

Abstract
Full-field optical coherence microscopy (FFOCM) is a high-resolution interferometric technique that is particularly attractive for biomedical imaging. Here we show that combining it with structured illumination fluorescence microscopy on one platform can increase its versatility since it enables co-localized registration of optically sectioned reflectance and fluorescence images. To demonstrate the potential of this dual modality, a fixed and labeled mouse retina was imaged. Results showed that both techniques can provide complementary information and therefore the system could potentially be useful for biomedical imaging applications.
OCIS codes: (180.3170) Interference microscopy, (180.2520) Fluorescence microscopy, (170.6900) Three-dimensional microscopy
1. Introduction
Optical coherence tomography (OCT) and Fluorescence microscopy are widely used techniques in biomedical imaging. Full-field optical coherence microscopy (FFOCM) [1,2] is a wide-field, high-resolution form of OCT. It utilizes relatively inexpensive spatially incoherent illumination source and an array detector to produce en face images. In contrast to other types of tissue microscopic imaging modalities, like confocal microscopy, FFOCM can achieve isotropic resolution of <1 µm in 3D [3]. FFOCM has been shown to be a useful technique to study microstructural morphology and reveal subcellular structures in a range of biological samples [4] deep within scattering tissues [5,6].
In general FFOCM, like other OCT techniques, can provide structural information without administration of any contrast dye. However, techniques like fluorescence microscopy can record images with enhanced contrast of specific molecules, if appropriate fluorescent label is used. Advantage of fluorescence microscopy implies that combining the two techniques can increase the versatility of each modality since each can provide supplementary information, as has been previously demonstrated [7]. Fluorescence imaging and OCT, for example, has recently been combined in our lab for intra-arterial imaging in vivo [8]. Furthermore, OCT (FFOCM) images of biomedical samples often have to be compared to gold standards, like histology or fluorescence imaging, in order to establish and validate the contents of the reflectance image. If a sample is imaged with FFOCM and fluorescence microscopy on two separate microscopes, it can be time consuming and there can be significant registration difficulties. The combination of FFOCM and fluorescence microscopy is natural—only minimal modifications are necessary, as the standard FFOCM configuration is already suitable to perform conventional wide-field fluorescence imaging—simply a set of appropriate filters and a more sensitive camera is needed. Both techniques have an advantage of being able to use spatially incoherent illumination sources, like Xenon or Mercury arc lamps, which are relatively inexpensive compared to laser sources. Moreover, such a spectrally broadband sources allows spectral tunability, which is attractive feature for fluorescence microscopy. Conventional wide-field fluorescence microscopy, however, does not provide optical sectioning, which results in the detection of out-of-focus light and, therefore, its application is mainly limited to thin samples. To address that problem, so that thicker tissues can be imaged, we have combined FFOCM with structured illumination microscopy technique [9,10]. For that, a grid inserted into the illumination path of the microscope was focused on a sample so as to produce one-dimensional sinusoidal excitation. Several images, with the grid translated by a defined distance, were recorded and processed mathematically [9], in order to extract an optically sectioned fluorescence image. To illustrate the performance of this dual imaging microscopy system for biomedical imaging, we measured its axial resolution and imaged fixed and stained mouse retina.
2. Methods
2.1. Full-field optical coherence microscopy
The FFOCM setup, shown in Fig. 1(a) , was based on the Linnink interference microscope configuration [2,11]. A broadband (central wavelength, λ = 0.6 µm and spectral bandwidth, Δλ = 0.2 µm, as measured in front of the camera) and spatially incoherent illumination was provided by a 75 W Xenon arc lamp source (Oriel 6263) coupled into a 1.00 mm diameter multimode fiber (Oriel 77519). Light from the fiber was collimated with a 10 × microscope objective. A 50:50 broadband beamsplitter divided the illumination equally to sample and reference arms. Each arm consisted of an identical 20 × water immersion microscope objective (Olympus, UMPlanFl, 0.5 NA). The reference arm contained absorptive neutral density (ND) filter in its focal plane to provide a weak reflection from its first surface while attenuating the reflection from the back surface. Light reflected from ND and sample was collimated by the same objectives and recombined with the beamsplitter. A 30 cm focal length, visible achromatic doublet lens (tube lens), was used to focus light on a CMOS camera (MV-D1024E-160-CL-12, PhotonFocus). A motorized translation stage (Picomotor 8302, New Focus) was used to move the sample along the optical axis.
Fig. 1.
