

Abstract
Epigallocatechin gallate (EGCG), the major polyphenol in green tea, acutely stimulates production of nitric oxide (NO) from vascular endothelium to reduce hypertension, and improve endothelial dysfunction in SHR rats. Herein, we explored additional mechanisms whereby EGCG may mediate beneficial cardiovascular actions. When compared with vehicle-treated controls, EGCG treatment (2.5 μM, 8 h) of human aortic endothelial cells (HAEC) caused a ~3-fold increase in hemeoxygenase-1 (HO-1) mRNA and protein with comparable increases in HO-1 activity. This was unaffected by pre-treatment of cells with wortmannin, LY294002, PD98059, or L-NAME (PI 3-kinase, MEK, and NO synthase inhibitors, respectively). Pre-treatment of HAEC with SB203580 (p38 MAPK inhibitor) or siRNA knockdown of p38 MAPK completely blocked EGCG-stimulated induction of HO-1. EGCG treatment also inhibited TNF-α-stimulated expression of VCAM-1 and decreased adhesion of monocytes to HAEC. siRNA knockdown of HO-1, p38 MAPK, or Nrf-2 blocked these inhibitory actions of EGCG. In HAEC transiently transfected with a human HO-1 promoter luciferase reporter (or an isolated Nrf-2 responsive region), luciferase activity increased in response to EGCG. This was inhibitable by SB203580 pre-treatment. EGCG-stimulated expression of HO-1 and Nrf-2 was blocked by siRNA knockdown of Nrf-2 or p38 MAPK. Finally, liver from mice chronically treated with EGCG had increased HO-1 and decreased VCAM-1 expression. Thus, in vascular endothelium, EGCG requires p38 MAPK to increase expression of Nrf-2 that drives expression of HO-1 resulting in increased HO-1 activity. Increased HO-1 expression may underlie anti-inflammatory actions of EGCG in vascular endothelium that may help mediate beneficial cardiovascular actions of green tea.
Keywords: Epigallocatechin gallate (EGCG), hemeoxygenase-1, p38 kinase, nuclear factor-E2-related factor-2, endothelium
INTRODUCTION
Epigallocatechin gallate (EGCG) is a polyphenol constituting ~30% of the solids in the green tea leaf (camillia senensis) [1]. In epidemiological studies, green tea consumption is associated with a dose-dependent decrease in the incidence of diabetes, hypertension, and cardiovascular morbidity and mortality [2–5]. However, mechanisms underlying beneficial actions of green tea to reduce metabolic and cardiovascular risk are poorly understood. Recently, we demonstrated that EGCG stimulates acute production of NO from primary endothelial cells using a signaling pathway involving fyn/PI3K/Akt/eNOS [6]. This pathway shares features in common with insulin signaling pathways regulating activation of eNOS and NO production in endothelial cells [7]. In addition, like insulin, EGCG inhibits synthesis and secretion of the vasoconstrictor ET-1 through activation of PI3K/Akt/FoxO1 [8, 9]. Similarly, EGCG inhibits gluconeogenesis in hepatocytes using a PI3K-dependent mechanism similar to metabolic actions of insulin [10]. Thus, EGCG mimics and/or augments metabolic and vasodilator actions of insulin and opposes synthesis and secretion of ET-1. This is predicted to simultaneously improve both metabolic and cardiovascular phenotypes in conditions characterized by reciprocal relationships between insulin resistance and endothelial dysfunction including diabetes, obesity, and their cardiovascular complications [11]. Indeed, chronic oral administration (3 wk) of EGCG to spontaneously hypertensive rats (SHR), a model with features of the human metabolic syndrome, simultaneously improves endothelial dysfunction, lowers blood pressure and circulating ET-1 levels, reduces insulin resistance, increases circulating adiponectin levels, and protects against myocardial ischemia/reperfusion injury [12]. These beneficial actions of EGCG are likely mediated, at least in part, by augmenting endothelial bioavailability of NO [12, 13].
Insulin resistance and endothelial dysfunction are caused by genetic and/or environmental factors including glucotoxicity, lipotoxicity and pro-inflammatory signaling that promote oxidative stress resulting in pathway-selective impairment in PI3K-dependent insulin signaling pathways in both metabolic and vascular tissues [7, 14]. Hemeoxygenase-1 (HO-1), an ubiquitous enzyme up regulated in response to oxidant stress and inflammatory stimuli, may play an important role in preserving vascular and metabolic homeostasis [15, 16]. HO catalyzes the oxidative degradation of heme to biliverdin and carbon monoxide [17, 18]. Among the three HO isoforms identified, HO-1 is inducible while HO-2 and HO-3, are constitutively expressed [19]. Insulin stimulates increases in expression of HO-1 in retinal pericytes, retinal endothelial cells, and retinal pigment epithelial cells through activation of IRS-1/PI3K/Akt-2 pathways that do not involve activation of p38 MAPK [20]. Induction of HO-1 in animal models of obesity, diabetes, insulin resistance, and hypertension is associated with improvement in insulin sensitivity, glucose tolerance, and blood pressure [21–23]. To elucidate additional mechanisms underlying putative salutary actions of green tea to simultaneously improve both cardiovascular and metabolic function, we characterized regulation of the heme-oxygenase-1 gene by EGCG in endothelial cells from peripheral vasculature.
MATERIALS and METHODS
Antibodies
Antibodies against phospho-p44/42 MAPK, phospho-p38 MAPK, and p38 MAPK were from Cell Signaling Technologies (Danvers, MA). Anti-HO-1 and -2 antibodies were from Assay Designs (Victoria, BC, Canada). Antibodies against Nrf-2 and VCAM-1 were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA).
Cell Culture
Human aortic endothelial cells (HAEC) (Lonza, Walkersville, MD) in primary culture were grown in endothelial growth medium EGM-MV (Lonza) as described [24]. All experiments were conducted on HAEC between passage 3 and 6. HAEC were grown in 60-mm dishes to 80% confluence and then serum starved for 1 h with endothelial basal medium (Lonza) followed by treatment with EGCG (Sigma; St. Louis, MO) at various concentrations and times as indicated in legends to figures. EGCG was prepared as a 50 mM stock in 50% dimethylsulfoxide (DMSO) and stored at −20 °C. On the day of use, this initial stock was diluted further to 10 mM with serum free media, and then this was diluted even further using serum free media to final concentrations as noted in the legends to the figures. For experiments using selective chemical inhibitors, HAEC were serum starved for 1 h followed by 1 h pre-treatment with PI 3-kinase inhibitors wortmannin (100 nM, Sigma) or LY294002 (50 μM, Sigma), MEK inhibitor PD98059 (25 μM, Sigma), p38 MAPK inhibitor SB203580 (10 μM, Sigma), or NOS inhibitor L-NAME (100 μM, Sigma). Then cells were treated with vehicle (DMSO) or EGCG (2.5 μM, 8 h). The concentrations of chemical inhibitors cited above have previously been shown to be selective and specific [6, 69].
Differential mRNA screening analysis and quantitative real-time PCR
For the screening analysis, HAEC were serum-starved 1 h before vehicle or EGCG treatment (10 μM, 8 h). Subsequently, RNA was isolated using total RNA and Turbo-DNase kits (Ambion, Inc.; Austin, TX). Isolated RNA was subjected to differential gene analysis using the GeneFishing kit (Seegene; Seoul, South Korea) according to the manufacturer’s instructions. For quantitative real-time PCR analysis, total RNA from HAEC was isolated using RNeasy mini kit (Qiagen, Valencia, CA). One μg of total RNA was reverse transcribed into cDNA using a High-Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA). All primers for mRNA analysis were obtained from Integrated DNA Technologies (Coralville, IA). Primers used for human HO-1 were 5′-ATT GCC AGT GCC ACC AAG TTC AAG-3′ (forward), and 5′-TCG CCA CCA GAA AGC TGA GTG TAA-3′ (reverse); for human HO-2: 5′-AAT GTG GAC AAT GCC CAG CAG TTC-3′ (forward), and 5′-TAC GCA TGT CTC CTT TCC CAT CGT-3′ (reverse); for human ET-1: 5′-AAA CAG CAG TCT TAG GCG CTG A-3′ (forward) and 5′-GAC ACA CTC TTT ATC CAT CAG GGA C-3′ (reverse); for human VCAM-1: 5′-TTG CTC AGA TTG GTG ACT CCG-3′ (forward), and 5′-AAA ACT CAC AGG GCT CAG GGT CAG-3′ (reverse); for human β-actin: 5′-CTG GCA CCC AGC ACA ATG AAG-3′ (forward), and 5′-TAG AAG CAT TTG CGG TGG ACG-3′ (reverse). Quantitative real-time PCR was performed using QuantiTect SYBR Green PCR Kit (Qiagen) and analyzed with ABI Prism 7900HT Sequence Detection System (Applied Biosystems; Foster City, CA). Quantitative PCR was performed as follows: step 1: 50 °C for 2 min, step 2: 95 °C for 15 min, and step 3: 95 °C for 15 sec, 59 °C for 30 sec, and 72 °C for 30 sec for 45 cycles. The data and calculation of cycle threshold were analyzed using SDS 2.2 software (Applied Biosystems). mRNA expression for HO-1, HO-2, and ET-1 was normalized to β-actin mRNA expression.
Immunoblotting
For immunoblotting experiment, cells were washed with ice-cold PBS and then cell lysates were prepared using lysis buffer containing 40 mM Tris.Cl pH 7.4, 10 mM EDTA pH 8.0, 120 mM NaCl, 0.1% NP-40, and complete protease inhibitor cocktail (Roche Applied Science; Indianapolis, IN). For each experiment, equal amounts of cell lysates (15 – 25 μg of total protein) were boiled with Laemmli sample buffer for 5 min. Then proteins were resolved by 10% SDS-PAGE (Invitrogen; Carlsbad, CA), transferred to nitrocellulose membranes, and subjected to immunoblotting according to standard methods. Results from multiple independent experiments were quantified by scanning densitometry using ImageQuant software (Molecular Dynamics, Piscataway, NJ).
