

Abstract
Objective
To determine whether N-carbamylglutamate reduces plasma levels of ammonia and glutamine and increases ureagenesis rate in patients with propionic acidemia
Patients and Methods
Identical four-hour studies were performed before and immediately after a 3-day trial of oral N-carbamylglutamate in 7 patients with propionic acidemia. An oral bolus of [13C]-sodium acetate was administered at the start of each study, and sequential blood samples were obtained to measure [13C]-urea, ammonia, urea and amino acids.
Results
With longitudinal mixed effects linear regression, peak [13C]urea increased following treatment with N-carbamylglutamate (from 2.2 μM to 3.8 μM; p < 0.0005). There were concomitant decreases in mean plasma ammonia (59 to 43 μM, p <0.0005)) and glutamine (552 to 331 μM, p <0.0005).
Conclusion
N-carbamylglutamate augments ureagenesis and decreases plasma ammonia and glutamine in patients with propionic acidemia. The drug may serve as an important therapeutic adjunct in the treatment of acute hyperammonemia in this disorder.
Keywords: N-acetyl-L-glutamate, acetylglutamate, carbamylglutamate, hyperammonemia, organic acidemia, urea cycle, stable isotopes, clinical trial
Introduction
The conversion of ammonia to urea by the liver requires the coordinated function of 6 enzymes and 2 mitochondrial membrane transporters [1]. An interruption in any of these steps can lead to an accumulation of toxic ammonia, which can result in mental retardation, coma, and even death[2-4]. The first and rate-limiting enzyme of ureagenesis is carbamyl-phosphate synthetase I (CPSI) (EC 6.3.4.16), which catalyzes the conversion of ammonia, bicarbonate and ATP to carbamyl-phosphate[5]. However, CPSI is active only in the presence of its obligate allosteric activator, N-acetylglutamate (NAG) (EC 2.3.1.1)[5]. NAG, in turn, is catalytically produced from acetyl-CoA and glutamate by NAG synthase (NAGS). A deficiency of NAG, seen in inherited NAGS defects [6], reduces flux through CPSI, thereby resulting in hyperammonemia.
Previous studies indicated that N-carbamylglutamate (NCG), a stable structural analog of NAG, activates CPSI[5]. Indeed, the administration of NCG to patients with NAGS deficiency normalizes blood ammonia concentrations[7-10] and restores ureagenesis[8, 11]. Moreover, in healthy individuals, the administration of NCG can promote carbamyl-phosphate synthesis and increase ureagenesis[12]. Thus, NCG administration may be beneficial in conditions characterized by a compromise of the NAGS or CPSI reaction, such as in some forms of CPSI deficiency, propionic acidemia (PA)[13], methylmalonic acidemia (MMA)[14], valproate administration[15], and hyperinsulinism-hyperammonemia syndrome[16]. In PA, a decrease in NAG synthesis results from competitive inhibition of NAGS by propionyl-CoA[17-19] or a reduction of hepatic acetyl-CoA or free coenzyme-A[19].
We previously described the effects of a 3-day course of NCG treatment in a patient with PA[8]. We measured ureagenesis by using mass spectrometry to monitor the conversion of administered [1-13C]acetate to [13C]urea. This method was demonstrated to be robust and free of significant risk. The affected patient responded to NCG treatment with improved ureagenesis, as evidenced by increased isotopic incorporation of 13C into urea[8].
Here we report our findings of the efficacy of NCG administration in a cohort of 7 patients. We found that this drug is likely to be an effective therapeutic adjunct for the management of hyperammonemia in PA.
Patients and Methods
We studied 7 patients aged 15 months-13 years, including a previously published case. The study design is shown in Fig. 1. All subjects presented with neonatal hyperammonemia and were documented to have a diagnosis of propionic acidemia by urine organic acid analysis. Molecular testing results were available for 3 patients. The study was approved by the Institutional Review Boards at the Children’s National Medical Center and the Children’s Hospital of Philadelphia. Informed consent was obtained for each individual before enrollment. This trial has been registered at Clinicaltrials.gov.
Figure 1.
