

Abstract
The pathophysiological mechanisms of both familial and sporadic Amyotrophic Lateral Sclerosis (ALS) are unknown, although growing evidence suggests that skeletal muscle tissue is a primary target of ALS toxicity. Skeletal muscle biopsies were performed on transgenic SOD1G93A mice, a mouse model of ALS, to determine genetic biomarkers of disease longevity. Mice were anesthetized with isoflurane, and three biopsy samples were obtained per animal at the three main stages of the disease. Transcriptional expression levels of seventeen genes, Ankrd1, Calm1, Col19a1, Fbxo32, Gsr, Impa1, Mef2c, Mt2, Myf5, Myod1, Myog, Nnt, Nogo A, Pax7, Rrad, Sln and Snx10, were tested in each muscle biopsy sample. Total RNA was extracted using TRIzol Reagent according to the manufacturer's protocol, and variations in gene expression were assayed by real-time PCR for all of the samples. The Pearson correlation coefficient was used to determine the linear correlation between transcriptional expression levels throughout disease progression and longevity. Consistent with the results obtained from total skeletal muscle of transgenic SOD1G93A mice and 74-day-old denervated mice, five genes (Mef2c, Gsr, Col19a1, Calm1 and Snx10) could be considered potential genetic biomarkers of longevity in transgenic SOD1G93A mice. These results are important because they may lead to the exploration of previously unexamined tissues in the search for new disease biomarkers and even to the application of these findings in human studies.
Introduction
According to Biomarkers Definitions Working Group, a biomarker, which must be objectively measured and evaluated, is “an indicator of normal biological processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention” as well as “an indicator of functional and structural changes in organs and cells”. Therefore, biomarkers can also be considered potential therapeutic molecular targets [1].
Amyotrophic Lateral Sclerosis (ALS) is one of the most common neurodegenerative disorders, and the search for molecular markers is increasing. This is especially true of the search for prognostic markers that might be involved in or promote the neurodegeneration process. These markers can then use to predict the outcome for a patient who is suffering from the disease. Because familial and sporadic ALS share clinical and pathological signs, understanding of the pathophysiological processes in familial ALS (FALS) would also provide a better understanding of the neurodegenerative mechanisms in sporadic ALS (SALS) [2]. FALS follows a predominantly autosomal dominant pattern; in SALS, genetic factors that occur sporadically contribute to its pathogenesis. In particular, mutations in the copper/zinc superoxide-dismutase-1 gene (SOD1) [3]–[5], Tar DNA-binding protein gene (TARDBP) and, most recently discovered, the DNA/RNA-binding proteins FUS (fused in sarcoma) or TLS (translocation in liposarcoma) produce the typical adult-onset ALS phenotype, suggesting that alterations in RNA processing may play a central role in ALS pathogenesis [6]–[13].
Interestingly, a wide range of molecules involved in different molecular pathways in ALS, such as excitotoxicity, inflammation and oxidative stress, have been described during the last three decades as possible biomarkers of the disease [14]–[23]. Many studies have been carried out to detect molecular markers for the diagnosis of ALS in tissues such as the brain, spinal cord, blood or cerebrospinal fluid (CSF).
Although skeletal muscle plays an important role in the neurophysiological diagnosis of ALS [24], [25], growing evidence supports the fact that it can be considered a primary target of ALS toxicity [22], [26], [27]. Under neurodegenerative conditions in ALS, muscle atrophy can result from lost connections to motor neurons in the neuromuscular junction; this loss of connections is likely due to an energetic deficit in the mutant muscle that leads to pathological conditions [27]. Consequently, skeletal muscle shares a direct connection with the nervous system and can clearly contribute to an alteration in the functional communication between muscle and nerve. Furthermore, the study of ALS markers in this tissue suggests that it may be possible to take muscle biopsy samples from live patients or animal models; however, this is not the case for brain and spinal cord samples. The availability of muscle biopsy samples might make it possible to carry out more accurate studies during disease progression, especially when markers of the disease have yet to be found.
The transgenic mice that have a G93A mutation in the SOD1 gene (SOD1G93A) are one of the best characterized animal models for ALS disease and present both clinical and pathological characteristics of ALS patients [14]. The aim of this study was to use this mouse model to search for potential genetic biomarkers of the disease that could be used to predict the animals' longevity based on their transcriptional expression during disease progression. Among the seventeen genes tested, only five, Mef2c, Gsr, Col19a1, Calm1 and Snx10, may be considered potential genetic biomarkers of longevity in ALS disease as there was a significant linear correlation between their transcriptional profile during disease progression and the longevity of the animals.
Results
Significant variation in gene expression profiles in transgenic SOD1G93A mice throughout disease progression
The profile expression pattern of the seventeen genes tested varied significantly throughout disease progression in the skeletal muscle of transgenic SOD1G93A mice when compared to control, age-matched and wild type mice; the exception was Calm1, though this gene had an irregular expression pattern throughout the three disease stages. The transcriptional expression levels of Sln, another gene involved in calcium homeostasis, exhibited a significant variation in expression, indicating dysfunctions in calcium influx and calcium-induced endocytosis [28] (Figure 1). Moreover, Ankrd1 and Col19a1 produced the most significant difference in transcriptional expression patterns, especially at the terminal stage of the disease. This indicates there was an increase in muscle differentiation throughout disease progression that was prompted by an upregulation of Col19a1 [29] and followed by an upregulation of Myog, Myod1, Myf5 and Mef2c. Interestingly, the only myogenic regulatory factor that displayed a downregulation was Pax7, which would suggest that the myogenic potential and therefore the capacity for tissue repair were significantly diminished as neurodegeneration progressed; this finding is in accordance with the results of previous studies [30]. Significantly, this neurodegenerative progression also induced an upregulation in the transcriptional expression levels of Ankrd1 and, to a lesser extent, Nogo A, Fbxo32 and Snx10, which have previously found to be altered under degenerative conditions [27], [31]–[35]. Furthermore, we observed a significant upregulation in genes related to metabolic processes, Impa1, Nnt, Rrad, Gsr and Mt2, which may be altered under neurodegenerative conditions due to the uncoupled metabolic pathways they are involved in [36]–[39].
Figure 1. Transcriptional expression levels of the sixteen genes varied significantly throughout disease progression in transgenic SOD1G93A mice.

Representative graphs showing the fold change in transcriptional levels of the seventeen genes tested in the skeletal muscle of transgenic SOD1G93A mice throughout disease progression with respect to the early symptomatic stage. Age-matched wild type mice were used as controls in each stage of the disease: early symptomatic (60 days, grey bar), symptomatic (90 days, blue bar) and terminal (120 days, pale blue bar) stages. The highest transcriptional expression levels were found in Ankrd1 and Col19a1, which at the terminal stage were almost 27 and 18 times higher than those observed at the early symptomatic stage, respectively. A significant upregulation of transcriptional levels was found in all of the genes, except for Calm1, despite its irregular profile pattern throughout disease progression.
Transcriptional expression of thirteen genes studied in muscle biopsies correlated in a linear fashion with longevity
The correlation study between gene expression profile and longevity focused on previously studied genes in transgenic SOD1G93A and age-matched wild type mice. We hypothesized that those genes whose transcriptional expression yielded statistical significance during the progression of the disease in transgenic SOD1G93A mice would be more likely to vary linearly through the disease stages and that they would correlate with longevity because they were involved in the neurodegenerative process of ALS. Therefore, the expression patterns of these genes could be used to predict longevity in transgenic SOD1G93A mice.