HO-1 Activity Assay
HAEC were treated as described in legends to figures and cell lysates were assayed for HO-1 activity using Human HO-1 ELISA Kit according to the manufacturer’s protocol. HO-1 activity was calculated as nmol of bilirubin (product) per μg of cell lysate protein per min. For quantification of multiple independent experiments, results were normalized to results from the vehicle treatment group without inhibitor pre-treatment and the data expressed as mean ± SEM.
siRNA Knockdown of p38 MAPK, HO-1, and Nrf-2
HAEC were transfected with siRNA specifically targeting human HO-1 (catalog #L-006372-00; gene HMOX-1; Dharmacon, Chicago, IL), human p-38 MAPK (catalog #L-003555-00; gene MAPK1; Dharmacon), Nrf-2 (catalog #L-003755-00; gene NFE2L2; Dharmacon), or non-targeting control siRNA (catalog #D-001810-10-20, Dharmacon) using Lipofectamine/PLUS Reagent (Invitrogen) for 4 h. Two days after transfection, cells were serum-starved for 1 h and then some groups of cells were treated with vehicle or EGCG (2.5 μM) in the absence or presence of TNF-α as described in legends to figures. Cells lysates were subjected to immunoblotting as described above or cells were fixed for immunohistocytochemistry.
Monocyte Adhesion Assay
HAEC were grown to 90% confluence in Lab-Tek chamber slides and serum-starved for 1 h in endothelial basal medium. Cells were then treated without or with TNF-α (10 ng/ml, PeproTech, USA) in the presence of vehicle or EGCG (2.5 μM) for 5 h. In some experiments, HAEC transfected with siRNA targeting HO-1, p-38, Nrf-2, or non-targeting control siRNA were serum-starved for 1 h followed by treatment with TNF-α vehicle (10 ng/ml) in the presence of vehicle or EGCG (2.5 μM) for 5 hours. U937 monocytes (American Type Culture Collection, Manassas, VA) were grown in DMEM medium containing 10% FBS and then labeled with 5 μM calcein-AM (Invitrogen, Molecular Probes, Inc. USA) for 15 min. Labeled U937 cells (6 × 105) were incubated for 15 min at 37 °C with confluent HAEC that were pre-treated with vehicle, EGCG (2.5 μM) and/or TNF-α (10 ng/ml) for 5 h. Co-cultured cells were washed 3 times with PBS and fixed in 2% folmaldehyde. Prior to visualization, cells were washed three times with PBS and treated with ProLong Gold anti-fade reagent (Invitrogen). Images of monocytes adhering to HAEC were obtained with an Olympus IX81 inverted microscope with attached CCD camera (Retiga Exi, Burnaby, BC, Canada).
HO-1 Promoter and ARE Activity
HAEC were grown to 80% confluence in 6-well plates, and each well of cells was co-transfected with 0.2 μg of HO-1 phLUC45 DNA and 10 ng of renilla luciferase reporter (pRL-TK, Promega; internal control) using Lipofectamine/PLUS reagent for 4 h according to the manufacturer’s protocol. HO-1 phLUC45 is a luciferase reporter construct containing the human HO-1 promoter driving a luciferase reporter gene (4.5 kb of the 5′ flanking region of human HO-1; kindly provided by Dr. Shigeki Shibahara, Department of Molecular Biology and Applied Physiology, Tohoku University School of Medicine, Sendai, Japan) [25]. Transfected cells were cultured in complete medium. 48 h post-transfection, cells were serum-starved for 1 h and then treated with vehicle, EGCG (2.5 μM), or insulin (100 nM, Humulin, Eli Lilly) in the absence or presence of the p38 MAPK inhibitor SB203580 (10 μM) for 6 h. Luciferase activity was determined using the Dual-luciferase Reporter Assay System (Progema) in conjunction with a Lumat LB9501 luminometer (Berthold Technologies, Oak Ridge, TN). HO-1 promoter driven luciferase activity was normalized to renilla luciferase activity.
For the antioxidant-response element (ARE) luciferase assay, HAEC were grown in 6-well dishes to 60–70% confluence in complete growth media. The cells were co-transfected with 1 μg of the luciferase reporter gene construct containing wildtype ARE (ARE-WT), a deletion mutant (ARE-Ti) lacking the ARE-core binding sequence, or a point mutant (ARE-GC) with mutations in the GC box of the ARE core sequence that prevents binding of ARE elements (kindly provided by Dr. Jeffrey A. Johnson, University of Wisconsin, Madison, WI) [26, 27] and 10 ng of renilla luciferase reporter (internal control) using Lipofectamine/PLUS reagent for 4 h according to the manufacturer’s protocol. 48 h after transfection, cells were serum starved for 1 h and then treated with vehicle or EGCG (2.5 μM) for 6 h. Luciferase activity was determined as described above.
Preparation and Analysis of Mouse Liver Homogenates and mRNA
All procedures in mice were approved by the NHLBI, NIH Animal Care and Use Committee. Mice (C57BL/6J) were treated with EGCG (75 mg/kg body weight daily) or vehicle 5 times a week (daily except for weekends) for 5 weeks. Liver samples from 3 EGCG-treated and 3 placebo-treated mice were homogenized in ice-cold PBS and then lysed in lysis buffer (40 mM Tris.Cl pH 7.4, 10 mM EDTA pH 8.0, 120 mM NaCl, 0.1% NP-40, and complete protease inhibitor mixture). Samples were subjected to immunoblotting as described above. Total RNA was isolated from other liver samples from the same mice using the RNeasy mini kit (Qiagen, Valencia, CA). Total RNA was subjected to quantitative PCR using a High-Capacity cDNA Archive Kit and SYBR green real-time PCR according to the manufacturer’s instructions (Qiagen). mRNA for HO-1, HO-2, ET-1, VCAM-1 and β-actin was amplified using primer sets as described above. Data and calculation of ΔΔ Ct (cycle threshold) were analyzed using SDS 2.2 software. HO-1, HO-2, ET-1, and VCAM-1 mRNA expression was normalized to β-actin mRNA expression.
Statistics
Data are expressed as mean ± S.E.M. of multiple independent experiments. Student’s t-tests (paired or unpaired) and one-way analysis of variance (ANOVA) followed by Dunnett’s multiple comparisons test was used to evaluate differences where appropriate. p values < 0.05 were considered to indicate statistical significance.
RESULTS
EGCG Stimulates Increased Expression of HO-1 mRNA and Protein in Vascular Endothelial Cells
We previously demonstrated that EGCG treatment of vascular endothelial cells stimulates production of the vasodilator NO through signaling pathways involving H2O2/Fyn/PI3K/Akt/eNOS [6] and inhibits synthesis and secretion of the vasoconstrictor ET-1 through activation of PI3K/Akt/FoxO1 [8]. To identify additional mechanisms whereby EGCG may mediate beneficial cardiovascular actions, we used differential gene expression analysis to screen for changes in gene expression in peripheral vascular endothelial cells in response to EGCG treatment. In our initial screen, among a number of genes whose expression significantly changed, we found that treatment of human aortic endothelial cells (HAEC) with EGCG (10 μM, 8 h) significantly induced expression of the gene for hemeoxygenase-1 (HO-1), an important enzyme in cellular antioxidant defenses (Fig. 1A). This preliminary result was subsequently confirmed by quantitative RT-PCR in HAEC (Fig. 1B, C). That is, EGCG treatment of HAEC (10 μM, 8 h) increased expression of HO-1 mRNA 2-fold without significantly affecting expression of the HO-2 isoform. As a positive control, ET-1 mRNA expression was appropriately suppressed by EGCG treatment consistent with our previous findings [8]. Interestingly, a lower concentration of EGCG (2.5 μM, 8 h) stimulated an even greater 3-fold increase in HO-1 mRNA without altering the previously observed effects on HO-2 and ET-1 (Fig. 1C).
Figure 1.

Treatment of endothelial cells with EGCG increased HO-1 mRNA expression. A. HAEC were serum-deprived for 2 h and then treated with vehicle or EGCG (10 μM, 8 h). Total RNA was isolated from these cells and subjected to differential gene analysis as described in Methods. M = marker lane. B. HAEC were serum-deprived for 1 h and then treated with vehicle or EGCG (10 μM, 8 h). One μg of total RNA was reverse transcribed and subjected to quantitative real-time PCR for HO-1, HO-2, ET-1, and β-actin using QuantiTect SYBR Green PCR with appropriate primer sets as described in Methods. HO-1, HO-2, and ET-1 mRNA expression were normalized to β-actin mRNA expression. Data shown are the mean ± SEM of 4 independent experiments (vehicle vs. EGCG treatment for HO-1, HO-2, and ET-1; p < 0.02, p > 0.18, and p < 0.0005, respectively). C. HAEC were treated as in panel B except various concentrations of EGCG were used as indicated. Total RNA was subjected to quantitative real-time PCR analysis as described in panel B. Data shown are the mean ± SEM of 4 independent experiments. EGCG treatment caused increased expression of HO-1 (but not HO-2) mRNA and attenuated ET-1 mRNA expression (p < 0.001 by ANOVA and Dunnett’s post-test).
With respect to protein levels, EGCG treatment of HAEC (2.5 μM, 8 h) increased expression of HO-1 protein ~1.5 fold without significantly altering HO-2 protein expression (Fig. 2). This effect of EGCG to increase expression of HO-1 protein was time-dependent (Fig. 3A). Moreover, corresponding to results with HO-1 mRNA expression (Fig. 1C), we observed a concentration-dependent effect of EGCG to stimulate HO-1 (but not HO-2) protein expression. The highest induction was observed with an EGCG concentration of 2.5 μM (Fig. 3B). Taken together, our results suggest that EGCG treatment of endothelial cells at concentrations in the low micromolar range (comparable to levels in human circulation after consumption of green tea [28]) specifically stimulated substantial increases in expression of HO-1 (but not HO-2) mRNA and protein.