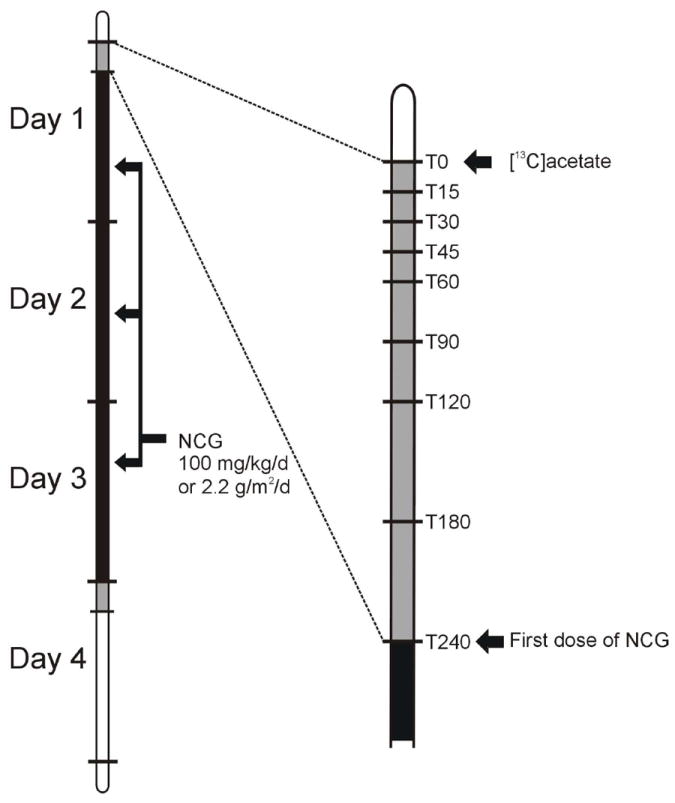
Overall trial timeline. Identical studies were performed before (‘day 1’) and immediately following (‘day 4’) a 3-day course of N-carbamylglutamate at a dose of 100 mg/kg/d or 2.2g/m2/d if ≥25 kg. Plasma samples for ammonia, urea, amino acids and [13C]urea were obtained at T0, 15, 30, 45, 60, 90, 120, 180, and 240 min.
An indwelling venous catheter was used for blood drawing in subjects who did not have a central venous line. For subjects without ready venous access, a peripherally inserted central catheter was introduced under conscious sedation (midazolam, fentanyl) by an experienced interventional radiologist. The PICC remained in place for the duration of the study, and was removed upon obtaining the last blood sample.
Following an overnight fast, a baseline sample (5 mL) of heparinized blood was obtained. At time 0, each subject received (either by ingestion or nasogastric tube) 0.33 mmol/kg, (27.5 mg/kg) of [1-13C] sodium acetate (98 atom % excess) dissolved in 60 mL of water. Blood samples (2-4 mL) were subsequently obtained at 15, 30, 45, 60, 75, 90, 120, 180, and 240 min. For subjects under 5 years, the times were restricted to 15, 30 50 and 120 minutes in order to limit total blood taken to less than 3 ml/kg body weight. Each specimen was placed in a pre-cooled, heparinized tube and immediately centrifuged at 4°C to separate the plasma, which was immediately frozen on dry ice and kept frozen at -70°C until analysis performed within 2 days after colelction. Each sample was analyzed for plasma ammonia and urea (RXL Dade Behring, Siemens Healthcare Diagnostics, Deerfield, IL), and quantitative amino acids (Biochrom, Cambridge, UK) and [13C]urea as previously described [8, 12].
All subjects underwent an identical study procedure before, and immediately after treatment for 3 days with NCG (Carbaglu®, Orphan Europe, Paris, 100 mg/kg/d, or 2.2g/m2/d if ≥ 25 kg body weight in four divided doses).
Subjects
Subject 1 was an 11 year-old female born at term following an uncomplicated pregnancy, labor and delivery. She presented at 2 days of life with hyperammonemia and acidosis. She was treated for presumptive organic acidemia until laboratory results confirmed the diagnosis of propionic acidemia. She developed seizures at two weeks of life and required anti-epileptic drugs until one year of age. She required multiple admissions for acute illness leading to metabolic decompensation. She had a mild developmental delay, and was fed by a gastrostomy tube. Her historic ammonia concentrations while stable ranged between 81 and 127 μM.
Subject 2 was an 8 year-old female born at term following uncomplicated pregnancy labor and delivery. She presented on the 3rd day of life with hypothermia, metabolic acidosis and hyperammonemia (808 μM). She was diagnosed with propionic acidemia by urinary organic acid and plasma acylcarnitine analyses. She required hemodialysis and ventilatory and vasopressor support. She also had a dilated cardiomyopathy in the newborn period, requiring medications, but this subsequently resolved. Her ammonia levels ranged chronically between 55 to 122 μM. She required over 30 admissions or emergency department visits for acute exacerbations of her condition. She had developmental delay, including compromised speech, and received nutrition via a gastrostomy tube.
Subject 3 was a 5 year-old male born at term following uncomplicated pregnancy, labor and delivery. At 2 weeks of age, he presented with vomiting and lethargy, and was found to have a mild metabolic acidosis and a plasma ammonia level of 382 μM. A diagnosis of propionic acidemia was subsequently made by plasma acylcarnitine and urine organic acid analyses. After the initial episode, he was admitted 14 times for acute illness, including 5 admissions for pancreatitis. He had a mild developmental delay, more accentuated in language. He required continuous feeding via a gastrostomy tube. He was found to be heterozygous for a c.183+3 G>C mutation in the beta-subunit gene of propionyl-CoA carboxylase (PCCB). He was also noted to be heterozygous for two undocumented sequence variants, both potentially pathogenic: c.734G>A and c.967-14 A>G. His chronic plasma ammonia concentration ranged from 36 to 110 μM.
Subject 4 was a 15 month female born at 34 weeks gestation after uncomplicated labor and delivery. Apgar scores were 8 at 1 minute and 9 at 5 minutes. At 10 days of age, following findings of lethargy and hypotonia, she was noted to have acidosis, ketosis and a plasma ammonia level of 459 μM. Peak ammonia in the neonatal period was 1349 μM.