The Pearson's correlation coefficients from each correlation study are shown in Table 1. The longevity of the mice ranged between 120 and 160 days. Importantly, we found that the transcriptional expression levels during disease progression of thirteen genes, Myf5, Mef2c, Gsr, Myod1, Col19a1, Calm1, Myog, Snx10, Pax7, Impa1, Mt2, Ankrd1 and Sln, correlated significantly and negatively with longevity (Table 1). This negative correlation with longevity implies that the animals that survived longer displayed lower transcriptional levels of these genes during disease progression at the early symptomatic stage. Interestingly, unlike the other genes studied, Mt2 and Nnt, displayed a positive correlation, which implied a longer survival when the transcriptional levels of Mt2 and Nnt increased during the progression of the disease at the early symptomatic stage. The highest Pearson's correlation coefficients were found when testing Myf5, Mef2c, Gsr, Myod1 and Col19a1, while the lowest coefficients were found for Calm1, Myog, Snx10, Pax7, Impa1, Mt2, Ankrd1 and Sln, though they were still statistically significant. However, the transcriptional expression levels of Nnt, Fbxo32, Rrad and Nogo A did not display a significant correlation with longevity. These results suggest that an increase in myogenic potential may compensate for the muscle damage induced by the neurodegenerative progression of the disease, and therefore, the animals that survived longer may exhibit a higher regenerative capacity than the animals that survived for less time. It is possible that the animals with a shorter survival time had exhausted their myogenic regenerative capacity by overexpressing the myogenic precursors Myf5, Mef2c, Myod1 and Col19a1 at the early stages. This hypothesis is supported by the results shown in Figure 1 that indicated a significantly diminished myogenic potential at the terminal stage of 120 days, which is the shortest survival time among animals included in the muscle biopsy study.
Table 1. Pearson's correlation coefficients and statistical significance in the seventeen genes studied in the skeletal muscle biopsies of transgenic SOD1G93A mice.
| Gene | Pearson's correlation coefficient, r | p value |
| MYF5 | −0,599 | 0,000 |
| MEF2C | −0,552 | 0,000 |
| GSR | −0,547 | 0,000 |
| MYOD1 | −0,527 | 0,000 |
| COL19A1 | −0,440 | 0,000 |
| CALM1 | −0,372 | 0,000 |
| MYOG | −0,339 | 0,001 |
| SNX10 | −0,330 | 0,002 |
| PAX7 | −0,292 | 0,007 |
| IMPA1 | −0,265 | 0,014 |
| MT2 | 0,256 | 0,018 |
| ANKRD1 | −0,244 | 0,001 |
| SLN | −0,242 | 0,024 |
| NNT | 0,208 | 0,056 |
| FBXO32 | −0,188 | 0,080 |
| RRAD | −0,166 | 0,129 |
| NOGO A | −0,161 | 0,137 |
Furthermore, expression levels of SOD1G93A were measured as their significant variation could affect disease onset and progression. No statistical differences were found among the relative expression levels of SOD1G93A in all the samples obtained from the three biopsies (Figure 2). This result confirmed the constant expression levels of SOD1G93A in all the studied biopsies, suggesting that the neurodegenerative progress of the disease induced the different gene expression profiles in the studied genes.
Figure 2. Transcriptional SOD1G93A fold change in muscle biopsy samples.

The transcriptional expression levels of SOD1G93A were measured in all the samples obtained from the muscle biopsies corresponding to early symptomatic (first biopsy), symptomatic (second biopsy) and terminal stages (third biopsy). No statistical differences were found in SOD1G93A levels along disease progression. SOD1G93A fold change in the symptomatic and terminal stages was calculated respect to the relative expression found in all the muscle samples extracted at the early symptomatic stage.
From these results, we hypothesized that some of the genes that presented a significant correlation with longevity were involved in the denervation process that is present not only in ALS but also in all other known neurodegenerative disorders. To test this hypothesis, we used a denervation mouse model. To avoid false positive results, we tested all seventeen genes.
The expression profiles of eleven genes varied significantly in the skeletal muscle of denervated mice
In this study, we identified the genes that were significantly altered in the skeletal muscle of denervated mice to differentiate the genes that are directly involved in the denervation process due to the disease from those that may be involved in the specific neurodegenerative process of ALS.
Among the seventeen genes studied, eleven displayed significantly different transcriptional levels in denervated mice compared to age-matched wild type mice. Similar to the results obtained from transgenic SOD1G93A mice during disease progression (Figure 1), the expression levels of Ankrd1, Rrad, Myog, Mt2, Myod1, Sln, Myf5, Pax7, Nogo A and Impa1 were significantly increased in denervated mice; Fbxo32, however, displayed a decreased expression profile (Figure 3). Though the upregulation of Fbxo32 has been associated with ALS [34], its downregulation appears to be related to an internal mechanism involved in reducing further loss of muscle proteins in denervation-induced muscle atrophy [40], which may explain our results. We also observed a downregulation of the transcriptional levels of Pax7 in transgenic SOD1G93A mice during disease progression (Figure 1), though in denervated mice, an overexpression of this gene was detected. This would suggest that mutant SOD1 toxicity might limit the capacity of mutant muscle to regenerate because the Pax7-expressing muscle progenitor pool in denervated muscles may remain less limited than in mutant muscles [30].
Figure 3. Eleven genes were related to the denervation process.

Fold change variation in the transcriptional levels of seventeen genes in the skeletal muscle of 74-day-old denervated mice. Wild type mice aged for 60 days were used as controls. Among the seventeen genes tested, the transcriptional levels of Ankrd1, Rrad, Myog, Mt2, Myod1, Sln, Myf5, Pax7, Nogo A, Impa1 and Fbox32 varied significantly in denervated mice compared to control mice. Fbox32 was the only gene that displayed a downregulated transcription level in denervated mice, which is probably due to its role in compensating for denervation-induced muscle atrophy via an internal mechanism.
Five potential genetic biomarkers of longevity in ALS
The results observed under denervation conditions show that among the thirteen genes that displayed a significant correlation between transcriptional level expression and longevity during disease progression, five of them, Mef2c, Gsr, Col19a1, Calm1 and Snx10 may be considered potential biomarkers of longevity in ALS disease. The linear regression plots for these genes are shown in Figure 4. These results support the hypothesis that myogenic potential is significantly favored in animals that display longer survival and that an increase in transcriptional expression of these five genes makes it is possible to predict shorter longevity in transgenic SOD1G93A mice.
Figure 4. Mef2c, Gsr, Col19a1, Calm1 and Snx10 are potential genetic biomarkers of longevity in ALS.

Linear correlation graphs of five potential genetic biomarkers of longevity. The graph shows the linear relation between the fold change in transcriptional levels of these genes throughout disease progression in skeletal muscle biopsies at the early symptomatic stage and the longevity of the animals at the terminal stage. The transcriptional levels of these genes can predict longevity in transgenic SOD1G93A mice.