Figure 2.

Treatment of endothelial cells with EGCG increased protein expression of HO-1 but not HO-2. A. HAEC were serum-starved for 1 h and then treated with vehicle or EGCG (10 μM, 8 h). Cell lysates were immunoblotted using antibodies against HO-1, HO-2, and β-actin as indicated. Representative results from two independent experiments are shown in each set of blots. B. Results from 6 independent experiments represented in Panel A were quantified by scanning densitometry and normalized to β-actin expression (mean ± SEM) (vehicle vs. EGCG treatment for HO-1 and HO-2 expression; p < 0.0001 and p > 0.60, respectively).
Figure 3.

Treatment of endothelial cells with EGCG increased expression of HO-1 protein, but not HO-2, in a time- and concentration-dependent manner. A. HAEC were serum-starved for 1 h and then treated with vehicle or EGCG (10 μM) for the indicated times. B. HAEC were serum-starved for 1 h and then treated with vehicle or EGCG (8 h) at the indicated concentrations. In panels A and B, cell lysates were immunoblotted using antibodies against HO-1, HO-2, or βactin. Representative blots from experiments repeated independently 4 times are shown in panels A and B. Data in bar graphs are mean ± SEM of 4 independent experiments quantified by scanning densitometry and normalized to β-actin expression. EGCG significantly increased the amount of HO-1 protein (but not HO-2) in a time-(p < 0.001 by ANOVA and Dunnett’s post-test) and concentration-dependent fashion (p < 0.05 by ANOVA and Dunnett’s post-test).
EGCG-stimulated Expression of HO-1 mRNA, Protein, and Activity in Vascular Endothelial Cells Requires activation of p38 MAPK but not PI 3-kinase
To help elucidate mechanisms by which EGCG treatment increased expression of HO-1 mRNA and protein, we used specific chemical inhibitors to block some signaling pathways known to be activated by EGCG [6, 8, 10]. After serum-starving HAEC for 1 h, cells were pre-treated without or with wortmannin (100 nM, PI3K inhibitor) or SB203580 (10 μM, p38 MAPK inhibitor) for 1 h and then treated without or with EGCG (2.5 μM, 6 h) (Fig. 4). As shown previously (Figs. 1 – 3), treatment of HAEC with EGCG in the absence of pathway-selective inhibitors significantly increased expression of HO-1 mRNA and decreased expression of ET-1 mRNA without significantly changing expression of HO-2 mRNA and protein (Fig. 4A, left group; Fig. 4B, lanes 1 and 2; Fig. 4C, left group). Moreover, EGCG stimulated a significant increase in intracellular HO-1 activity (Fig. 4D, left group). Similar results for HO-1 expression were observed in the presence of wortmannin pre-treatment where EGCG-stimulated inhibition of ET-1 mRNA expression was blocked as expected (Fig. 4A, middle group; Fig. 4B, lanes 3 – 4; Fig. 4C, D, middle groups) [8]. Thus, activation of PI3K was not required for EGCG to stimulate increased expression of HO-1 mRNA and protein and HO-1 activity in peripheral vascular endothelial cells. By contrast, in cells pretreated with SB203580, EGCG treatment of HAEC was unable to significantly induce expression of HO-1 mRNA, protein, or HO-1 activity (Fig. 4A, right group; Fig. 4B, lanes 5 – 6; Fig. 4C, D, right groups). To demonstrate the specificity of our chemical inhibitors under these experimental conditions, we showed that wortmannin pre-treatment did not block EGCG-stimulated phosphorylation of either p44/p42 MAPK or p38 MAPK while pre-treatment with SB208530 blocked EGCG-stimulated phosphorylation of p38 MAPK but not p44/p42 MAPK (Fig. 4B and 4C). Similar to wortmannin pre-treatment, pre-treatment of HAEC with PD98059 (25 μM, MEK inhibitor), LY 294002 (50 μM, another PI 3-kinase inhibitor), or L-NAME (100 μM, NOS inhibitor) did not significantly affect the ability of EGCG to stimulate increased expression of HO-1 protein (Fig. 5).
Figure 4.

EGCG-stimulated increases in HO-1 mRNA, protein, and enzyme activity required p38 MAPK, but not PI3K, activity. HAEC were serum-starved for 1 h, then pre-treated without or with pathway-selective chemical inhibitors (wortmannin or SB208530) for 1 h as described in Methods. This was followed by treatment without or with EGCG (2.5 μM, 6 h). A. One μg of total RNA was reverse-transcribed and subjected to quantitative real-time PCR for HO-1, HO-2, ET-1, and β-actin using QuantiTect SYBR Green PCR with appropriate primer sets as described in Methods. HO-1, HO-2, and ET-1 mRNA expression were normalized to β-actin mRNA expression. Data shown are mean ± SEM of 4 independent experiments. In the absence of pre-treatment with inhibitors, when compared with vehicle-treatment, EGCG increased expression of HO-1 (p < 0.0002) but not HO-2 mRNA (p > 0.25) while decreasing expression of ET-1 mRNA (p < 0.0001). In the presence of wortmannin pre-treatment, EGCG stimulated increased expression of both HO-1 and ET-1 mRNA (p < 0.01), but did not change expression of HO-2 (p > 0.28). In the presence of SB203580, EGCG treatment did not increase expression of either HO-1 or HO-2 mRNA (p > 0.25) but did decrease expression of ET-1 mRNA (p < 0.0005). B. Cell lysates were immunoblotted using antibodies against HO-1, HO-2, phospho-p44/42 MAPK, phospho-p38 MAPK, and β-actin as indicated. Immunoblots shown in panels B and C are representative results from at least 3 independent experiments. C. Results from 4 independent experiments represented in panel B were quantified by scanning densitometry and normalized to β-actin expression (mean ± SEM). In the absence of pre-treatment with inhibitors, when compared with vehicle-treatment, EGCG stimulated increased expression of HO-1 (p < 0.0007) but not HO-2 (p > 0.29). Pre-treatment of cells with wortmannin had no effect on EGCG-induced expression of HO-1 (p > 0.18). Pre-treatment of cells with SB208530 diminished EGCG-induced expression of HO-1 (p < 0.001). D. Cell lysates from cells treated as above were used to determine HO-1 activity. Data shown are mean ± SEM of 3 independent experiments. In the absence of pre-treatment with inhibitors, when compared with vehicle treatment, EGCG stimulated increased activity of HO-1 (p < 0.008). In the presence of wortmannin pre-treatment, EGCG also stimulated increased activity of HO-1 (p < 0.05). Pre-treatment of cells with SB203580 blocked EGCG-stimulated increases in HO-1 activity (p > 0.9). Wort = wortmannin; SB = SB208530. In the absence of pre-treatment with inhibitors or with wortmannin pre-treatment, when compared with vehicle-treatment, EGCG stimulated increased phosphorylation of both p44/42 MAPK and p38 MAPK (p < 0.001). With SB pre-treatment, when compared with vehicle treatment, EGCG stimulated increased phosphorylation of p44/42 MAPK (p < 0.05) but did not alter phosphorylation of p38 MAPK (p > 0.33).
Figure 5.
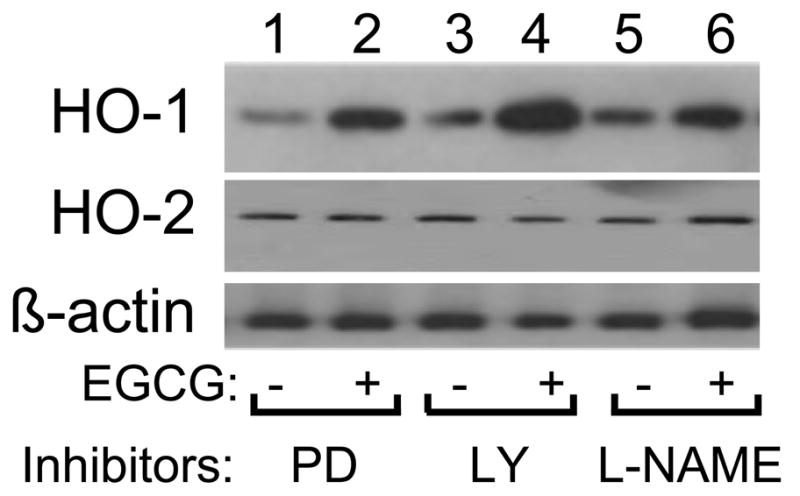
EGCG-stimulated increases in HO-1 protein expression did not require MEK, PI3K, or NOS activity. HAEC were serum-starved for 1 h and then pre-treated without or with various pathway-selective chemical inhibitors for 1 h (PD = PD98059, 25 μM; LY = LY294002, 50 μM; L-NAME, 100 μM). This was followed by treatment without or with EGCG (2.5 μM, 6 h). Cell lysates were immunoblotted using antibodies against HO-1, HO-2, and β-actin as indicated. Immunoblots shown are representative results from at least 3 independent experiments.
