Observer, Zeiss) via a polarization-maintaining optical fibre (HP460, Thorlabs, Newton, USA) and custom-built TIRF module. In the alternative setup, the three single-wavelength lasers were coupled to the TIRF module via a polarization-maintaining wavelength multiplexer (Oz Optics, Ottawa, Canada). Using an image splitter (Optosplit III, Cairn Research, Faversham, UK), the resultant fluorescence was split into three wavelength bands, one for each fluorophore, which were imaged simultaneously onto different regions of the electron-multiplying CCD (iXon+, Andor, Belfast, UK) (Fig. 2 ).
Fig. 2.
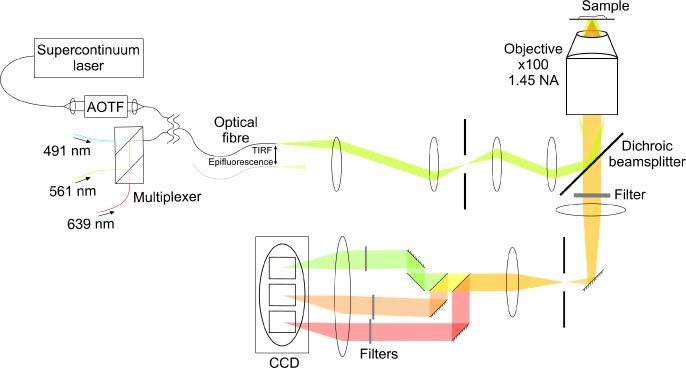
Multicolour total internal reflection microscope, with either a supercontinuum laser or multiplexed single-wavelength lasers as the excitation source.
3. Single molecule imaging
We demonstrated multicolour single molecule detection of signalling proteins on live cells using the human T47D breast cancer cell line. 0.2 nM of each of EGF-Alexa 488, EGF-Alexa 546 and EGF-Atto 647N were all added to the same cell sample, incubated for 15 minutes, washed and imaged immediately, i.e. before ErbB receptor aggregation in coated pits and cell death. Using the supercontinuum, the AOTF channels indicated above and a multiband filter set (LF405/488/561/635; Semrock, Rochester, USA) in the microscope, the total power out of the objective was 0.98 mW for the 475-495 nm range, 0.95 mW for the 550-570 nm range and 0.78 mW for the 635-645 nm range. These powers are similar to those reported by others for single molecule imaging in cells [5,7,13]. The position of the achromatic lens used to couple the beam into the fibre was set to maximise the transmitted power <500 nm as the supercontinuum output is lower in this wavelength range and Alexa 488 is the dimmest of the three dyes. The image splitter contained FF560 and FF650 dichroic beamsplitters and FF525/50, HQ590/60M and FF697/75 clean-up filters (Semrock or Chroma, Brattleboro, USA) in the 3 channels.
Figure 3 shows that the supercontinuum laser is a suitable source for imaging single molecules in live cells. Typical images of the different splitter channels are shown in Fig. 3a. Molecules were identified and their intensites over time determined using custom-written software [14] (Fig. 3b). Careful filter selection is imperative with the supercontinuum because other (unwanted) excitation wavelengths appear in the spectrum when multiple AOTF channels are selected, albeit at a very low intensity (and hence are not seen in Fig. 1a). The fluorescence intensity over time shows characteristics typical of single molecules, including two-step photobleaching (Alexa 488 and Atto 647N) and blinking (Alexa 546). Note that it is significantly more challenging to image single molecules in live cells than in vitro because the signal-to-noise ratio (SNR) is much poorer - the molecules are not spatially fixed, the evanescent field is less uniform and there is an autofluorescence component to the signal. In the 660-695 nm channel, the SNR is much higher, which we attribute to both the superior brightness of Atto 647N and the reduction in autofluorescence with increasing wavelength.
Fig. 3.