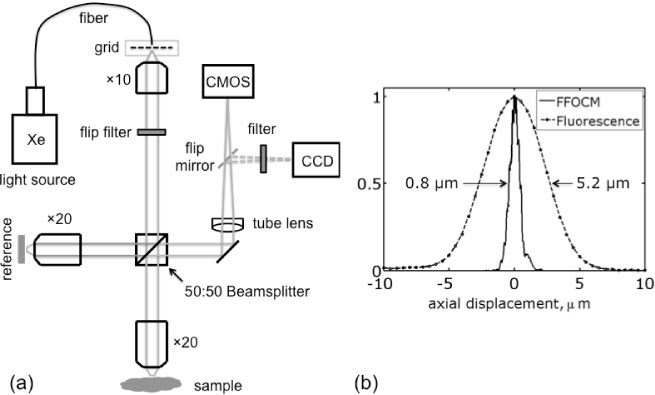
(a) A schematic diagram of dual imaging microscope. The filter and mirror are flipped-in to switch between FFOCM and fluorescence microscopy. A spatial intensity grid is added to implement structured illumination fluorescence microscopy technique. (b) Comparison of measured optical sectioning performance for FFOCM and fluorescence structured illumination microscopy.
The two beams interfered on the camera only if their optical path length difference was within the temporal coherence length of the illumination source. The path length difference between the two beams was modulated by either moving ND filter on its own, or together with the objective lens, using a piezo actuator (AE0203D04F, Thorlabs). Image acquisition by the camera was synchronized with the beam modulation using a DAQ card. To reconstruct an optically sectioned image, a coherent part of the signal was extracted by recording four images over a single modulation period and processed by a simple mathematical algorithm, described in [2,12]. For biological imaging, typically 70 images (512 × 512), acquired with 200 Hz acquisition speed, were averaged in order to increase signal to noise ratio (S/N) [2]. These acquisition parameters resulted in a total acquisition time of over 1 sec for one axially sectioned FFOCM image and sensitivity of 75 dB. All FFOCM images were represented on logarithmic scale. To measure the optical sectioning strength of FFOCM, two identical mirrors were placed in the reference and sample arms and the mirror in the latter was scanned along the optical axis. The microscope was aligned so that the beams coming from both arms were collinear. The resultant optical sectioning curve, derived from the acquired image stack, is shown in Fig. 1(b). The full width half-maximum (FWHM) of the curve was determined to be 0.8 µm, which is consistent with previously reported values for similar high-resolution FFOCM systems [2,12].
2.2. Structured illumination fluorescence microscopy
To switch from FFOCM to structured illumination fluorescence microscopy, a grid and a bandpass excitation filter (540/50 nm) were inserted into the illumination path, and a flipper mirror was used to direct fluorescence to a CCD camera (Cascade II:512, Photometrics), as shown in Fig. 1(a). A longpass 570 nm emission filter in front of the camera was used to filter out the excitation light. For accurate grid translation, which is important in producing artifact-free images [13], a closed-loop piezo actuator (DRV120, Thorlabs) driven by a piezo controller (BPC201, Thorlabs) was used. In order to remove grid structure from a sample, and thus reconstruct optically sectioned image, at least three images have to be recorded, as explained in [9]. However, here we used four images since it produced less grid artifacts in the reconstructed image. The grid illumination on the sample had a spatial frequency, ν of 0.35 µm−1 and a normalized spatial frequency νn = νλ/NA of 0.38. To characterize optical sectioning strength, a layer of 0.5 µm yellow-green fluorescent FluoSpheres beads (Invitrogen) spread on a coverslip, was used. The measured optical sectioning curve is shown in Fig. 1(b). The FWHM values of the measured and calculated [14,15] optical sectioning curves were estimated to be of 5.2 µm and 4.8 µm, respectively. The mismatch between the values can be largely attributed to the optical aberrations.
Lateral resolution for fluorescence microscopy and FFOCM should essentially be the same and, following Rayleigh formula (0.61 × λ/NA), was estimated to be 0.67 µm.
2.3. Mouse retina sample
We received paraformaldehyde fixed C57B6/J mouse retinas with oxygen-induced retinopathy from The Schepens Eye Research Institute, prepared following procedure described in [16]. They were stained for endothelial cells with isolectin GS-IB4 Alexa Fluor® 594 (Molecular Probes). We used this model to demonstrate morphology of blood vessels seen by fluorescence in conjunction with the microstructure of the retina provided by FFOCM.