Since pathway-selective chemical inhibitors may have non-specific actions, we used siRNA knockdown to help definitively assess the role of p38 MAPK in EGCG-stimulated HO-1 expression. As with previous experiments (Figs. 2, 3, 4), in HAEC transfected with non-targeting control siRNA, EGCG treatment (2.5 μM, 8 h) significantly increased expression of HO-1 protein (Fig. 6A, lanes 1 – 4; Fig. 6B). By contrast, in HAEC transfected with siRNA specifically targeting p38 MAPK, EGCG-stimulated induction of HO-1 was completely blocked (Fig. 6A, lanes 5 – 8; Fig. 6B). In this experimental preparation, transfecting HAEC with siRNA targeting p38 MAPK (but not non-targeting control siRNA) significantly and substantially reduced expression of p38 MAPK protein as expected (Fig. 6A, C). Taken together, our results strongly suggest that EGCG-stimulated increases in expression and activity of HO-1 in HAEC required activation of p38 MAPK but not other signaling pathways activated by EGCG.
Figure 6.

siRNA knockdown of p38 MAPK blocked EGCG-stimulated increases in HO-1 expression. A. HAEC were transfected with scrambled siRNA (control) or siRNA specifically targeting p38-MAPK as described in Methods. 48 h after transfection, cells were serum-starved for 1 h and then treated with vehicle or EGCG (2.5 μM, 8 h). Whole cell lysates were subjected to immunoblotting using antibodies against HO-1, p38 MAPK, and β-actin as indicated. Immunoblots shown represent 2 independent experiments that were repeated independently 3 times (total of 6 independent experiments). B. Results for HO-1 expression from 6 independent experiments represented in panel A were quantified by scanning densitometry and normalized to β-actin expression (mean ± SEM). In cells transfected with scrambled control siRNA, when compared with vehicle treatment, EGCG stimulated increased expression of HO-1 (p < 0.0001). By contrast, in cells transfected with siRNA targeting p38-MAPK, this effect of EGCG was completely blocked (p > 0.23). C. Results for p38-MAPK expression from 6 independent experiments represented in panel A were quantified by scanning densitometry and normalized to β-actin expression (mean ± SEM). When compared with cells transfected with scrambled (control) siRNA, cells transfected with siRNA targeting p38-MAPK had a reduction of p38-MAPK expression of ~40% in the absence or presence of EGCG treatment (p < 0.0001). When compared with vehicle treatment, EGCG did not alter expression of p38-MAPK in cells transfected with either scrambled (control) siRNA or siRNA targeting p38-MAPK (p > 0.17).
EGCG-stimulated Activation of the Human HO-1 promoter and a Nrf-2-responsive Promoter Requires p38 MAPK Activity
In the present study, we found that the ability of EGCG to stimulate increased expression of HO-1 requires p38 MAPK (Figs. 4, 6). Therefore, we used the p38 MAPK inhibitor SB208530 to examine mechanisms by which EGCG stimulates activation of the human HO-1 promoter. In HAEC transiently transfected with phHOLuc45 (4.5 kb of the human HO-1 promoter driving a luciferase reporter [25]), EGCG-stimulated activation of the HO-1 promoter was completely blocked by pre-treatment of cells with SB203580 (Fig. 7A). Similarly, in HAEC transiently transfected with a luciferase reporter construct driven by the Nrf-2-responsive Antioxidant Response Element (ARE-WT, constructed from a 34 bp fragment from the ARE region of the 5′-flanking promoter region of the human HO-1 gene [29]), EGCG-stimulated activation of ARE-WT was completely blocked by pre-treatment of cells with SB208530 (Fig. 7B, left group). Mutants of ARE-WT lacking the core DNA binding sequence (ARE-Ti [30]) or with point mutations in the GC box of the ARE core binding region for Nrf-2 (ARE-GC [26]) were both unresponsive to EGCG treatment, in either the absence or presence of SB208530 (Fig. 7B, center and right groups). Taken together, these data strongly suggest that EGCG increases expression of HO-1 by p38 MAPK-dependent induction of Nrf-2.
Figure 7.

Activation of the human HO-1 promoter and Nrf-2 responsive promoter constructs in response to EGCG required p38 MAPK activity. A. HAEC were co-transfected with a luciferase reporter construct driven by the human HO-1 promoter (phHOLUC-45) and a renilla luciferase construct (internal control). 48 h after transfection, cells were serum-starved for 1 h and then pre-treated without or with SB203580 (10 μM) for 1 h followed by treatment with vehicle or EGCG (2.5 μM, 6 h). Luciferase activity normalized to renilla luciferase activity was assessed in cell lysates as described in Methods. Data shown are mean ± SEM of 4 independent experiments. In the absence of SB, EGCG stimulated increased HO-1 promoter activity (when compared with vehicle; p < 0.001). This effect was completely blocked by SB pre-treatment (p > 0.05). B. HAEC were transfected with ARE-WT, ARE-Ti, or ARE-GC mutant-Luc reporter constructs along with a renilla luciferase construct. Forty eight h after transfection, cells were serum starved for 1 h and then treated with vehicle or EGCG (2.5 μM, 6 h). Luciferase activity normalized to renilla luciferase activity was assessed in cell lysates as described in Methods. Data shown are mean ± SEM of 3 independent experiments. In cells transfected with ARE-WT, EGCG treatment increased luciferase activity (when compared with vehicle treatment, p < 0.001) and SB inhibited this effect of EGCG (p < 0.01). In cells transfected with either ARE-Ti or ARE-GC, EGCG had no significant effect to alter luciferase activity in either the absence or presence of SB (when compared with vehicle treatment, p > 0.05)
To more completely define the roles of p38 MAPK and Nrf-2 in EGCG-stimulated expression of HO-1, we next examined effects of EGCG treatment on Nrf-2 and HO-1 expression in HAEC treated without or with EGCG in the absence or presence of Nrf-2 or p38 MAPK knockdown by siRNA (Fig. 8). Treatment of HAEC with EGCG (2.5 μM. 8 h) simultaneously increased Nrf-2 and HO-1 protein expression (Figs. 8A and 8B). When compared with control cells transfected with non-targeting siRNA, in HAEC transfected with siRNA targeting Nrf-2, EGCG treatment no longer increased expression of either Nrf-2 or HO-1 (Fig. 8C). Similar results were observed with siRNA knockdown of p38 MAPK (Fig. 8D). Taken together, our results suggest that both p38 MAPK and Nrf-2 are required for EGCG to stimulate increased expression of HO-1 in vascular endothelial cells. Moreover, p38 MAPK activity is required for EGCG to stimulate increased expression of Nrf-2. Therefore, one mechanism for EGCG to increase expression of HO-1 likely involves activation of p38 MAPK that subsequently induces expression of Nrf-2. Nrf-2 binding to the ARE region of the HO-1 promoter then increases expression of HO-1 mRNA and protein resulting in an increase in total cellular HO-1 activity.
Figure 8.

EGCG-stimulated increases in expression of HO-1 in endothelial cells required Nrf-2 and p38 MAPK. A. HAEC were serum starved for 1 h and then treated without or with EGCG (2.5 μM, 8 h). Nuclear extracts were prepared as described in Methods and then immunoblotted using antibodies against Nrf-2, HO-1 and β-actin. B. Results from 3 independent experiments represented in panel A were quantified by scanning densitometry and normalized to β-actin expression (mean ± SEM). EGCG stimulated increased Nrf-2 expression (p < 0.001). C and D. HAEC were transiently transfected with scrambled siRNA (control) or siRNA targeting Nrf-2 or p38 MAPK-1 as described in Methods. 48 h after transfection, cells were serum-starved for 1 h and then treated with vehicle or EGCG (2.5 μM, 8 h). Nuclear extracts were immunoblotted using antibodies against HO-1, Nrf-2 and β-actin. Representative blots are shown for experiments that were repeated independently 3 times.
EGCG Treatment of Vascular Endothelial Cells Opposes Increased Expression of VCAM-1 and Monocyte Adhesion Caused by Exposure to TNF-α
Pro-inflammatory cytokines including TNF-α contribute to endothelial dysfunction, in part, by increasing expression of cellular adhesion molecules resulting in increased adhesion of monocytes to vascular endothelium [31, 32]. One potential mechanism for EGCG to improve cardiovascular health may be to oppose actions of pro-inflammatory cytokines in vascular endothelium. To evaluate this possibility, we treated serum-starved HAEC without or with EGCG (2.5 μM, 5 h) in the absence or presence of TNF-α (10 ng/ml, 5 h) (Fig. 9). In the absence of EGCG treatment, exposure to TNF-α resulted in a ~ 5-fold increase in VCAM-1 expression (Figs. 9A and 9B; c.f., lanes 1 and 3), and increased adhesion of monocytes to HAEC (Fig. 9C; c.f., 1st and 3rd columns). Interestingly, treating HAEC simultaneously with EGCG and TNF-α nearly completely abrogated these pro-atherogenic actions of TNF-α (Figs. 9A and 9B; c.f., lanes 3 and 4; Fig. 9C; c.f., 3rd and 4th columns). To evaluate whether these salutary effects of EGCG were mediated by increased expression of HO-1, we used siRNA to specifically knockdown HO-1, p38 MAPK, or Nrf-2 (transcription factor implicated in HO-1 expression [30, 33, 34]) in HAEC treated with TNF-α (10 ng/ml, 5 h) in the absence or presence of EGCG (2.5 μM, 5 h) (Fig. 10). In HAEC transfected with non-targeting control siRNA and treated with TNF-α, EGCG treatment reduced expression of VCAM-1 and increased expression of HO-1 while reducing monocyte adhesion to HAEC (Fig. 10C, columns 1 and 2) without significantly altering expression of HO-2 (Figs. 10A and 10B, lanes 1 and 2) (when compared with HAEC transfected with non-targeting control siRNA and treated with TNF-α in the absence of EGCG). Importantly, in HAEC transfected with siRNA targeting HO-1, treatment of cells with EGCG did not increase HO-1 expression and EGCG no longer blocked the ability of TNF-α to increase expression of VCAM-1 (Fig. 10A and 10B, lanes 3 and 4). Moreover, siRNA knockdown of HO-1 also prevented the ability of EGCG to inhibit monocyte adhesion to HAEC in response to TNF-α (Fig. 10C, columns 3 and 4). With respect to monocyte adhesion, similar results were observed with siRNA knockdown of p38 MAPK or Nrf-2 (Fig. 10C columns 5 – 8). Therefore, the ability of EGCG to oppose anti-atherogenic actions of TNF-α in HAEC requires the induction of HO-1 that is likely mediated by p38 MAPK and Nrf-2.