Discussion
Since Amyotrophic Lateral Sclerosis (ALS) was discovered and described in 1869 as a neurodegenerative disease characterized by motor neuron death and muscular atrophy, a wide range of biomarkers have been examined in the search for a therapeutic target. Although ALS shares altered molecular pathways with other neurodegenerative diseases, such as Alzheimer's, Huntington's or Parkinson's disease, there is an obvious need for specific ALS molecular markers that will allow an easier and earlier prognosis and/or diagnosis.
Because the skeletal muscle may be considered a primary target of ALS toxicity [22], [26], [27], we initially tested the gene expression profiles of seventeen genes using skeletal muscle from transgenic SOD1G93A mice at different stages of disease progression. Interestingly, all the genes displayed significant changes in transcriptional expression throughout the three main stages of the disease, except for Calm1, though this was probably due to the high variability in its expression observed throughout the disease (Figure 1). An increase in the level of Calm1 expression under degenerative conditions in mutant muscle, especially during the terminal stage, could activate expression of Mef2c and thereby activate a regenerative myogenic pathway. Similarly, increasing levels of Col19a1, Mef2c, Myf5, Myog and Myod1 were also observed as a regenerative response to the muscle damage [41], [42]. However, the decreasing levels of Pax7, especially at the terminal stage (Figure 1), would indicate that myogenic regeneration was diminishing at the same time that muscle damage was increasing in this animal model [30]. According to our results, the upregulation of the transcriptional expression levels of Ankrd1, Nogo A, Fbxo32 and Snx10, suggests that muscle damage increases throughout disease progression in transgenic SOD1G93A mice [27], [31]–[35].
Furthermore, our results support those of a recent study in another mouse model for the disease, transgenic SOD1G86R mice, which suggested that the aberrant expression of Rrad in response to oxidative stress may be pathologically relevant [38]. The increasing transcriptional levels of Gsr and Mt2 in transgenic SOD1G93A mice could activate the upregulation of Rrad expression, together with Impa1 and Nnt (Figure 1), prompting an alteration of the muscle excitation-contraction coupling regulated by Rrad [38]. This alteration of the muscle could also be negatively affected by decreasing Sln expression levels, as Sln regulates relaxation-contraction cycles [43].
We hypothesized from these results that one or more of these genes, especially Ankrd1 and Col19a1, which showed the highest significant upregulation during the terminal stage (Figure 1), might represent a potential prognostic biomarker of the disease. To elucidate this, biopsy muscle samples from a balanced number of male and female transgenic SOD1G93A mice were obtained at three disease stages: the early symptomatic (75 days), symptomatic (105 days) and endpoint stages. In this last stage, the longevity of each animal was different, allowing us carry out a correlation study between longevity and the transcriptional expression profiles of each gene during disease progression.
Interestingly, the expression of twelve genes, Myf5, Mef2c, Gsr, Myod1, Col19a1, Calm1, Myog, Snx10, Pax7, Impa1, Ankrd1 and Sln, during the early symptomatic stage of the disease correlated significantly and negatively with longevity; Mt2 was the exception (Table 1). This negative correlation implies that the higher the expression level of these genes during the progression of the disease, the shorter the survival of the animal. Importantly, the Myf5, Mef2c, Gsr, Myod1 and Col19a1 genes displayed the highest Pearson's coefficients with longevity, suggesting that animals displaying a downregulation in expression of these genes at the early symptomatic stage may exhibit a higher regenerative capacity to compensate for muscle damage, as they survived longer than animals that overexpressed these genes. Therefore, because myogenic potential was significantly diminished at 120 days in transgenic SOD1G93A mice (Figure 1), the ability to maintain this potential and compensate for muscle damage must closely relate to survival of the animal.
Additionally, Mt2 and Nnt expression levels were positively correlated with longevity, significantly for Mt2 expression, suggesting that the upregulation of the t expression levels of these genes during disease progression could be induced by the neurodegenerative progress of the disease. However, Mt2 and Rrad have also been shown, potentially, to be deregulated after denervation in mouse skeletal muscle [38], [44]. Furthermore, it has been observed that the expression of Nogo A in ALS skeletal muscle promotes denervation in transgenic SOD1G86R mice [32]. Because the process of denervation is crucial to inducing muscle atrophy in ALS, we tested the transcriptional expression of seventeen genes in the skeletal muscle of denervated mice to determine whether these genes are related to the denervation process itself; denervation is present in all known neurodegenerative disorders, including ALS.
Among the seventeen genes studied, the expression profile of eleven varied significantly in the skeletal muscle of 74-day-old denervated mice from that in age-matched wild type control mice (Figure 3). Our results support previously published studies that found significant variation in the expression levels of Rrad, Mt2 and Nogo A, together with Ankrd1, Myog, Myod1, Sln, Myf5, Pax7, Impa1 and Fbxo32, in denervated mice. The highest expression profiles were observed for Ankrd1, Rrad, Myog and Mt2, which were approximately 20 to 30 times higher than those of controls. This was especially significant for Ankrd1 expression. These results, along with those of previous studies [30], [32], [38], [44], suggest that after 74 days under denervation conditions, skeletal muscle exhibits damage. This is evidenced by deregulated levels of Ankrd1, Fbxo32, Nogo A and Rrad causing induction of muscle differentiation and a subsequent increase in expression levels of Myog, Myod1, Myf5, Pax7 and Mef2c (although this is not significant for Mef2c) or overexpression of Mt2, Sln and Impa1 in response to increasing oxidative stress levels and alterations in calcium and glucose homeostasis, previously described as characteristics of ALS [22]. In particular, the downregulation of Fbxo32 may suggest that an internal mechanism is involved in reducing further loss of muscle proteins during chronic degeneration [40]. Although they were not significant, the decrease in the expression levels of Calm1, Nnt and Snx10 may also represent such a mechanism due to their role in catabolic and anabolic processes that lead to reduced muscle mass [40]. Similarly, the increase in expression of Col19a1 and Gsr, though not significant, suggests that there may be concomitant activation of a second regulatory mechanism that acts as an effective scavenger of reactive oxygen species [45] or that favors skeletal myogenesis [29].
Taken together, these results along with the significant Pearson correlation coefficients observed in muscle biopsies suggest that Mef2c, Gsr, Col19a1, Calm1 and Snx10 may be considered potential genetic biomarkers of longevity in a mouse model of ALS (Figure 4), and the level of their expression in skeletal muscle may predict the longevity of transgenic SOD1G93A mice.
In summary, the first step of the complex neurodegenerative process in ALS remains to be elucidated. However, studying the skeletal muscle as an ALS target tissue that is more accessible than other tissues, such as the spinal cord or CSF, opens the door to the discovery of new biomarkers that may lead to more accurate knowledge of the disease. Our studies suggest that the transcriptional levels of Mef2c, Gsr, Col19a1, Calm1 and Snx10 are closely related to the neurodegenerative process of ALS in the skeletal muscle, in such a way that they can predict longevity in a mouse model for the disease. These observations could help promote further studies exploring new tissues, and the translation of these results to human samples may lead to the discovery of new biomarkers and therefore new potential therapeutic targets.