3. Results
Figure 2 shows x-z (axial) cross-section of wide-field (a), structured illumination (b), and FFOCM (c) images, which clearly demonstrate the optical sectioning performance differences between the techniques. Different retinal layers can be identified in the FFOCM image (Fig. 2(c)), such as ganglion cell layer (GCL), inner plexiform layer (IPL) and others. Similar high resolution FFOCM image has been demonstrated previously in [17]. In addition, the FFOCM images clearly enable visualization of ganglion cell bodies (arrows in Fig. 2(c)), which are located in the ganglion cell layer. The structured illumination fluorescence image, on the other hand, clearly shows axial distribution of the blood vessels, shown in Fig. 2(b).
Fig. 2.
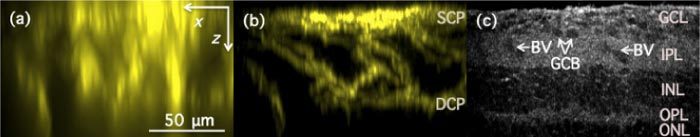
x-z (axial) images of fixed and stained mouse retina as acquired with conventional wide-field fluorescence (a), structured illumination fluorescence (b) and full-field optical coherence (c) microscopy techniques. Images (a)&(b) were constructed as the maximum value pixel projection from 512 x-z sections of the reconstructed images. Capillary plexi: SCP –superficial capillary plexus; DCP—deep capillary plexus; Retinal layers: GCL—Ganglion cell layer; IPL—Inner plexiform layer; INL—Inner nuclear layer; OPL—Outer plexiform layer; ONL—Outer nuclear layer; Arrows indicate: BV—Blood vessel; GCB—Ganglion cell bodies.
The blood vessels are known to form three distinct capillary plexi in retina [18], but only two of them are seen in our images—the superficial capillary plexus (SCP) and the deep capillary plexus (DCP), because of age of mice used in this study and their treatment [18]. Capillary plexi can also be identified in x-y (lateral) images in Fig. 3 and Fig. 4 . Under certain circumstances blood vessels can sometimes be identified in FFOCM images, but their appearance depend on their location in retinal layer and frequently they cannot be differentiated from the surroundings.
Fig. 3.
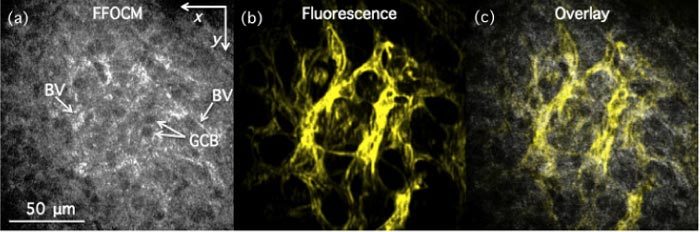
x-y (lateral) images of fixed mouse retina with labeled endothelial cells as acquired with FFOCM (a) and structured illumination fluorescence microscopy (b) in ganglion cell layer. (c) Overlay of (a) and (b). GCB—Ganglion cell bodies; BV—blood vessels.
Fig. 4.

x-y overlay of FFOCM (gray) and fluorescence (yellow) images recorded in different fixed mouse retinal layers with labeled endothelial cells (Media 1 (2.9MB, MOV) ).
For example, blood vessels indicated by arrows in Fig. 2(c), appeared as black voids in the inner plexiform layer (IPL), as is also evident in the x-y (lateral) overlay image in Fig. 4(a). On the other hand, blood vessels appeared as higher intensity structures in Fig. 3(a) (two of them are indicated by arrows), as can also be seen in Fig. 4(b). We were only able to make these determinations after comparing FFOCM data to the fluorescence images recorded from exactly the same place. 3D rendering of the data was also performed to highlight the volumetric image capabilities of the multimodality technique. Figure 5 shows snapshots at three different angles of a 3D data set that was created by fusing the fluorescence data into the FFOCM volume (rendered in OsiriX 3.5). The blood vessel three-dimensional structure, forming the two distinct capillary plexi, can thus be better appreciated.
Fig. 5.
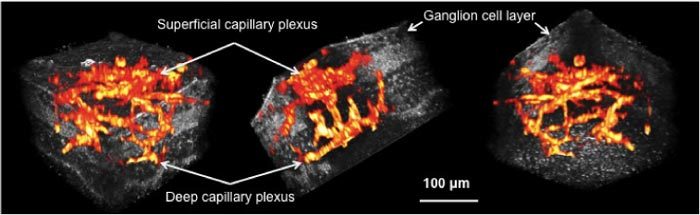
3D cutaway view (Media 2 (4.7MB, MOV) ) of the mouse retina acquired by the dual-modality microscope (gray—FFOCM signal, red-yellow—fluorescence signal).