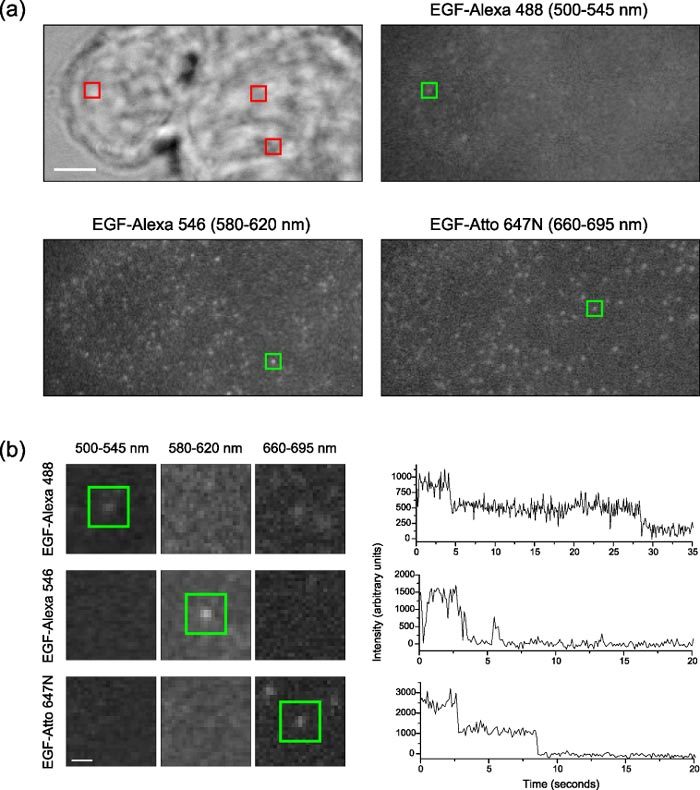
Multicolour single molecule imaging with supercontinuum excitation. (a) White light transmission and single molecule fluorescence images of live T47D cells labelled with EGF-Alexa 488, EGF-Alexa 546 and EGF-Atto 647N, focused at the basolateral membrane. Images were acquired at 10 Hz. Scale bar = 5 µm. (Note: Image registration was performed on the acquired sub-images, which were then remapped to a common reference space to produce the images shown here.) The green squares indicate the molecules analysed in (b), whose locations within the cell are indicated by the red squares. (b) Detail images of the areas around the three analysed molecules in each wavelength band and plots of the intensities of the molecules over time. Scale bar = 1 µm.
Similar results are obtained when single wavelength lasers are used. Figure 4 shows that the characteristic bleaching and blinking behaviour of single molecules is again seen. For example, the fluorescence intensity from Alexa 546 drops to zero in two equal steps, indicating the presence of two colocalised molecules. In this case, the intensities of the 491, 561 and 639 nm lasers out of the objective were 4.1, 1.85 and 4.0 mW respectively; all other experimental details were the same.
Fig. 4.
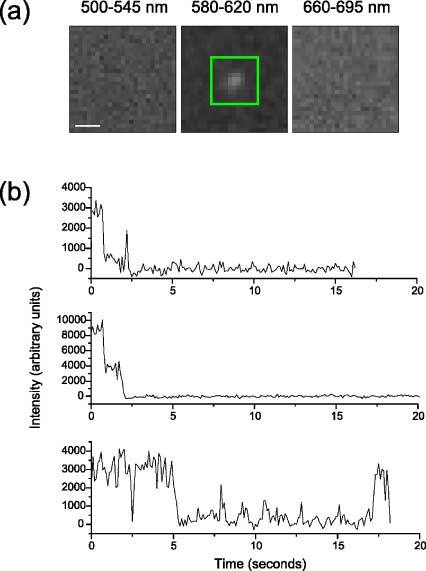
Multicolour single molecule imaging with multiplexed single-wavelength laser excitation. (a) Detail images of the area around two colocalised EGF-Alexa 546 molecules on live T47D cells, in each wavelength band. Images were acquired at 10 Hz. Scale bar = 1 µm. (b) Intensities of typical EGF-Alexa 488, EGF-Alexa 546 and EGF-Atto 647N molecules.
We can also use the supercontinuum to show the binding of the EGF ligand to its receptor (Fig. 5 ). We used the Chinese hamster ovary (CHO) cell line, expressing ErbB1 labelled with EGFP. 0.2 nM EGF-Cy3.5 was added to the live cells for 15 minutes, then washed twice with PBS and imaged immediately. The ~490 and ~580 nm excitation powers out of the objective were 1.12 and 2.05 mW respectively. We used a FITC/TxRed filter set (Semrock) in the microscope and a Q565LP dichroic beamsplitter (Chroma) with FF525/50 and FF620/52 filters (Semrock) in the image splitter. The detail images show that the ligand and receptor are colocalised. The Cy3.5 shows frequent blinking behaviour between two intensities before bleaching completely, revealing that it is a single molecule, but the EGFP intensity trace does not show single-step photobleaching. This is to be expected with live cells as the receptors are free to move. However, it is probable that the fluorescence is due to a single EGFP molecule, because its intensity is comparable to other molecules within the complete image (data not shown), and the decline in intensity over several frames at the end of the trace can be attributed to movement of the complex in the z-axis.