Materials and Methods
Transgenic SOD1G93A mice
Inbred B6SJL SOD1G93A mice (The Jackson Laboratory, Bar Harbor, ME, USA) were used in this study because they provide a suitable ALS disease model. These mice carry a G93A mutation (substitution of Glycine to Alanine at residue 93) in the human gene superoxide dismutase 1 (SOD1). Hemizygous mutants, obtained by crossing a mutant male with a wild-type (WT) female, were used for all of the experiments. The offspring were identified by PCR amplification of DNA extracted from tail tissue as described in The Jackson Laboratory protocol for genotyping hSOD1 transgenic mice (http://jaxmice.jax.org/pub-cgi/protocols.sh?objtype=protocol,protocolid=523). The animals were housed in the Unidad Mixta de Investigación of the University of Zaragoza, in accordance with international guidelines for the use of laboratory animals. Food and water were available ad libitum. Routine microbiological monitoring did not reveal evidence of infection with common murine pathogens. All of the experimental procedures were approved by the ethics committees of our institutions and followed the international guidelines for the use of laboratory animals, particularly the guidelines for the preclinical in vivo evaluation of pharmacologically active drugs for ALS/MND.
Search for gene targets as potential biomarkers of disease
Based on a previous microarray study of wild type and transgenic SOD1G93A mice (data not shown) and previously published results suggesting several possible molecular markers in the skeletal muscle of an ALS mouse model [46], seventeen genes were tested as potential biomarkers of ALS disease (Table 2). Because skeletal muscle was the target tissue, the majority of these genes are involved in muscle physiology and differentiation. Some are involved in metabolic and anabolic processes. Neuromuscular junction dismantlement has been described as the primary pathogenic event in transgenic SOD1 mice [22]. Among the genes involved in this neurodegenerative process, cardiac ankyrin repeat domain 1 (Ankrd1) plays an important role in skeletal muscle plasticity and appears to be a general marker of muscle damage when it is upregulated [47], while reticulon 4 (Rtn4, also known as Nogo A) accelerates the progressive failure of motor neuron innervation [31], [48]–[50]. Other targets lead more directly to muscle atrophy when they are upregulated in muscle; these include F-box only protein (Fbxo32), which enhances proteolysis, and sorting nexin 10 (Snx10), which promotes vacuolization in mammalian cells [35], [40], [51].
Table 2. Functional role and related molecular pathway of the seventeen genes studied.
| GENE NAME | SYMBOL | GENE ID | FUNCTION | MOLECULAR PATHWAY |
| ankyrin repeat domain 1 (cardiac muscle) | Ankrd1 | 107765 | marker of muscle damage | |
| muscle plasticity | ||||
| F-box only protein 32 | Fbxo32 | 67731 | promotes skeletal muscle atrophy | MUSCLE DAMAGE |
| reduction of its gene expression levels in spinal cord injury disorders | ||||
| sorting nexin 10 | Snx10 | 71982 | regulation of endosome homeostasis | |
| paired box gene 7 | Pax7 | 18509 | muscle development | |
| myogenic differentiation 1 | Myod1 | 17927 | myogenesis, muscle differentiation | MUSCLE DIFFERENTIATION |
| myogenic factor 5 | Myf5 | 17877 | regulator of regenerative myogenesis and homeostasis, muscle regeneration | AND REGENERATION |
| myocyte enhancer factor 2C | Mef2c | 17260 | maintenance sarcomere integrity, muscle differentiation | |
| myogenin | Myog | 17928 | differentiation of muscle cells | |
| collagen, type XIX, alpha 1 | Col19a1 | 12823 | esophageal muscle development and function | MAINTENANCE MUSCLE INTEGRITY |
| reticulon 4 | NOGO A | 68585 | inhibitor axonal regeneration | AND MUSCLE REINNERVATION |
| calmodulin 1 | Calm1 | 12313 | calcium signal modulator | |
| endocitosis mediator at nerve terminal | CALCIUM HOMEOSTASIS | |||
| sarcolipin | Sln | 66402 | regulator calcium transport and muscle relaxation-contraction cycles | |
| inositol (myo)-1(or 4)-monophosphatase 1 | Impa1 | 55980 | inositol homeostasis | |
| activated target of calbindin | ||||
| nicotinamide nucleotide transhydrogenase | Nnt | 18115 | glucose homeostasis | GLUCOSE METABOLISM |
| ras-related associated with diabetes | Rrad | 56437 | glucose tolerance and insuline sensitivity | |
| regulation intracellular calcium signalling | ||||
| glutathione reductase | Gsr | 14782 | oxidative stress metabolism | |
| metallothionein 2 | Mt2 | 17750 | metal binding and free radical scavenging properties | OXIDATIVE STRESS |
| oxidative stress metabolIsm, zinc homeostasis |
The dismantling of the neuromuscular junction is a sign of muscle denervation, which might be a consequence of skeletal muscle hypermetabolism. Hypermetabolism leads to a constant energy deficit in transgenic mice, which precedes amyotrophy and muscle denervation [52]. Furthermore, glucose metabolism and calcium homeostasis are altered in this animal model [27]. In particular, possible genes related to glucose metabolism include inositol (myo)-1(or 4)-monophosphatase 1 (Impa1), which plays an important role in motor coordination and is directly involved in glucose metabolism [36]; nicotinamide nucleotide transhydrogenase (Nnt), which leads to appropriate glucose homeostasis in these mice when it is downregulated [37]; and ras-related associated with diabetes (Rrad), which promotes altered lipid metabolism and deregulates glucose uptake [53]. Rrad is also involved in disease progression, insofar as the accumulation of reactive oxygen species is coincident with its upregulation [38]. In skeletal muscle, calmodulin 1 (Calm1) and sarcolipin (Sln) are needed to reach an adequate calcium influx, which is necessary to either maintain synaptic transmission in the neuromuscular junction, in the case of calmodulin1 [54], or to regulate muscle relaxation–contraction cycles, in the case of sarcolipin [43].
Oxidative stress has also been described in ALS [22]. In particular, glutathione reductase (Gsr) [39] and metallothionein 2 (Mt2) [55] represent the most studied enzymes involved in this process. In fact, their deficiency results in muscle atrophy and oxidative injury.
In contrast to degenerative processes, regenerative processes tend to compensate for the induced unbalance. In skeletal muscle, many genes are involved in regenerative pathways, either by maintaining the integrity of the tissue and favoring skeletal myogenesis, as is the case for collagen, type XIX, alpha 1 (Col19a1) [29], or by promoting muscle differentiation and regeneration, as is the case for the paired box gene (Pax7), myogenic differentiation 1 (Myod1), myogenic factor 5 (Myf5), myocyte enhancer factor 2C (Mef2c) and myogenin (Myog) [30], [56]–[60].
Extraction of muscle samples in wild type and transgenic SOD1G93A animals
Hemizygous SOD1G93A mice and age-matched nontransgenic wild-type control mice at the early symptomatic, symptomatic and terminal stages (n = 10 transgenic SOD1G93A mice and 10 wild type mice, balanced males and females, per stage) were used to study gene expression throughout disease progression. All of the animals were sacrificed by intraperitoneal (i.p.) injection of sodium pentobarbital (100 mg/Kg) following the guidelines from the report of the American Veterinary Medical Association Panel on Euthanasia, J. Am. Vet. Med. Assoc., 2007 (http://icwdm.org/Publications/pdf/ControlMethods/Euthanasia/AVMA2007report.pdf.
All surgical material was sterilized before dissection. After dissection, skeletal muscle of the hind limbs was immediately frozen in liquid nitrogen and stored at −80°C. Samples were collected by simple random sampling using the Statistical Package for the Social Sciences (SPSS) 15.0.