Figure 9.

EGCG treatment of endothelial cells impaired TNF-α-stimulated expression of VCAM-1 and monocyte adhesion. A. HAEC were serum-starved for 1 h and then treated with vehicle or TNF-α (10 ng/ml, 5 h) in the absence or presence of EGCG (2.5 μM). Whole cell lysates were subjected to immunoblotting using antibodies against VCAM-1, HO-1, and β-actin as indicated. A representative set of blots from experiments that were repeated independently 6 times is shown. B. Results from 6 independent experiments represented in panel A were quantified by scanning densitometry and normalized to β-actin expression (mean ± SEM). In the absence of TNF-α, EGCG treatment did not alter expression of VCAM-1 (when compared with vehicle treatment, p > 0.19). In the absence of EGCG, TNF-α treatment significantly increased expression of VCAM-1 (when compared with vehicle-treatment, p < 0.0001). In the presence of EGCG, TNF-α induced expression of VCAM-1 was significantly reduced (when compared with the absence of EGCG, p < 0.003). C. U937 monocytic cells (6 × 105) were labeled as described in Methods and then incubated for 30 min at 37 °C with confluent HAEC treated with various combinations of EGCG, TNF-α, and vehicle as indicated (same conditions as Panel A). Chambered wells were then washed 3 times with PBS and cells were fixed in 2% folmaldehyde. Labeled monocytes adhering to HAEC were visualized using an epifluorescent microscope as described in Methods (upper panel). Phase contrast view of cells is shown in lower panel.
Figure 10.

siRNA knockdown of HO-1 blocked the inhibitory action of EGCG on TNF-α-stimulated VCAM-1 expression and monocyte adhesion in endothelial cells. A. HAEC were transiently transfected with scrambled siRNA (control) or siRNA targeting HO-1 as described in Methods. 48 h after transfection, cells were serum-starved for 1 h and then treated with vehicle or TNF-α (10 ng/ml) for 5 h in the presence or absence of EGCG (2.5 μM). Cell lysates were then subjected to immunoblotting using antibodies against VCAM-1, HO-1, HO-2, and β-actin as indicated. Immunoblots shown are representative of experiments that were repeated independently 4 times. B. Results for VCAM-1 expression from 4 independent experiments represented in panel A were quantified by scanning densitometry and normalized to β-actin expression (mean ± SEM). In cells transfected with control scrambled siRNA, EGCG treatment diminished TNF-α stimulated VCAM-1 expression (p < 0.001). In cells transfected with HO-1 siRNA, EGCG treatment failed to modulate TNF-α induced VCAM-1 expression with, (p > 0.05) (by ANOVA and Bonferroni’s post-test). C. HAEC were transfected with scrambled siRNA (control) or siRNA specifically targeting HO-1, p38-MAPK, or nrf-2. 48 h after transfection, cells were serum-starved for 1 h and then treated with vehicle or TNF-α (10 ng/ml) for 5 h in the presence or absence of EGCG (2.5 μM). Labeled U937 monocytes (6 × 105) were incubated for 30 min at 37 °C with confluent HAEC pre-treated with EGCG and/or TNF-α. Co-cultured cells were washed 3 times with PBS and fixed in 2% folmaldehyde. Images of monocytes adhering to HAECs were visualized using an epifluorescent microscope (upper panel). Phase contrast view of cells is shown in lower panel.
In Vivo Effects of EGCG Treatment on HO-1 and VCAM-1 Expression in Mice
HO-1 is generally expressed in tissues where heme metabolism takes place [35] and is highly inducible in response to oxidative stress in many organs including liver [16, 36, 37]. To determine if effects of EGCG to increase HO-1 expression and inhibit VCAM-1 expression in primary endothelial cells have physiological correlates in vivo, we treated mice (C57BL/6J) with vehicle control or purified EGCG (75 mg/kg body weight daily) 5 times per week for 5 weeks as described in Methods. Liver homogenates from 3 EGCG-treated and 3 vehicle-treated mice were processed and assessed for HO-1, HO-2, ET-1, and VCAM-1 mRNA expression using quantitative RT-PCR (Fig. 11A). Similar to our acute in vitro experiments in vascular endothelial cells, chronic in vivo treatment of mice with EGCG caused an increase in HO-1 mRNA expression in liver without changing HO-2 mRNA expression (ET-1 mRNA expression was also decreased). Liver samples were immunoblotted using antibodies against HO-1 and VCAM-1. Gratifyingly, expression of HO-1 protein increased while expression of VCAM-1 protein decreased in livers from mice treated in vivo with EGCG (when compared with livers from placebo-treated mice) (Figs. 11B, 11C). Therefore, chronic treatment of rodents with EGCG at doses sufficient to raise circulating EGCG levels into the micromolar range [38] had significant effects to increase HO-1 expression and decrease VCAM-1 expression in liver. These results raise the possibility that mechanisms of action for EGCG to acutely induce HO-1 and oppose pro-inflammatory events in vascular endothelial cells have correlates in vivo that may be physiologically relevant to cardiovascular and metabolic health benefits of green tea consumption.
Figure 11.

Increased expression of HO-1 mRNA and protein and decreased expression of VCAM-1 in the liver of mice treated chronically with oral EGCG. Mice (C57BL/6J) were given EGCG (75 mg/kg body weight, n = 3) or vehicle (n = 3) by gavage 5 times per week (daily except for weekends) for 5 weeks. A. Total RNA extracted from liver homogenates from 3 mice in each group was subjected to real time quantitative PCR for HO-1, HO-2, ET-1, and β-actin. Results shown are mean ± SEM of mRNA expression for HO-1, HO-2, and ET-1 normalized to β-actin from 3 mice each in the vehicle- and EGCG-treatment groups. Mice treated with EGCG had higher expression of HO-1 mRNA and lower expression of ET-1 mRNA in liver than mice treated with vehicle (p < 0.02). By contrast, EGCG treatment of mice did not alter expression of HO-2 mRNA in liver when compared with vehicle-treated mice (p > 0.43). B. Liver homogenates of vehicle- and EGCG-treated mice were immunoblotted using antibodies against HO-1, VCAM-1 and β-actin as indicated (results from three mice from each group are shown). C. Results represented in panel B (n = 3 for each group) were quantified by scanning densitometry and normalized to β-actin expression (mean ± SEM). Livers from mice treated with EGCG had higher expression of HO-1 (p < 0.013) and decreased expression of VCAM-1 (p < 0.015) when compared with livers from control mice treated with vehicle.
DISCUSSION
The green tea polyphenol EGCG mediates biological actions in vascular endothelium, liver, and other insulin targets that may contribute to beneficial metabolic and cardiovascular effects of drinking green tea [2–6, 8, 10, 12, 39, 40]. In the present study, we identified and characterized the ability of EGCG to induce expression of HO-1 in endothelial cells resulting in anti-inflammatory actions that may oppose atherogenesis and improve endothelial dysfunction.
EGCG stimulates increased HO-1 expression in endothelial cells via p38 MAPK and Nrf-2
We identified HO-1 as a candidate gene for regulation of cardiovascular and metabolic function that was induced in response to EGCG treatment. Low micromolar concentrations of EGCG specifically increased expression of HO-1, but not HO-2, mRNA and protein in a time-dependent manner in human vascular endothelial cells (maximal effect at 2.5 μM EGCG). We found that induction of HO-1 protein in response to EGCG treatment in HAEC required p38 MAPK, but not PI3K, MEK, ERK (p44/42 MAPK), or NOS. Moreover, expression of Nrf-2, a transcription factor that regulates HO-1 expression [29, 30, 41–44] by binding to ARE regions of the human HO-1 promoter [29, 30] was also increased by EGCG treatment of HAEC in a p38 MAPK-dependent fashion. siRNA knockdown of Nrf-2 prevented induction of HO-1 in response to EGCG in HAEC. Finally, EGCG-stimulated increases in HO-1 promoter activity and ARE-driven reporter constructs also required p38 MAPK.
A variety of protein kinase signaling pathways including Akt, MAPK, PKA, and PKG are implicated in regulation of HO-1 expression in various tissues [45, 46]. Cell-specific regulation of transcription factors including nuclear factor-κB (NF-κB), heat-shock factor, activator protein-1, and Nrf-2 increase HO-1 mRNA and protein expression [45, 46]. Nrf-2, a redox-sensitive leucine-zipper transcription factor activated by oxidants, binds to antioxidant response elements (AREs) in promoter regions of genes involved in cellular antioxidant responses including HO-1 [45–47]. In the basal state, Nrf-2 is cytoplasmic and bound to Keap1, an actin binding protein that keeps Nrf-2 in an inactive form [48]. Keap1 recruits ubiquitin ligases that target Nrf-2 to degradation. Modification of Nrf2 by oxidative stress or phosphorylation leads to dissociation from Keap1 and translocation to the nucleus where it binds to AREs to enhance HO-1 promoter activity [49, 50]. Nrf-2 has a short half-life (~30 min) and is regulated in part through changes in stability [51]. Our data suggest that EGCG-induced increases in Nrf-2 expression via p38 MAPK may limit Nrf-2 degradation. Interestingly, carnosal, a polyphenol in rosemary and curcuminoids enhance cellular Nrf-2 expression in PC12 (pheochromocytoma cell line) and MIN6 (pancreatic β-cell line), respectively [43, 52]. However, PI3K/Akt signaling, not p38 MAPK signaling, plays a major role in carnosal- and curcuminoid-induced increases in cellular Nrf2 and HO-1 expression [43, 52]. By contrast, even though we found that EGCG (2.5 μM) activates Akt and ERK, inhibition of p38 MAPK kinase (but not Akt or ERK) was sufficient to completely suppress EGCG-stimulated increases in Nrf2 and HO-1 expression in vascular endothelial cells.