4. Discussion and Conclusions
The ability of the dual-modality imaging to provide a co-localized FFOCM and fluorescence signal can be beneficial for imaging biomedical samples. The two techniques provide complementary information, which can be used to obtain molecular information in the context of tissue microstructure or for validating OCT images. For example, here we showed that appearance of blood vessels in FFOCM images depend on their location in the retinal layer. Simultaneous FFOCM and fluorescence imaging was, however, not possible in the current configuration because of fluorescence spectral overlap with the broadband illumination, necessary to achieve high axial resolution FFOCM images. To enable simultaneous imaging, a near-infrared fluorescence dye could be used instead of the visible, in conjunction with a dichroic beamsplitter between the 50:50 beamsplitter and the objective lens in the sample arm (and a dispersion compensating glass in the reference arm). Fluorescence, reflected from the dichroic beamsplitter, could then be focused onto a CCD by a separate tube lens. The use of the dichroic beamsplitter would also enable a more efficient fluorescence collection compared to the current configuration, where half of the signal is lost because of the 50:50 beamsplitter. However, the grid would also be present in FFOCM images, which perhaps could be eliminated by, for example, summing up FFOCM images recorded at the different grid positions. Alternatively, infrared FFOCM [5,19] and visible fluorescence microscopy could be implemented by using two separate (visible and infrared) illumination sources, combined by the help of another appropriate dichroic mirror. A grid, inserted only in the visible illumination path, would not show up in the FFOCM images. Possible endoscopic microscopy applications have been previously reported for FFOCM [20,21] and structured illumination fluorescence microscopy [22,23], showing that this dual-modality technique can potentially be used for in vivo imaging.
Acknowledgments
We acknowledge financial support from the Air Force Office of Scientific Research under grant FA9550-10-1-0537 and the NIH under grants R01HL076398, R01HL093717, R01CA103769, R01DK091923.
References and links
- 1.Beaurepaire E., Boccara A. C., Lebec M., Blanchot L., Saint-Jalmes H., “Full-field optical coherence microscopy,” Opt. Lett. 23(4), 244–246 (1998). 10.1364/OL.23.000244 [DOI] [PubMed] [Google Scholar]
- 2.Dubois A., Vabre L., Boccara A. C., Beaurepaire E., “High-resolution full-field optical coherence tomography with a Linnik microscope,” Appl. Opt. 41(4), 805–812 (2002). 10.1364/AO.41.000805 [DOI] [PubMed] [Google Scholar]
- 3.Dubois A., Moneron G., Grieve K., Boccara A. C., “Three-dimensional cellular-level imaging using full-field optical coherence tomography,” Phys. Med. Biol. 49(7), 1227–1234 (2004). 10.1088/0031-9155/49/7/010 [DOI] [PubMed] [Google Scholar]
- 4.Jain M., Shukla N., Manzoor M., Nadolny S., Mukherjee S., “Modified full-field optical coherence tomography: A novel tool for rapid histology of tissues,” J Pathol Inform 2(1), 28 (2011). 10.4103/2153-3539.82053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oh W. Y., Bouma B. E., Iftimia N., Yun S. H., Yelin R., Tearney G. J., “Ultrahigh-resolution full-field optical coherence microscopy using InGaAs camera,” Opt. Express 14(2), 726–735 (2006). 10.1364/OPEX.14.000726 [DOI] [PubMed] [Google Scholar]
- 6.Ben Arous J., Binding J., Léger J. F., Casado M., Topilko P., Gigan S., Boccara A. C., Bourdieu L., “Single myelin fiber imaging in living rodents without labeling by deep optical coherence microscopy,” J. Biomed. Opt. 16(11), 116012 (2011). 10.1117/1.3650770 [DOI] [PubMed] [Google Scholar]
- 7.Beaurepaire E., Moreaux L., Amblard F., Mertz J., “Combined scanning optical coherence and two-photon-excited fluorescence microscopy,” Opt. Lett. 24(14), 969–971 (1999). 10.1364/OL.24.000969 [DOI] [PubMed] [Google Scholar]