Extraction of biopsies from skeletal muscle of transgenic SOD1G93A mice
Forty-eight transgenic SOD1G93A male and female mice were used to study the correlation of gene expression with longevity (n = 24 mice per sex). Three muscle biopsies from the Gluteus superficialis muscle were obtained per mouse, from a different hind limb each time, at three different ages that coincided with the early symptomatic stage (75 days), symptomatic stage (105 days) and terminal stage (endpoint age). This innovative procedure allowed the study of gene expression in the same animal; it was possible to keep the animal alive at different disease stages throughout the study because the Gluteus superficialis muscle is easily to reach and its manipulation does not prevent the mouse from moving properly after each surgery. All surgical material was sterilized before the extraction. Twenty minutes before starting the extraction, the analgesic Meloxicam 2 mg/kg (Metacam© AINES Cox-2) was administered subcutaneously to the animal. Once the gluteus superficialis muscle was localized, the zone was shaved and then disinfected with 70% alcohol and povidone iodide.
Each mouse was anesthetized by administration of isoflurane (4–5%) and a constant flux of 1.5–2% isoflurane maintained using a facemask. The lack of motor reflexes was confirmed before starting the surgery. The body temperature was maintained at a constant level with a thermal blanket, and a moisturizer gel (Lubrithal©) was applied on the eyes to prevent corneal damage during the extraction.
Once the animal was prepared for surgery, an incision of <1 cm was made on the zone where the gluteus superficialis is localized. The connective tissue around this muscle was carefully removed. A small biopsy of the muscle, with a weight of approximately 3 mg (≈1 mm2), was transferred to an eppendorf tube containing RNAlater® solution (AM7021, Ambion, Madrid, Spain) to preserve the extracted tissue. The zone was closed with staples (EZ 9 mm clip) and rehydrated with physiological serum 0.9%, and a scar gel (Aloe vet©) was applied to promote cicatrisation of the wound. The mouse was placed back into its cage,, and it's body temperature was maintained using an infrared lamp until it awoke. The cage contained hydrated food and paper to facilitate the recovery of the animal.
For 24 hours following surgery, the mouse was tested two times per day to check coordination and to ensure the viability of the next biopsy. One week following surgery, the staples were removed. This methodology was repeated in each animal for the three selected stages so that the three biopsy samples were obtained from each mouse were of from the same kind of muscle [61]. The final biopsy sample was extracted at the endpoint for each animal. The mice were sacrificed when they were unable to right themselves within 30 s after being placed on their side; this point was considered the survival endpoint according to the guidelines for preclinical testing and colony management [62], [63].
Extraction of muscle samples in denervated animals
Six male wild-type mice of the B6CLJ strain were anesthetized (pentobarbital 30 mg/kg, i.p.) at the age of 60 days, and muscle denervation was performed following the methodology described previously [30]. After surgical denervation, the animals were sacrificed by cervical dislocation at 74 days of age, and the gastrocnemius muscles were dissected and frozen immediately in liquid nitrogen.
RNA extraction, synthesis of cDNA and real time PCR assay
RNAlater® solution was removed from the biopsy samples, and total RNA was extracted using the RNeasy Micro Kit protocol (74004, Qiagen-Izasa, Barcelona, Spain), which included treatment with Dnase I solution to eliminate genomic DNA.
Tissue samples from the skeletal muscle of transgenic SOD1G93A, wild type, denervated, heterozygous and hemizygous SMA mice were pulverized in liquid nitrogen in a cold mortar. In this group of samples, total RNA was extracted using TRIzol Reagent according to the manufacturers' protocol (Invitrogen S.A., Prat de Llobregat, Spain). RNA was treated to eliminate genomic DNA using the Turbo DNA-free™ kit (AM1907, Ambion, Madrid, Spain).
RNA extracted from both groups of samples was processed for reverse transcription (RT) using the SuperScript™ First-Strand Synthesis System kit (12371-019, Invitrogen S.A., Prat de Llobregat, Spain). Gene expression variations in all of the samples were assayed by real-time PCR. PCR reactions were carried out in a StepOne™ Real-Time PCR System (4387925, Applied Biosystems, Madrid, Spain). Primer and probe mixtures for each gene of interest were supplied by Applied Biosystems (Madrid, Spain) (Table 3).
Table 3. Taqman® probe and primer mixtures used in gene expression assays.
| NAME | GEN SYMBOL | ACCESION NUMBER | PROBE LOCATION | PART NUMBER | ORGANISM |
| Ankyrin repeat domain 1 (cardiac muscle) | Ankrd1 | NM_013468.3 | Exon 8–9 | Mm00496512_m1 | Mus Musculus |
| Calmodulin 1 | Calm | NM_009790.4 | Exon 2–3 | Mm00486655_m1 | Mus Musculus |
| Collagen, type XIX, alpha 1 | Col19a1 | NM_007733.2 | Exon 2–3 | Mm00483576_m1 | Mus Musculus |
| F-box only protein 32 | Fbxo32 | NM_026346.2 | Exon 5–6 | Mm01207878_m1 | Mus Musculus |
| Glutathione reductase | Gsr | NM_010344.4 | Exon 12–13 | Mm00833903_m1 | Mus Musculus |
| Inositol (myo)-1(or 4)-monophosphatase 1 | Impa1 | NM_018864.5 | Exon 7–8 | Mm00497770_m1 | Mus Musculus |
| Metallothionein 2 | Mt2 | NM_008630.2 | Exon 3-3 | Mm00809556_s1 | Mus Musculus |
| Myocyte enhancer factor 2C | Mef2c | NM_025282.2 | Exon 1–2 | Mm00600423_m1 | Mus Musculus |
| Myogenic differentiation 1 | Myod1 | NM_010866.2 | Exon 1–2 | Mm00440387_m1 | Mus Musculus |
| Myogenic factor 5 | Myf5 | NM_008656.5 | Exon 2–3 | Mm00435125_m1 | Mus Musculus |
| Myogenin | Myog | NM_008656.5 | Exon 1–2 | Mm00446194_m1 | Mus Musculus |
| Nicotinamide nucleotide transhydrogenase | Nnt | NM_031189.2 | Exon 5–6 | Mm00435154_m1 | Mus Musculus |
| Paired box gene 7 | Pax7 | NM_011039.2 | Exon 4–5 | Mm00834079_m1 | Mus Musculus |
| Ras-related associated with diabetes | Rrad | NM_019662.2 | Exon 2–3 | Mm00451053_m1 | Mus Musculus |
| Reticulon 4 | Rtn4 | NM_194052.2 | Exon 2–3 | Mm00445861_m1 | Mus Musculus |
| Sarcolipin | Sln | NM_025540.2 | Exon 1–2 | Mm00481536_m1 | Mus Musculus |
| Sorting nexin 10 | Snx10 | NM_001127349.1 | Exon 1–2 | Mm00511049_m1 | Mus Musculus |
| Actin, beta, cytoplasmic | Actb (β-actin) | NM_008084.2 | Exon 3 | 4352932E | Mus Musculus |
| Glyceraldehyde-3-phosphate dehydrogenase | Gapdh | NM_007393.1 | Exon 6 | 4352933E | Mus Musculus |
| 18S ribosomal RNA (18S rRNA) | 18S | X03205.1 | _ | Hs99999901_s1 | Homo sapiens |
| Superoxide dismutase 1 | Sod1 | NM_000454.4 | Exon 2–3 | Hs00916176_m1 | Homo sapiens |
Reactions were performed in a final volume of 5 µL with 1× TaqMan® Fast Universal PCR Master Mix (4352042, No AmpErase® UNG, Applied Biosystems, Madrid, Spain), 1× of the primer and TaqMan® MGB probe mix for each studied gene and 2 µL of 10× diluted cDNA per reaction. Three endogenous genes (18S rRNA, GAPDH, and β-actin) were used for normalization following the methodology described previously [64]. All reactions were performed in triplicate and all reaction efficiencies of the primer/probe sets were close to 100%. Thermal cycling parameters were as follows: incubation at 95°C, 20 seconds, and 40 cycles of 95°C for 1 second and 60°C for 20 seconds.