p38 MAPK is implicated in HO-1 induction in various cell types [20, 46]. However, insulin, peroxynitrite, and celecoxib utilize the PI3K/Akt pathway to increase HO-1 in an Nrf-2 dependent fashion in endothelial cells [20, 53, 54]. Therefore, relative contributions of PI3K/Akt, ERK, and p38 MAPK pathways to HO-1 regulation depend on specific cell types and stimuli for HO-1 induction. TGF-β- and cadmium-induced HO-1 expression requires p38 MAPK, but not ERK, in pulmonary epithelial cells (A549) or breast cancer cells (MCF-7), respectively [42, 55]. By contrast, both p38 MAPK and ERK are required to mediate cadmium- and NO-induced HO-1 expression in gastric cancer cells and HeLa cells, respectively [56, 57]. With respect to carnosal-induced HO-1 expression, both PI3K/Akt and p38 MAPK are required [43]. Regulation of HO-1 by insulin in endothelial cells requires PI3K/Akt, but not p38 MAPK [20]. Thus, stimulation of HO-1 expression may require a specific set of signaling pathways in a cell and compound-specific fashion. In some studies, very high concentrations of EGCG (50 μM) that may be cytotoxic activate both PI3K/Akt and MAPK pathways to regulate HO-1 expression in endothelial cells and lymphoblasts (EGCG, 20 μM) [33, 58]. In our study, stimulatory effects of EGCG were observed at low-micromolar concentrations that are achievable in vivo after green tea consumption (< 5 μM). Thus, findings from in vitro studies using high concentrations of EGCG may represent toxic and/or stress responses with limited therapeutic relevance. Data from our study are the first to demonstrate in a single experimental system of primary endothelial cells that EGCG regulates HO-1 expression through a mechanism requiring p38 MAPK upstream of Nrf2 that then binds and activates the HO-1 promoter.
EGCG suppresses pro-inflammatory actions of TNF-α in endothelial cells through upregulation of HO-1
TNF-α is a pro-inflammatory cytokine that promotes endothelial dysfunction and atherogenesis, in part, by increasing expression of cell surface adhesion molecules including VCAM-1 in vascular endothelium [59]. This leads to increased monocyte adhesion to endothelium, a necessary step in atherogenesis [60, 61]. We found that treatment of HAEC with EGCG (2.5 μM) substantially inhibited induction of VCAM-1 by TNF-α that prevented TNF-α-stimulated increases in monocyte adhesion to endothelial cells. Using siRNA to knockdown specific genes, we demonstrated that these anti-atherogenic actions of EGCG required p38 MAPK, Nrf-2, and HO-1.
The cell surface integrin that binds VCAM-1 is selectively expressed in monocytes and a subset of lymphocytes [62] [63]. Therefore, endothelial VCAM-1 expression plays a key role in mediating monocyte and lymphocyte adhesion in atherosclerotic lesions. Loss of laminar blood flow and pro-atherogenic factors including cytokines, oxidized LDL, and hyperinsulinemia in the presence of insulin resistance all stimulate VCAM-1 expression [63]. Activation of signaling pathways including, PI3K/Akt, ERK, p38 MAPK, c-Src, and ROS modulate redox-sensitive transcription factors to mediate cytokine-induced VCAM-1 expression in endothelium [64]. EGCG at high concentrations (> 50 μM) decreases angiotensin II- (Ang II) and cytokine- (TNF-α or IL-1β) stimulated VCAM-1 expression in endothelial cells [32, 65]. However, in these previous studies, intracellular mechanisms mediating anti-inflammatory actions of EGCG were not elucidated. In the present study, we show that low micromolar concentrations of EGCG activated a signaling cascade involving p38 MAPK/Nrf-2/HO-1 in the endothelium to antagonize pro-atherogenic actions of TNF-α. This is consistent with previous studies demonstrating that adenovirus-mediated over-expression of Nrf-2 is sufficient to suppress TNF-α-stimulated VCAM-1 expression in endothelial cells [66]. Moreover, products of HO-1 activity including bilirubin and CO contribute to reducing VCAM-1 expression in endothelium [67, 68]. Thus, EGCG-induced expression of HO-1 in endothelium may oppose cytokine stimulated VCAM-1 expression, in part, through its enzymatic products.
Actions of EGCG on HO-1 expression in vivo
To determine if effects of EGCG to increase HO-1 expression in primary endothelial cells also occur in vivo, we examined livers from mice chronically treated with EGCG or vehicle (placebo). We chose to examine liver because it is an easily accessible tissue known to express HO-1 in a regulated manner. Gratifyingly, livers from mice treated with EGCG had significantly increased expression of HO-1 that corresponded to decreased expression of VCAM-1. Given our results from endothelial cells, it seems plausible that increased expression of HO-1 due to EGCG treatment in vivo may be responsible for diminished expression of VCAM-1. Therefore, anti-atherogenic mechanisms we elucidated in primary endothelial cells may be relevant to in vivo therapy with EGCG and/or beneficial health effects of drinking green tea that reduce cardiovascular and metabolic morbidity and mortality.
Acknowledgments
We gratefully acknowledge the valuable gift of the human HO-1 promoter luciferase reporter construct (phLUC45) from Dr. Shigeki Shibahara, Department of Molecular Biology and Applied Physiology, Tohoku University School of Medicine, Sendai, Japan. We also thank Dr. Jeffery A. Johnson, University of Wisconsin, Madison, for providing us with the wild type and mutant ARE-luciferase reporter constructs used in this study. We thank Dr. Yukihiko Hara from TheaphenonR Tea Solutions, Hara Office Inc. Tokyo, Japan for providing us with purified EGCG.
This work was supported in part by the Intramural Research Program, National Center for Complementary and Alternative Medicine, National Institutes of Health (M.J.Q.) and NHLBI (J.H.C.); in part by a Clinical Research Award from the American Diabetes Association (M.J.Q.); and in part by a grant from the Office of Dietary Supplements, National Institutes of Health (P.P.).
Footnotes
DISCLOSURES
The authors have nothing to disclose
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Graham HN. Green tea composition, consumption, and polyphenol chemistry. Prev Med. 1992;21:334–350. doi: 10.1016/0091-7435(92)90041-f. [DOI] [PubMed] [Google Scholar]
- 2.Kuriyama S, Shimazu T, Ohmori K, Kikuchi N, Nakaya N, Nishino Y, Tsubono Y, Tsuji I. Green tea consumption and mortality due to cardiovascular disease, cancer, and all causes in Japan: the Ohsaki study. Jama. 2006;296:1255–1265. doi: 10.1001/jama.296.10.1255. [DOI] [PubMed] [Google Scholar]
- 3.Stangl V, Lorenz M, Stangl K. The role of tea and tea flavonoids in cardiovascular health. Mol Nutr Food Res. 2006;50:218–228. doi: 10.1002/mnfr.200500118. [DOI] [PubMed] [Google Scholar]
- 4.Iso H, Date C, Wakai K, Fukui M, Tamakoshi A. The relationship between green tea and total caffeine intake and risk for self-reported type 2 diabetes among Japanese adults. Ann Intern Med. 2006;144:554–562. doi: 10.7326/0003-4819-144-8-200604180-00005. [DOI] [PubMed] [Google Scholar]
- 5.Polychronopoulos E, Zeimbekis A, Kastorini CM, Papairakleous N, Vlachou I, Bountziouka V, Panagiotakos DB. Effects of black and green tea consumption on blood glucose levels in non-obese elderly men and women from Mediterranean Islands (MEDIS epidemiological study) Eur J Nutr. 2008;47:10–16. doi: 10.1007/s00394-007-0690-7. [DOI] [PubMed] [Google Scholar]
- 6.Kim JA, Formoso G, Li Y, Potenza MA, Marasciulo FL, Montagnani M, Quon MJ. Epigallocatechin gallate, a green tea polyphenol, mediates NO-dependent vasodilation using signaling pathways in vascular endothelium requiring reactive oxygen species and Fyn. J Biol Chem. 2007;282:13736–13745. doi: 10.1074/jbc.M609725200. [DOI] [PubMed] [Google Scholar]
- 7.Muniyappa R, Montagnani M, Koh KK, Quon MJ. Cardiovascular actions of insulin. Endocr Rev. 2007;28:463–491. doi: 10.1210/er.2007-0006. [DOI] [PubMed] [Google Scholar]