Fig. 5.
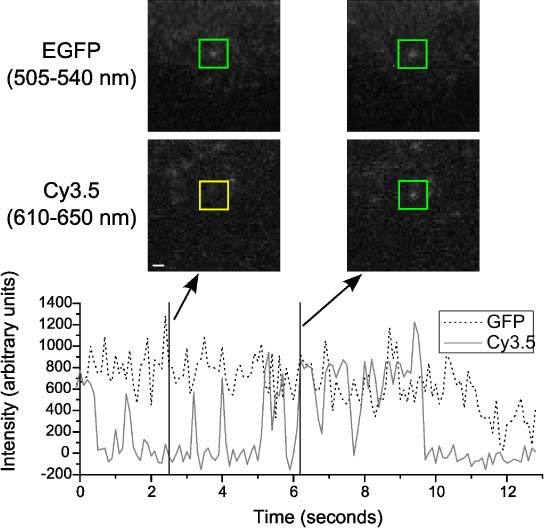
Multicolour single molecule colocalisation imaging with supercontinuum laser excitation. Intensities v time of colocalised ErbB1-EGFP and EGF-Cy3.5 molecules on live CHO cells and detail images of the area around them, in each wavelength band, at the times indicated. The highlighting squares are green at time points when fluorescence features are detected and yellow when they are not. Images were acquired at 10 Hz. Scale bar = 1 µm.
These results also demonstrate the benefits of the supercontinuum source for imaging a wide range of fluorophores. EGFP has very similar spectral properties to Alexa 488 and the 491 nm laser used earlier could be used to excite it equally well. Cy3.5, however, is typically excited at 561 nm because that is a readily available compact laser and commercial filter sets are optimised for it. Unfortunately, the absorption peak of Cy3.5 is at 580 nm. The absorption at 561 nm is ~40% less, which may be sufficient for ensemble fluorescence imaging, but is not ideal for the low SNR found with single molecule detection. The advantage of the supercontinuum laser in imaging dyes such as Cy3.5 is that they can be excited optimally, i.e. at their absorption peak. The trivial changes to the setup that were required to switch from imaging the sample in Fig. 3 to that in Fig. 5 were the replacement of the filter cubes in the microscope and the image splitter and the selection of different AOTF wavelengths.
4. Conclusion
We have demonstrated that supercontinuum lasers provide a flexible excitation source for single molecule fluorescence imaging and provide sufficient intensity for single molecule detection in live cells. They reduce the compromises that have to be made between lasers, dyes and filter sets by providing optimum excitation wavelengths. We have shown that we can determine the intensities of single molecules and follow them through time. These could be used to measure diffusion rates, stoichiometries, clustering and separations of multiple signalling receptors and ligands in live cells. By tagging different members of the ErbB receptor family with different fluorophores, we will be able to study the patterns of association between ErbB1, ErbB2 and ErbB3, for example.
In this paper, we have used a computer-controlled AOTF so that the setup is simple to use for a typical microscopist. Higher incident powers could be achieved using bandpass filters to select wavelengths from the laser, but at the expense of flexibility and ease of use. However, the lower excitation intensities used here are typical for imaging single molecules on cells, rather than in vitro. Many experiments with live cells require molecules to be observed continuously over tens of seconds, which is only possible if the incident power is low enough that most of the fluorophore molecules do not photobleach during this period.
Although a supercontinuum source has previously been incorporated into evanescent field microscopes, both in TIRF [15] and waveguide [16] modes, the excitation power at the sample was insufficient for single molecule detection and the ability to simultaneously excite and detect multiple wavelengths was not exploited.
We note that we do not exploit the fact that the supercontinuum source is pulsed with a repetition rate of 80 MHz. Replacement of the CCD with a gated imager would enable a versatile single molecule FLIM microscope, combining the ideas of [15] and [17].
Acknowledgments
Funding for this work was provided by BBSRC grant BB/G006911/1.