Gene expression analysis in total skeletal muscle samples of transgenic SOD1G93A and denervated mice
Changes in the RNA expression levels of each gene were studied in the skeletal muscles samples of mice, balanced for males and females. The corresponding ΔCT values of each extracted sample were normalized with geometric media of the selected housekeeping genes as described above. Age-matched wild type mice were used as controls when studying the disease progression in transgenic SOD1G93A mice. Sixty-day-old wild-type mice were used as controls when studying the expression patterns of the selected genes in denervated mice.
The ΔΔCT method was used to determine relative changes in transcriptional expression. Statistical analyses of the data were performed using the fold change 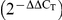 as previously described [64].
as previously described [64].
Longevity correlation analysis of muscle biopsy samples
For each gene studied, the ΔΔCT method was used to determine relative changes in gene expression, as previously described. Differences in the threshold cycles between the ΔCT value at 75 days and at 105 days/the endpoint stage were calculated in all the tested genes for each animal under study. Because the first muscle biopsy sample was obtained when the animals showed early disease symptoms (75 days), ΔCT 75 days was taken as a reference parameter to calculate the variation in the relative gene expression at the symptomatic (105 days) and endpoint stages with respect to the early symptomatic stage (75 days). Using this methodology, the corresponding fold changes were calculated using the equation 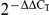 . In this way, for each animal, three fold change values were obtained for each gene and were plotted to calculate the slope. The linear regression between each slope value for each animal and the age at which the animal died was studied. The Pearson correlation coefficient was used to determine the linear correlation between the slopes obtained for each studied gene and the longevity data.
. In this way, for each animal, three fold change values were obtained for each gene and were plotted to calculate the slope. The linear regression between each slope value for each animal and the age at which the animal died was studied. The Pearson correlation coefficient was used to determine the linear correlation between the slopes obtained for each studied gene and the longevity data.
Statistical analysis
Statistical analyses were carried out using the non-parametric Mann-Whitney test to evaluate the variability in gene expression. The Pearson correlation coefficient was used to determine the linear correlation in wild type and transgenic SOD1G93A mice with longevity throughout disease progression. All of the values were expressed as the mean ± S.E.M. The statistical significance threshold was set at p<0.05. The software used for the statistical analysis was SPSS 15.0.
Acknowledgments
We wish to thank Eduardo Romanos (Unidad de Valoración Funcional, Aragon's Institute of Health Sciences (IACS)) for technical support.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This study was supported by grants from Caja Navarra: “Tú decides, tú eliges”, PI071133 and PI10/01787 from the Fondo de Investigación Sanitaria of Spain and by Carlos III Health Institute (ISCIII) (PI07/1283 and EC08/00049). We gratefully appreciate the support received from the Foundation for the Improvement in ALS Research in Spain “FUNDELA” and from the benefit act “No llores, no te rindas” in Barcelona. The Biomedical Research Centre Network (CIBER) for Rare Diseases is an initiative of the ISCIII. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Azuaje F. Bioinformatics and Biomarker Discovery. “Omic” data analysis for personalized medicine. West Sussex, UK: Wiley-Blackwell; 2010. [Google Scholar]
- 2.Siddique N, Siddique T. Genetics of Amyotrophic Lateral Sclerosis. Phys Med Rehabil Clin N Am. 2008;19:429–vii. doi: 10.1016/j.pmr.2008.05.001. doi: 10.1016/j.pmr.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hensley K, Mhatre M, Mou S, Pye QN, Stewart C, et al. On the relation of oxidative stress to neuroinflammation: lessons learned from the G93A-SOD1 mouse model of amyotrophic lateral sclerosis. Antioxid. Redox. Signal. 2006;8:2075–2087. doi: 10.1089/ars.2006.8.2075. [DOI] [PubMed] [Google Scholar]
- 4.Cluskey S, Ramsden DB. Mechanism of neurodegeneration in amyotrophic lateral sclerosis. J Clin Pathol. 2001:54. [PMC free article] [PubMed] [Google Scholar]
- 5.Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, et al. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science. 1998;281:1851–1854. doi: 10.1126/science.281.5384.1851. [DOI] [PubMed] [Google Scholar]
- 6.Wijesekera LC, Leigh PN. Amyotrophic Lateral Sclerosis. Orph J Rare Dis. 2009;4:3. doi: 10.1186/1750-1172-4-3. doi: 10.1186/1750-1172-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baek WS, Desai NP. ALS: pitfalls in the diagnosis. Pract Neurol. 2007;7:74–81. [PubMed] [Google Scholar]
- 8.Turner MR, Kiernan MC, Nigel P, et al. Biomarkers in amyotrophic lateral sclerosis. The Lancet. 2009;8:94–109. doi: 10.1016/S1474-4422(08)70293-X. [DOI] [PubMed] [Google Scholar]
- 9.Kwiatkowski TJJ, Bosco DA, LeClerc AL, Tamrazian E, Vanderburg CR, et al. Mutations in the FUS/TLS Gene on Chromosome 16 Cause Familial Amyotrophic Lateral Sclerosis. Science. 2009;323:1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- 10.Vance C, Rogelj B, Hortobágyi T, De Vos KJ, Nishimura AL, et al. Mutations in FUS, an RNA processing protein, cause Familial Amyotrophic Lateral Sclerosis type 6. Science. 2009;323:1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deng HX, Zhai H, Bigio EH, Yan J, Fecto F, et al. FUS-immunoreactive inclusions are a common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosis. Ann Neurol. 2010;67:739–748. doi: 10.1002/ana.22051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liscic RM, Grinberg LT, Zidar J, Gitcho MA, Cairns NJ. ALS and FTLD: two faces of TDP-43 proteinopathy. European Journal of Neurology. 2008;15:772–780. doi: 10.1111/j.1468-1331.2008.02195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lagier-Tourenne C, Cleveland DW. Rethinking ALS: The FUS about TDP-43. Cell. 2009;136:1001–1004. doi: 10.1016/j.cell.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 15.Sumiyoshi H, Mor N, Lee SY, Doty S, Henderson S, et al. Esophageal muscle physiology and morphogenesis require assembly of a collagen XIX–rich basement membrane zone. J Cell Biol. 2004;166:591–600. doi: 10.1083/jcb.200402054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mitsumoto H, Santella RM, Liu X, Bogdanov M, Zipprich J, et al. Oxidative stress biomarkers in sporadic ALS. Amyotrophic Lateral Sclerosis. 2008;9:177–183. doi: 10.1080/17482960801933942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ryberg H, Bowser R. Protein biomarkers for amyotrophic lateral sclerosis. Expert Rev Proteomics. 2008;5:249–262. doi: 10.1586/14789450.5.2.249. [DOI] [PubMed] [Google Scholar]