- 8.Reiter CE, Kim JA, Quon MJ. Green tea polyphenol epigallocatechin gallate reduces endothelin-1 expression and secretion in vascular endothelial cells: roles for AMP-activated protein kinase, Akt, and FOXO1. Endocrinology. 2010;151:103–114. doi: 10.1210/en.2009-0997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen H, Lin AS, Li Y, Reiter CE, Ver MR, Quon MJ. Dehydroepiandrosterone stimulates phosphorylation of FoxO1 in vascular endothelial cells via phosphatidylinositol 3-kinase- and protein kinase A-dependent signaling pathways to regulate ET-1 synthesis and secretion. J Biol Chem. 2008;283:29228–29238. doi: 10.1074/jbc.M802906200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Collins QF, Liu HY, Pi J, Liu Z, Quon MJ, Cao W. Epigallocatechin-3-gallate (EGCG), a green tea polyphenol, suppresses hepatic gluconeogenesis through 5′-AMP-activated protein kinase. J Biol Chem. 2007;282:30143–30149. doi: 10.1074/jbc.M702390200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim JA, Montagnani M, Koh KK, Quon MJ. Reciprocal relationships between insulin resistance and endothelial dysfunction: molecular and pathophysiological mechanisms. Circulation. 2006;113:1888–1904. doi: 10.1161/CIRCULATIONAHA.105.563213. [DOI] [PubMed] [Google Scholar]
- 12.Potenza MA, Marasciulo FL, Tarquinio M, Tiravanti E, Colantuono G, Federici A, Kim JA, Quon MJ, Montagnani M. EGCG, a green tea polyphenol, improves endothelial function and insulin sensitivity, reduces blood pressure, and protects against myocardial I/R injury in SHR. Am J Physiol Endocrinol Metab. 2007;292:E1378–1387. doi: 10.1152/ajpendo.00698.2006. [DOI] [PubMed] [Google Scholar]
- 13.Lorenz M, Wessler S, Follmann E, Michaelis W, Dusterhoft T, Baumann G, Stangl K, Stangl V. A constituent of green tea, epigallocatechin-3-gallate, activates endothelial nitric oxide synthase by a phosphatidylinositol-3-OH-kinase-, cAMP-dependent protein kinase-, and Akt-dependent pathway and leads to endothelial-dependent vasorelaxation. J Biol Chem. 2004;279:6190–6195. doi: 10.1074/jbc.M309114200. [DOI] [PubMed] [Google Scholar]
- 14.Kim JA, Koh KK, Quon MJ. The union of vascular and metabolic actions of insulin in sickness and in health. Arterioscler Thromb Vasc Biol. 2005;25:889–891. doi: 10.1161/01.ATV.0000164044.42910.6b. [DOI] [PubMed] [Google Scholar]
- 15.Maines MD. The heme oxygenase system: update 2005. Antioxid Redox Signal. 2005;7:1761–1766. doi: 10.1089/ars.2005.7.1761. [DOI] [PubMed] [Google Scholar]
- 16.Maines MD. The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol. 1997;37:517–554. doi: 10.1146/annurev.pharmtox.37.1.517. [DOI] [PubMed] [Google Scholar]
- 17.Morse D, Choi AM. Heme oxygenase-1: from bench to bedside. Am J Respir Crit Care Med. 2005;172:660–670. doi: 10.1164/rccm.200404-465SO. [DOI] [PubMed] [Google Scholar]
- 18.Maines MD. Heme oxygenase: function, multiplicity, regulatory mechanisms, and clinical applications. Faseb J. 1988;2:2557–2568. [PubMed] [Google Scholar]
- 19.Maines MD, Trakshel GM, Kutty RK. Characterization of two constitutive forms of rat liver microsomal heme oxygenase. Only one molecular species of the enzyme is inducible. J Biol Chem. 1986;261:411–419. [PubMed] [Google Scholar]
- 20.Geraldes P, Yagi K, Ohshiro Y, He Z, Maeno Y, Yamamoto-Hiraoka J, Rask-Madsen C, Chung SW, Perrella MA, King GL. Selective regulation of heme oxygenase-1 expression and function by insulin through IRS1/phosphoinositide 3-kinase/Akt-2 pathway. J Biol Chem. 2008;283:34327–34336. doi: 10.1074/jbc.M807036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li M, Kim DH, Tsenovoy PL, Peterson SJ, Rezzani R, Rodella LF, Aronow WS, Ikehara S, Abraham NG. Treatment of obese diabetic mice with a heme oxygenase inducer reduces visceral and subcutaneous adiposity, increases adiponectin levels, and improves insulin sensitivity and glucose tolerance. Diabetes. 2008;57:1526–1535. doi: 10.2337/db07-1764. [DOI] [PubMed] [Google Scholar]
- 22.Nicolai A, Li M, Kim DH, Peterson SJ, Vanella L, Positano V, Gastaldelli A, Rezzani R, Rodella LF, Drummond G, Kusmic C, L’Abbate A, Kappas A, Abraham NG. Heme oxygenase-1 induction remodels adipose tissue and improves insulin sensitivity in obesity-induced diabetic rats. Hypertension. 2009;53:508–515. doi: 10.1161/HYPERTENSIONAHA.108.124701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kruger AL, Peterson SJ, Schwartzman ML, Fusco H, McClung JA, Weiss M, Shenouda S, Goodman AI, Goligorsky MS, Kappas A, Abraham NG. Up-regulation of heme oxygenase provides vascular protection in an animal model of diabetes through its antioxidant and antiapoptotic effects. J Pharmacol Exp Ther. 2006;319:1144–1152. doi: 10.1124/jpet.106.107482. [DOI] [PubMed] [Google Scholar]
- 24.Chen H, Montagnani M, Funahashi T, Shimomura I, Quon MJ. Adiponectin stimulates production of nitric oxide in vascular endothelial cells. J Biol Chem. 2003;278:45021–45026. doi: 10.1074/jbc.M307878200. [DOI] [PubMed] [Google Scholar]
- 25.Udono-Fujimori R, Takahashi K, Takeda K, Furuyama K, Kaneko K, Takahashi S, Tamai M, Shibahara S. Expression of heme oxygenase-1 is repressed by interferon-gamma and induced by hypoxia in human retinal pigment epithelial cells. Eur J Biochem. 2004;271:3076–3084. doi: 10.1111/j.1432-1033.2004.04241.x. [DOI] [PubMed] [Google Scholar]
- 26.Lee JM, Moehlenkamp JD, Hanson JM, Johnson JA. Nrf2-dependent activation of the antioxidant responsive element by tert-butylhydroquinone is independent of oxidative stress in IMR-32 human neuroblastoma cells. Biochem Biophys Res Commun. 2001;280:286–292. doi: 10.1006/bbrc.2000.4106. [DOI] [PubMed] [Google Scholar]
- 27.Moehlenkamp JD, Johnson JA. Activation of antioxidant/electrophile-responsive elements in IMR-32 human neuroblastoma cells. Arch Biochem Biophys. 1999;363:98–106. doi: 10.1006/abbi.1998.1046. [DOI] [PubMed] [Google Scholar]
- 28.Yang CS, Chen L, Lee MJ, Balentine D, Kuo MC, Schantz SP. Blood and urine levels of tea catechins after ingestion of different amounts of green tea by human volunteers. Cancer Epidemiol Biomarkers Prev. 1998;7:351–354. [PubMed] [Google Scholar]
- 29.Kweon MH, Adhami VM, Lee JS, Mukhtar H. Constitutive overexpression of Nrf2-dependent heme oxygenase-1 in A549 cells contributes to resistance to apoptosis induced by epigallocatechin 3-gallate. J Biol Chem. 2006;281:33761–33772. doi: 10.1074/jbc.M604748200. [DOI] [PubMed] [Google Scholar]
- 30.Na HK, Kim EH, Jung JH, Lee HH, Hyun JW, Surh YJ. (−)-Epigallocatechin gallate induces Nrf2-mediated antioxidant enzyme expression via activation of PI3K and ERK in human mammary epithelial cells. Arch Biochem Biophys. 2008;476:171–177. doi: 10.1016/j.abb.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 31.Picchi A, Gao X, Belmadani S, Potter BJ, Focardi M, Chilian WM, Zhang C. Tumor necrosis factor-alpha induces endothelial dysfunction in the prediabetic metabolic syndrome. Circ Res. 2006;99:69–77. doi: 10.1161/01.RES.0000229685.37402.80. [DOI] [PubMed] [Google Scholar]
- 32.Ludwig A, Lorenz M, Grimbo N, Steinle F, Meiners S, Bartsch C, Stangl K, Baumann G, Stangl V. The tea flavonoid epigallocatechin-3-gallate reduces cytokine-induced VCAM-1 expression and monocyte adhesion to endothelial cells. Biochem Biophys Res Commun. 2004;316:659–665. doi: 10.1016/j.bbrc.2004.02.099. [DOI] [PubMed] [Google Scholar]
- 33.Andreadi CK, Howells LM, Atherfold PA, Manson MM. Involvement of Nrf2, p38, B-Raf, and nuclear factor-kappaB, but not phosphatidylinositol 3-kinase, in induction of hemeoxygenase-1 by dietary polyphenols. Mol Pharmacol. 2006;69:1033–1040. doi: 10.1124/mol.105.018374. [DOI] [PubMed] [Google Scholar]
- 34.Na HK, Surh YJ. Modulation of Nrf2-mediated antioxidant and detoxifying enzyme induction by the green tea polyphenol EGCG. Food Chem Toxicol. 2008;46:1271–1278. doi: 10.1016/j.fct.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 35.Willis D, Moore AR, Frederick R, Willoughby DA. Heme oxygenase: a novel target for the modulation of the inflammatory response. Nat Med. 1996;2:87–90. doi: 10.1038/nm0196-87. [DOI] [PubMed] [Google Scholar]
- 36.Cosso L, Maineri EP, Traverso N, Rosatto N, Pronzato MA, Cottalasso D, Marinari UM, Odetti P. Induction of heme oxygenase 1 in liver of spontaneously diabetic rats. Free Radic Res. 2001;34:189–191. doi: 10.1080/10715760100300171. [DOI] [PubMed] [Google Scholar]
- 37.Selzner N, Rudiger H, Graf R, Clavien PA. Protective strategies against ischemic injury of the liver. Gastroenterology. 2003;125:917–936. doi: 10.1016/s0016-5085(03)01048-5. [DOI] [PubMed] [Google Scholar]