References and links
- 1.Yarden Y., Sliwkowski M. X., “Untangling the ErbB signalling network,” Nat. Rev. Mol. Cell Biol. 2(2), 127–137 (2001). 10.1038/35052073 [DOI] [PubMed] [Google Scholar]
- 2.P. R. Selvin and T. Ha, Single Molecule Techniques: a Laboratory Manual (Cold Spring Harbor Laboratory Press, New York, 2007). [Google Scholar]
- 3.Sako Y., “Imaging single molecules in living cells for systems biology,” Mol. Syst. Biol. 2, 56 (2006). 10.1038/msb4100100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Axelrod D., “Total internal reflection fluorescence microscopy in cell biology,” Traffic 2(11), 764–774 (2001). 10.1034/j.1600-0854.2001.21104.x [DOI] [PubMed] [Google Scholar]
- 5.Hern J. A., Baig A. H., Mashanov G. I., Birdsall B., Corrie J. E. T., Lazareno S., Molloy J. E., Birdsall N. J. M., “Formation and dissociation of M1 muscarinic receptor dimers seen by total internal reflection fluorescence imaging of single molecules,” Proc. Natl. Acad. Sci. U.S.A. 107(6), 2693–2698 (2010). 10.1073/pnas.0907915107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ritchie K., Shan X. Y., Kondo J., Iwasawa K., Fujiwara T., Kusumi A., “Detection of non-Brownian diffusion in the cell membrane in single molecule tracking,” Biophys. J. 88(3), 2266–2277 (2005). 10.1529/biophysj.104.054106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dunne P. D., Fernandes R. A., McColl J., Yoon J. W., James J. R., Davis S. J., Klenerman D., “DySCo: quantitating associations of membrane proteins using two-color single-molecule tracking,” Biophys. J. 97(4), L5–L7 (2009). 10.1016/j.bpj.2009.05.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beausang J. F., Schroeder H. W., III, Nelson P. C., Goldman Y. E., “Twirling of actin by myosins II and V observed via polarized TIRF in a modified gliding assay,” Biophys. J. 95(12), 5820–5831 (2008). 10.1529/biophysj.108.140319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeRocco V. C., Anderson T., Piehler J., Erie D. A., Weninger K., “Four-color single-molecule fluorescence with noncovalent dye labeling to monitor dynamic multimolecular complexes,” Biotechniques 49(5), 807–816 (2010). 10.2144/000113551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vogelsang J., Cordes T., Forthmann C., Steinhauer C., Tinnefeld P., “Controlling the fluorescence of ordinary oxazine dyes for single-molecule switching and superresolution microscopy,” Proc. Natl. Acad. Sci. U.S.A. 106(20), 8107–8112 (2009). 10.1073/pnas.0811875106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Owen D. M., Auksorius E., Manning H. B., Talbot C. B., de Beule P. A. A., Dunsby C., Neil M. A. A., French P. M. W., “Excitation-resolved hyperspectral fluorescence lifetime imaging using a UV-extended supercontinuum source,” Opt. Lett. 32(23), 3408–3410 (2007). 10.1364/OL.32.003408 [DOI] [PubMed] [Google Scholar]
- 12.Wildanger D., Rittweger E., Kastrup L., Hell S. W., “STED microscopy with a supercontinuum laser source,” Opt. Express 16(13), 9614–9621 (2008). 10.1364/OE.16.009614 [DOI] [PubMed] [Google Scholar]
- 13.Sako Y., Minoghchi S., Yanagida T., “Single-molecule imaging of EGFR signalling on the surface of living cells,” Nat. Cell Biol. 2(3), 168–172 (2000). 10.1038/35004044 [DOI] [PubMed] [Google Scholar]
- 14.Rolfe D. J., McLachlan C. I., Hirsch M., Needham S. R., Tynan C. J., Webb S. E. D., Martin-Fernandez M. L., Hobson M. P., “Automated multidimensional single molecule fluorescence microscopy feature detection and tracking,” Eur. Biophys. J. 40(10), 1167–1186 (2011). 10.1007/s00249-011-0747-7 [DOI] [PubMed] [Google Scholar]
- 15.Blandin P., Lévêque-Fort S., Lécart S., Cossec J. C., Potier M. C., Lenkei Z., Druon F., Georges P., “Time-gated total internal reflection fluorescence microscopy with a supercontinuum excitation source,” Appl. Opt. 48(3), 553–559 (2009). 10.1364/AO.48.000553 [DOI] [PubMed] [Google Scholar]
- 16.Agnarsson B., Ingthorsson S., Gudjonsson T., Leosson K., “Evanescent-wave fluorescence microscopy using symmetric planar waveguides,” Opt. Express 17(7), 5075–5082 (2009). 10.1364/OE.17.005075 [DOI] [PubMed] [Google Scholar]
- 17.Knemeyer J. P., Herten D. P., Sauer M., “Detection and identification of single molecules in living cells using spectrally resolved fluorescence lifetime imaging microscopy,” Anal. Chem. 75(9), 2147–2153 (2003). 10.1021/ac026333r [DOI] [PubMed] [Google Scholar]