- 18.Turner MR, Kiernan MC, Leigh PN, Talbot K. Biomarkers in amyotrophic lateral sclerosis. Lancet Neurol. 2009;8:94–109. doi: 10.1016/S1474-4422(08)70293-X. [DOI] [PubMed] [Google Scholar]
- 19.Pradat PF, Dib M. Biomarkers in amyotrophic lateral sclerosis: facts and future horizons. Mol Diagn Ther. 2009;13:115–125. doi: 10.1007/BF03256320. [DOI] [PubMed] [Google Scholar]
- 20.Pradat PF. New biological and radiological markers in amyotrophic lateral sclerosis. Presse Med. 2009;38:1843–1851. doi: 10.1016/j.lpm.2009.01.022. [DOI] [PubMed] [Google Scholar]
- 21.Wijesekera LC, Leigh PN. Amyotrophic lateral sclerosis. Orph J Rare Dis. 2009;4:3. doi: 10.1186/1750-1172-4-3. doi: 10.1186/1750-1172-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Musaró A. State of the art and the dark side of amyotrophic lateral sclerosis. World J Biol Chem. 2010;1:62–68. doi: 10.4331/wjbc.v1.i5.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Carvalho M, Swash M. Amyotrophic lateral sclerosis: an update. Curr Opin Neurol. 2011;24(5):497–503. doi: 10.1097/WCO.0b013e32834916a9. [DOI] [PubMed] [Google Scholar]
- 24.Douglass CP, Kandler RH, Shaw PJ. An evaluation of neurophysiological criteria used in the diagnosis of motor neuron disease. J Neurol Neurosurg Psychiatry. 2010;81:646–649. doi: 10.1136/jnnp.2009.197434. [DOI] [PubMed] [Google Scholar]
- 25.Burgunder JM, Schöls L, Baets J, Andersen P, Gasser T, et al. EFNS guidelines for the molecular diagnosis of neurogenetic disorders: motoneuron, peripheral nerve and muscle disorders. Eur J Neurol. 2011;18(2):207–217. doi: 10.1111/j.1468-1331.2010.03069.x. doi: 10.1111/j.1468-1331.2010.03069.x. [DOI] [PubMed] [Google Scholar]
- 26.Dobrowolny G, Aucello M, Rizzuto E, Beccafico S, Mammucari C, et al. Skeletal muscle is a primary target of SOD1G93A-mediated toxicity. Cell Metabolism. 2008;8:425–436. doi: 10.1016/j.cmet.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 27.Dupuis L, Loeffler JP. Sclérose latérale amyotrophique, junction neuromusculaire et _raline énergétique. Medecine/Sciences. 2008;24:1077–1082. doi: 10.1051/medsci/200824121077. [DOI] [PubMed] [Google Scholar]
- 28.Babu GP, Bhupathy P, Timofeyev V, Petrashevskaya NN, Reiser PJ, et al. Ablation of sarcolipin enhances sarcoplasmic reticulum calcium transport and atrial contractility. PNAS. 2007;104:17867–17872. doi: 10.1073/pnas.0707722104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sumiyoshi H, Mor N, Lee SY, Doty S, Henderson S, et al. Esophageal muscle physiology and morphogenesis require assembly of a collagen XIX–rich basement membrane zone. J Cell Biol. 2004;166:591–600. doi: 10.1083/jcb.200402054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Manzano R, Toivonen JM, Oliván S, Calvo AC, Moreno-Igoa M, et al. Altered Expression of Myogenic Regulatory Factors in the Mouse Model of Amyotrophic Lateral Sclerosis. Neurodegenerative Dis. 2011;8(5):386–396. doi: 10.1159/000324159. doi: 10.1159/000324159. [DOI] [PubMed] [Google Scholar]
- 31.Dupuis L, González de Aguilar JL, di Scala F, Rene F, de Tapia M, et al. Nogo provides a molecular marker for diagnosis of Amyotrophic Lateral Sclerosis. Neurobiol Dis. 2002;10:358–365. doi: 10.1006/nbdi.2002.0522. [DOI] [PubMed] [Google Scholar]
- 32.Jokic N, González de Aguilar JL, Dimou L, Lin S, Fergani A. EMBO Reports. 2006;7:1162–1167. doi: 10.1038/sj.embor.7400826. doi: 10.1038/sj.embor.7400826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakamura K, Nakada C, Takeuchi K, Osaka M, Shomori K, et al. Altered expression of cardiac ankyrin repeat protein and its homologue, ankyrin repeat protein with PEST and proline-rich region, in atrophic muscles in amyotrophic lateral sclerosis. Pathobiology. 2002–2003;70:197–203. doi: 10.1159/000069329. [DOI] [PubMed] [Google Scholar]
- 34.Léger B, Vergani L, Sorarù G, Hespel P, Derawe W, et al. Human skeletal muscle atrophy in amyotrophic lateral sclerosis reveals a reduction in Akt and an increase in atrogin-1. The FASEB J. 2006;20:583–585. doi: 10.1096/fj.05-5249fje. doi 10.1096/fj.05-5249fje. [DOI] [PubMed] [Google Scholar]
- 35.Qin B, He M, Chen X, Pei D. Sorting nexin 10 induces giant vacuoles in mammalian cells. J Biol Chem. 2006;281:36891–36896. doi: 10.1074/jbc.M608884200. [DOI] [PubMed] [Google Scholar]
- 36.Berggård T, Szczepankiewicz O, Thulin E, Linse S. Myo-inositol monophosphatase is an activated target of calbindin D28k. J Biol Chem. 2002;277:41954–41959. doi: 10.1074/jbc.M203492200. [DOI] [PubMed] [Google Scholar]
- 37.Freeman HC, Hugill A, Dear NT, Ashcroft FM, Cox RD. Deletion of nicotinamide nucleotide transhydrogenase: A new quantitive trait locus accounting for glucose intolerance in C57BL/6J mice. Diabetes. 2006;55:2153–2156. doi: 10.2337/db06-0358. [DOI] [PubMed] [Google Scholar]
- 38.Halter B, Gonzalez de Aguilar JL, Rene F, Petri S, Fricker B, et al. Oxidative stress in skeletal muscle stimulates early expression of Rad in a mouse model of amyotrophic lateral sclerosis. Free Rad Biol Med. 2010;48:915–923. doi: 10.1016/j.freeradbiomed.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 39.Dudley RWR, Khairallah M, Mohammed S, Lands L, Des Rosiers C, et al. Dynamic responses of the glutathione system to acute oxidative stress in dystrophic mouse (mdx) muscles. Am J Physiol Regul Integr Comp Physiol. 2006;291:R704–R710. doi: 10.1152/ajpregu.00031.2006. [DOI] [PubMed] [Google Scholar]