- 38.Lambert JD, Lee MJ, Diamond L, Ju J, Hong J, Bose M, Newmark HL, Yang CS. Dose-dependent levels of epigallocatechin-3-gallate in human colon cancer cells and mouse plasma and tissues. Drug Metab Dispos. 2006;34:8–11. doi: 10.1124/dmd.104.003434. [DOI] [PubMed] [Google Scholar]
- 39.Wolfram S. Effects of green tea and EGCG on cardiovascular and metabolic health. J Am Coll Nutr. 2007;26:373S–388S. doi: 10.1080/07315724.2007.10719626. [DOI] [PubMed] [Google Scholar]
- 40.Kao YH, Chang HH, Lee MJ, Chen CL. Tea, obesity, and diabetes. Mol Nutr Food Res. 2006;50:188–210. doi: 10.1002/mnfr.200500109. [DOI] [PubMed] [Google Scholar]
- 41.Alam J, Stewart D, Touchard C, Boinapally S, Choi AM, Cook JL. Nrf2, a Cap’n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J Biol Chem. 1999;274:26071–26078. doi: 10.1074/jbc.274.37.26071. [DOI] [PubMed] [Google Scholar]
- 42.Alam J, Wicks C, Stewart D, Gong P, Touchard C, Otterbein S, Choi AM, Burow ME, Tou J. Mechanism of heme oxygenase-1 gene activation by cadmium in MCF-7 mammary epithelial cells. Role of p38 kinase and Nrf2 transcription factor. J Biol Chem. 2000;275:27694–27702. doi: 10.1074/jbc.M004729200. [DOI] [PubMed] [Google Scholar]
- 43.Martin D, Rojo AI, Salinas M, Diaz R, Gallardo G, Alam J, De Galarreta CM, Cuadrado A. Regulation of heme oxygenase-1 expression through the phosphatidylinositol 3-kinase/Akt pathway and the Nrf2 transcription factor in response to the antioxidant phytochemical carnosol. J Biol Chem. 2004;279:8919–8929. doi: 10.1074/jbc.M309660200. [DOI] [PubMed] [Google Scholar]
- 44.Shen G, Xu C, Hu R, Jain MR, Nair S, Lin W, Yang CS, Chan JY, Kong AN. Comparison of (−)-epigallocatechin-3-gallate elicited liver and small intestine gene expression profiles between C57BL/6J mice and C57BL/6J/Nrf2 (−/−) mice. Pharm Res. 2005;22:1805–1820. doi: 10.1007/s11095-005-7546-8. [DOI] [PubMed] [Google Scholar]
- 45.Alam J, Cook JL. How many transcription factors does it take to turn on the heme oxygenase-1 gene? Am J Respir Cell Mol Biol. 2007;36:166–174. doi: 10.1165/rcmb.2006-0340TR. [DOI] [PubMed] [Google Scholar]
- 46.Ryter SW, Alam J, Choi AM. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev. 2006;86:583–650. doi: 10.1152/physrev.00011.2005. [DOI] [PubMed] [Google Scholar]
- 47.Ishii T, Itoh K, Takahashi S, Sato H, Yanagawa T, Katoh Y, Bannai S, Yamamoto M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J Biol Chem. 2000;275:16023–16029. doi: 10.1074/jbc.275.21.16023. [DOI] [PubMed] [Google Scholar]
- 48.Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang HC, Nguyen T, Pickett CB. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J Biol Chem. 2002;277:42769–42774. doi: 10.1074/jbc.M206911200. [DOI] [PubMed] [Google Scholar]
- 50.McMahon M, Itoh K, Yamamoto M, Hayes JD. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J Biol Chem. 2003;278:21592–21600. doi: 10.1074/jbc.M300931200. [DOI] [PubMed] [Google Scholar]
- 51.Nguyen T, Sherratt PJ, Huang HC, Yang CS, Pickett CB. Increased protein stability as a mechanism that enhances Nrf2-mediated transcriptional activation of the antioxidant response element. Degradation of Nrf2 by the 26 S proteasome. J Biol Chem. 2003;278:4536–4541. doi: 10.1074/jbc.M207293200. [DOI] [PubMed] [Google Scholar]
- 52.Pugazhenthi S, Akhov L, Selvaraj G, Wang M, Alam J. Regulation of heme oxygenase-1 expression by demethoxy curcuminoids through Nrf2 by a PI3-kinase/Akt-mediated pathway in mouse beta-cells. Am J Physiol Endocrinol Metab. 2007;293:E645–655. doi: 10.1152/ajpendo.00111.2007. [DOI] [PubMed] [Google Scholar]
- 53.Li MH, Cha YN, Surh YJ. Peroxynitrite induces HO-1 expression via PI3K/Akt-dependent activation of NF-E2-related factor 2 in PC12 cells. Free Radic Biol Med. 2006;41:1079–1091. doi: 10.1016/j.freeradbiomed.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 54.Hamdulay SS, Wang B, Birdsey GM, Ali F, Dumont O, Evans PC, Haskard DO, Wheeler-Jones CP, Mason JC. Celecoxib activates PI-3K/Akt and mitochondrial redox signaling to enhance heme oxygenase-1-mediated anti-inflammatory activity in vascular endothelium. Free Radic Biol Med. 48 :1013–1023. doi: 10.1016/j.freeradbiomed.2010.01.017. [DOI] [PubMed] [Google Scholar]
- 55.Ning W, Song R, Li C, Park E, Mohsenin A, Choi AM, Choi ME. TGF-beta1 stimulates HO-1 via the p38 mitogen-activated protein kinase in A549 pulmonary epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2002;283:L1094–1102. doi: 10.1152/ajplung.00151.2002. [DOI] [PubMed] [Google Scholar]
- 56.Chen K, Maines MD. Nitric oxide induces heme oxygenase-1 via mitogen-activated protein kinases ERK and p38. Cell Mol Biol (Noisy-le-grand) 2000;46:609–617. [PubMed] [Google Scholar]
- 57.Liu ZM, Chen GG, Ng EK, Leung WK, Sung JJ, Chung SC. Upregulation of heme oxygenase-1 and p21 confers resistance to apoptosis in human gastric cancer cells. Oncogene. 2004;23:503–513. doi: 10.1038/sj.onc.1207173. [DOI] [PubMed] [Google Scholar]
- 58.Wu CC, Hsu MC, Hsieh CW, Lin JB, Lai PH, Wung BS. Upregulation of heme oxygenase-1 by Epigallocatechin-3-gallate via the phosphatidylinositol 3-kinase/Akt and ERK pathways. Life Sci. 2006;78:2889–2897. doi: 10.1016/j.lfs.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 59.Zhang H, Park Y, Wu J, Chen X, Lee S, Yang J, Dellsperger KC, Zhang C. Role of TNF-alpha in vascular dysfunction. Clin Sci (Lond) 2009;116:219–230. doi: 10.1042/CS20080196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ross R. Cell biology of atherosclerosis. Annu Rev Physiol. 1995;57:791–804. doi: 10.1146/annurev.ph.57.030195.004043. [DOI] [PubMed] [Google Scholar]
- 61.Iiyama K, Hajra L, Iiyama M, Li H, DiChiara M, Medoff BD, Cybulsky MI. Patterns of vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 expression in rabbit and mouse atherosclerotic lesions and at sites predisposed to lesion formation. Circ Res. 1999;85:199–207. doi: 10.1161/01.res.85.2.199. [DOI] [PubMed] [Google Scholar]
- 62.Ross R. Atherosclerosis--an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 63.Galkina E, Ley K. Vascular adhesion molecules in atherosclerosis. Arterioscler Thromb Vasc Biol. 2007;27:2292–2301. doi: 10.1161/ATVBAHA.107.149179. [DOI] [PubMed] [Google Scholar]
- 64.Collins T, Read MA, Neish AS, Whitley MZ, Thanos D, Maniatis T. Transcriptional regulation of endothelial cell adhesion molecules: NF-kappa B and cytokine-inducible enhancers. Faseb J. 1995;9:899–909. [PubMed] [Google Scholar]
- 65.Chae YJ, Kim CH, Ha TS, Hescheler J, Ahn HY, Sachinidis A. Epigallocatechin-3-O-gallate inhibits the angiotensin II-induced adhesion molecule expression in human umbilical vein endothelial cell via inhibition of MAPK pathways. Cell Physiol Biochem. 2007;20:859–866. doi: 10.1159/000110446. [DOI] [PubMed] [Google Scholar]
- 66.Chen XL, Dodd G, Thomas S, Zhang X, Wasserman MA, Rovin BH, Kunsch C. Activation of Nrf2/ARE pathway protects endothelial cells from oxidant injury and inhibits inflammatory gene expression. Am J Physiol Heart Circ Physiol. 2006;290:H1862–1870. doi: 10.1152/ajpheart.00651.2005. [DOI] [PubMed] [Google Scholar]
- 67.Mazzone GL, Rigato I, Ostrow JD, Bossi F, Bortoluzzi A, Sukowati CH, Tedesco F, Tiribelli C. Bilirubin inhibits the TNFalpha-related induction of three endothelial adhesion molecules. Biochem Biophys Res Commun. 2009;386:338–344. doi: 10.1016/j.bbrc.2009.06.029. [DOI] [PubMed] [Google Scholar]
- 68.Song H, Bergstrasser C, Rafat N, Hoger S, Schmidt M, Endres N, Goebeler M, Hillebrands JL, Brigelius-Flohe R, Banning A, Beck G, Loesel R, Yard BA. The carbon monoxide releasing molecule (CORM-3) inhibits expression of vascular cell adhesion molecule-1 and E-selectin independently of haem oxygenase-1 expression. Br J Pharmacol. 2009;157:769–780. doi: 10.1111/j.1476-5381.2009.00215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Formoso G, Chen H, Kim JA, Montagnani M, Consoli A, Quon MJ. Dehydroepiandrosterone mimics acute actions of insulin to stimulate production of both nitric oxide and endothelin 1 via distinct phosphatidylinositol 3-kinase- and mitogen-activated protein kinase-dependent pathways in vascular endothelium. Mol Endocrinol. 2006;20:1153–1163. doi: 10.1210/me.2005-0266. [DOI] [PubMed] [Google Scholar]