- 8.Yoo H., Kim J. W., Shishkov M., Namati E., Morse T., Shubochkin R., McCarthy J. R., Ntziachristos V., Bouma B. E., Jaffer F. A., Tearney G. J., “Intra-arterial catheter for simultaneous microstructural and molecular imaging in vivo,” Nat. Med. 17(12), 1680–1684 (2011). 10.1038/nm.2555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neil M. A., Juškaitis R., Wilson T., “Method of obtaining optical sectioning by using structured light in a conventional microscope,” Opt. Lett. 22(24), 1905–1907 (1997). 10.1364/OL.22.001905 [DOI] [PubMed] [Google Scholar]
- 10.Neil M. A., Squire A., Juškaitis R., Bastiaens P. I., Wilson T., “Wide-field optically sectioning fluorescence microscopy with laser illumination,” J. Microsc. 197(1), 1–4 (2000). 10.1046/j.1365-2818.2000.00656.x [DOI] [PubMed] [Google Scholar]
- 11.Vabre L., Dubois A., Boccara A. C., “Thermal-light full-field optical coherence tomography,” Opt. Lett. 27(7), 530–532 (2002). 10.1364/OL.27.000530 [DOI] [PubMed] [Google Scholar]
- 12.Dubois A., Grieve K., Moneron G., Lecaque R., Vabre L., Boccara C., “Ultrahigh-resolution full-field optical coherence tomography,” Appl. Opt. 43(14), 2874–2883 (2004). 10.1364/AO.43.002874 [DOI] [PubMed] [Google Scholar]
- 13.Cole M. J., Siegel J., Webb S. E., Jones R., Dowling K., Dayel M. J., Parsons-Karavassilis D., French P. M., Lever M. J., Sucharov L. O., Neil M. A., Juškaitis R., Wilson T., “Time-domain whole-field fluorescence lifetime imaging with optical sectioning,” J. Microsc. 203(3), 246–257 (2001). 10.1046/j.1365-2818.2001.00894.x [DOI] [PubMed] [Google Scholar]
- 14.Webb S. E. D., Gu Y., Leveque-Fort S., Siegel J., Cole M. J., Dowling K., Jones R., French P. M. W., Neil M. A. A., Juškaitis R., Sucharov L. O. D., Wilson T., Lever M. J., “A wide-field time-domain fluorescence lifetime imaging microscope with optical sectioning,” Rev. Sci. Instrum. 73(4), 1898–1907 (2002). 10.1063/1.1458061 [DOI] [Google Scholar]
- 15.Karadaglić D., Wilson T., “Image formation in structured illumination wide-field fluorescence microscopy,” Micron 39(7), 808–818 (2008). 10.1016/j.micron.2008.01.017 [DOI] [PubMed] [Google Scholar]
- 16.Connor K. M., Krah N. M., Dennison R. J., Aderman C. M., Chen J., Guerin K. I., Sapieha P., Stahl A., Willett K. L., Smith L. E., “Quantification of oxygen-induced retinopathy in the mouse: a model of vessel loss, vessel regrowth and pathological angiogenesis,” Nat. Protoc. 4(11), 1565–1573 (2009). 10.1038/nprot.2009.187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Le Gargasson J. F., Grieve K., Paques M., Dubois A., Boccara C., “Imaging of ocular tissues using ultrahigh resolution full-field optical coherence tomography,” Invest. Ophthalmol. Vis. Sci. 45, E-Abstract 2784 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Stahl A., Connor K. M., Sapieha P., Chen J., Dennison R. J., Krah N. M., Seaward M. R., Willett K. L., Aderman C. M., Guerin K. I., Hua J., Löfqvist C., Hellström A., Smith L. E. H., “The mouse retina as an angiogenesis model,” Invest. Ophthalmol. Vis. Sci. 51(6), 2813–2826 (2010). 10.1167/iovs.10-5176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dubois A., Moneron G., Boccara C., “Thermal-light full-field optical coherence tomography in the 1.2 µm wavelength region,” Opt. Commun. 266(2), 738–743 (2006). 10.1016/j.optcom.2006.05.016 [DOI] [Google Scholar]
- 20.Latrive A., Boccara A. C., “In vivo and in situ cellular imaging full-field optical coherence tomography with a rigid endoscopic probe,” Biomed. Opt. Express 2(10), 2897–2904 (2011). 10.1364/BOE.2.002897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oh W. Y., Bouma B. E., Iftimia N., Yelin R., Tearney G. J., “Spectrally-modulated full-field optical coherence microscopy for ultrahigh-resolution endoscopic imaging,” Opt. Express 14(19), 8675–8684 (2006). 10.1364/OE.14.008675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karadaglić D., Juškaitis R., Wilson T., “Confocal endoscopy via structured illumination,” Scanning 24(6), 301–304 (2002). 10.1002/sca.4950240604 [DOI] [PubMed] [Google Scholar]
- 23.Bozinovic N., Ventalon C., Ford T., Mertz J., “Fluorescence endomicroscopy with structured illumination,” Opt. Express 16(11), 8016–8025 (2008). 10.1364/OE.16.008016 [DOI] [PubMed] [Google Scholar]