- 40.Léger B, Sense R, Al-Khodairy AW, Dériaz O, Gobelet C, et al. Atrogin-1, murf1, and foxo, as well as phosphorylated gsk-3β and 4e-bp1 are reduced in skeletal muscle of chronic spinal cord–injured patients. Muscle & Nerve. 2009;40:69–78. doi: 10.1002/mus.21293. [DOI] [PubMed] [Google Scholar]
- 41.Borselli C, Storrie H, Benesch-Lee F, Shvartsman D, Cezar C. Functional muscle regeneration with combined delivery of angiogenesis and myogenesis factors. PNAS. 2010;107:3287–3292. doi: 10.1073/pnas.0903875106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sakuma K, Yamaguchi A. The functional role of calcineurin in hypertrophy, regeneration, and disorders of skeletal muscle. J Biomed Biotech. 2010 doi: 10.1155/2010/721219. doi: 10.1155/2010/721219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vasu VT, Ott S, Hobson B, Rashidi V, Oommen S, et al. Sarcolipin and ubiquitin carboxy-terminal hydrolase 1 mRNAs are over-expressed in skeletal muscles of α-tocopherol deficient mice. Free Radic Res. 2009;43:106–116. doi: 10.1080/10715760802616676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Magnusson C, Svensson A, Christerson U, Tågerud S. Denervation-induced alterations in gene expression in mouse skeletal muscle. Eur J Neurosci. 2005;21:577–580. doi: 10.1111/j.1460-9568.2005.03855.x. doi: 10.1111/j.1460-9568.2005.03855.x. [DOI] [PubMed] [Google Scholar]
- 45.Limon-Pacheco JH, Gonsebatt ME. The glutathione system and its regulation by neurohormone melatonin in the central nervous system. Cent Nerv Syst Agents Med Chem. 2010;10:287–297. doi: 10.2174/187152410793429683. [DOI] [PubMed] [Google Scholar]
- 46.Gonzalez de Aguilar JL, Niederhauser-Wiederkehr C, Halter B, de Tapia M, di Scala F, et al. Gene profiling of skeletal muscle in an amyotrophic lateral sclerosis mouse model. Physiol Genomics. 2008;32:207–218. doi: 10.1152/physiolgenomics.00017.2007. [DOI] [PubMed] [Google Scholar]
- 47.Laure L, Suel L, Roudaut C, Bourg N, Ouali A, et al. Cardiac ankyrin repeat protein is a marker of skeletal muscle pathological remodelling. FEBS J. 2009;276:669–684. doi: 10.1111/j.1742-4658.2008.06814.x. [DOI] [PubMed] [Google Scholar]
- 48.Fergani A, Dupuis L, Jokic N, Larmet Y, de Tapia M, et al. Reticulons as markers of neurological diseases: focus on amyotrophic lateral sclerosis. Neurodegen Dis. 2005;2:185–194. doi: 10.1159/000089624. [DOI] [PubMed] [Google Scholar]
- 49.Pradat PF, Bruneteau G, Gonzalez de Aguilar JL, Dupuis L, Jokic N, et al. Muscle Nogo-A expression is a prognostic marker in lower motor neuron syndromes. Ann Neurol. 2007;62:15–20. doi: 10.1002/ana.21122. [DOI] [PubMed] [Google Scholar]
- 50.Yan R, Shi Q, Hu X, Zhou X. Reticulon proteins: emerging players in neurodegenerative diseases. Cell Mol Life Sci. 2006;63:877–889. doi: 10.1007/s00018-005-5338-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lagirand-Cantaloube J, Cornille K, Csibi A, Batonnet-Pichon S, Leibovitch MP, et al. Inhibition of atrogin-1/MAFbx mediated MyoD proteolysis prevents skeletal muscle atrophy in vivo. PLos One. 2009;4:e4973. doi: 10.1371/journal.pone.0004973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dupuis L, Loeffler JP. Neuromuscular junction destruction during amyotrophic lateral sclerosis: insights from transgenic mice. Curr Op Pharm. 2009;9:341–346. doi: 10.1016/j.coph.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 53.Ilany J, Bilan PJ, Kapur S, Caldwell JS, Patti ME, et al. Overexpression of Rad in muscle worsens diet-induced insulin resistance and glucose intolerance and lowers plasma triglyceride level. PNAS. 2006;103:4481–4486. doi: 10.1073/pnas.0511246103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu XS, McNeil BD, Xu J, Fan J, Xue L, et al. Ca2+ and calmodulin initiate all forms of endocytosis during depolarization at a nerve terminal. Nat Neurosci. 2009;12:1003–1010. doi: 10.1038/nn.2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.DeRuisseau LR, Recca DM, Mogle JA, Zoccolillo M, DeRuisseau KC. Metallothionein deficiency leads to soleus muscle contractile dysfunction following acute spinal cord injury in mice. Am J Physiol Regul Integr Comp Physiol. 2009;297:R1795–R1802. doi: 10.1152/ajpregu.00263.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.White RB, Ziman MR. Genome-wide discovery of Pax7 target genes during development. Physiol Genomics. 2008;33:41–49. doi: 10.1152/physiolgenomics.00256.2007. [DOI] [PubMed] [Google Scholar]
- 57.Zhao P, Caretti G, Mitchell S, McKeehan WL, Boskey AL, et al. Fgfr4 is required for effective muscle regeneration in vivo: Delineation of a MyoD-Tead2-Fgfr4 transcriptional pathway. J Biol Chem. 2006;281:429–438. doi: 10.1074/jbc.M507440200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gayraud-Morel B, Chrétien F, Flamant P, Gomès D, Zammit PS, et al. A role for the myogenic determination gene Myf5 in adult regenerative myogenesis. Develop Biol. 2007;312:13–28. doi: 10.1016/j.ydbio.2007.08.059. [DOI] [PubMed] [Google Scholar]
- 59.Dodou E, Xu SM, Black BL. Mef2c is activated directly by myogenic basic helix-loop-helix proteins during skeletal muscle development in vivo. Mechanism Develop. 2003;120:1021–1032. doi: 10.1016/s0925-4773(03)00178-3. [DOI] [PubMed] [Google Scholar]
- 60.Cao Y, Kumar RM, Penn BH, Berkes CA, Kooperberg C, et al. Global and gene-specific analyses show distinct roles for Myod and Myog at a common set of promoters. The EMBO J. 2006;25:502–511. doi: 10.1038/sj.emboj.7600958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Murillo S, Calvo AC, Osta R, et al. Biopsia muscular en ratón. 2009. National Congress of the Spanish Society for the Sciences of the Laboratory Animals. Salamanca.
- 62.Ludolph A, Bendotti C, Blaugrund E, Chio A, Greensmith L, et al. Guidelines for preclinical animal research in ALS/MND: A consensus meeting. Amyotrophic Lateral Sclerosis. 2010;11:38–45. doi: 10.3109/17482960903545334. [DOI] [PubMed] [Google Scholar]
- 63.Leitner M, Menzies S, Lutz C. Working with ALS Mice. Guidelines for preclinical testing & colony management. Cambridge and Maine: Prize4Life and The Jackson Laboratory; 2010. pp. 1–21. [Google Scholar]
- 64.Calvo AC, Moreno-Igoa M, Manzano R, Ordovás L, Yagüe G, et al. Determination of protein and RNA expression levels of common housekeeping genes in a mouse model of neurodegeneration. Proteomics. 2008;8:4338–4343. doi: 10.1002/pmic.200701091. [DOI] [PubMed] [Google Scholar]