The diagnosis of propionic acidemia was confirmed via urine organic acid analysis. She required hemodialysis during the first episode. She has since had three episodes of hyperammonemia and acidosis requiring admission to the hospital. Her chronic ammonia concentrations ranged between 77 to 94 μM.
Subject 5 was a 13 year-old female born at term by a caesarean section. At 4 days of age, she presented with lethargy, acidosis, and a plasma ammonia level of 735 μM. The diagnosis of propionic acidemia was confirmed via urine organic acid analysis. She has had over 100 admissions to the hospital, primarily for lethargy and acidosis. One random plasma ammonia concentration was 47 μM. Molecular genetic tests are still pending, but her sister, subject 6, was found to be a homozygote for a mutation in the PCCB gene (see below).
Subject 6 was a 9 year-old sister of subject 5. She was born at 38 weeks gestation by a planned caesarean section. She was prospectively diagnosed in the newborn period due to her affected sister. She has been admitted to the hospital nearly 200 times, primarily for lethargy, vomiting and hyperammonemia. She was found to be homozygous for a known deleterious mutation in the PCCB gene, c.1218_1231delins12 (deletion of 14 bp, and insertion of 12 bp)[20]. Her chronic ammonia levels ranged between 94 and 171 μM.
Subject 7 was a 6 year-old male, born at term, who presented at 4 days of age with severe hyperammonemia (1200 μM) and metabolic acidosis requiring hemodialysis. The diagnosis of propionic acidemia was established by urine organic acid analysis. He was admitted to the hospital on multiple occasions during the first 3 years of life, typically for acute exacerbations of his disorder, but once for acute pancreatitis. During these episodes, his plasma ammonia rose to 3 to 6 times the upper limit of normal. His development was significantly delayed: he was ambulatory but with no speech. He was fed exclusively by a gastrostomy tube. Mutation analysis revealed that he was homozygous for the frame-shift G216fs mutation in the alpha-subunit of the propionyl-CoA carboxylase A gene. A single random plasma ammonia concentration was 74 μM. Results from his study have been previously reported by our group[8].
Statistical analysis
To evaluate the biochemical effects of NCG treatment, we used longitudinal mixed effects linear regression in Stata 10 (xtmixed). We used it first to compare grand mean levels of metabolites in the same participants when treated and not treated with NCG. The method allows us to perform a paired (pre vs post NCG) analysis and also to adjust variance estimates for the correlation of multiple measurements on the same subject within treatment condition. Thereafter, we used the same model to evaluate the detailed change over time within treatment condition. The model treated each participant as a random effect allowing him/her to have a different starting point prior to each treatment condition. In addition, the model allowed for nonlinear effects over time and different relationships (interactions) in time by treatment condition when estimating metabolic effects. Before we implemented the above parametric analyses, we evaluated the normality and variance homogeneity assumptions and when necessary implemented data transformations to ensure the data met these assumptions. Following analyses involving data transformations, the results were back transformed to return the estimates to their original scale and units. We set 95% confidence intervals (CI) around each estimate, as well as around group differences, and considered those differences to be statistically significant if the 95% CI failed to include zero.
Results
13CO2 and [13C]Urea
Fig. 2A illustrates model estimates of 13CO2 +/- 95% CI in venous blood for all 7 subjects following an enteral bolus of [1-13C]acetate. Data are expressed as atom % excess, relative to the baseline (t0) value. The results show that that the 13CO2 curves are similar, with 95% CI virtually superimposed, and thus the plasma levels of the labeling precursor (13CO2) do not differ in the pre- and post-NCG experiments.
Figure 2.
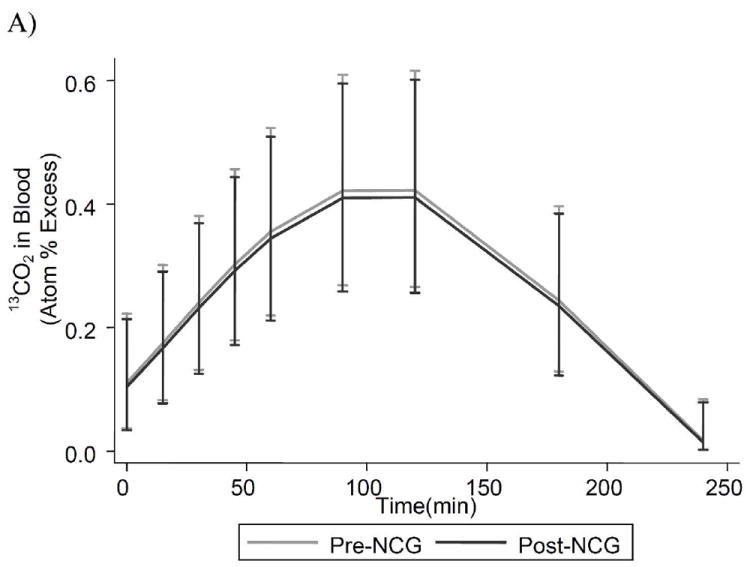
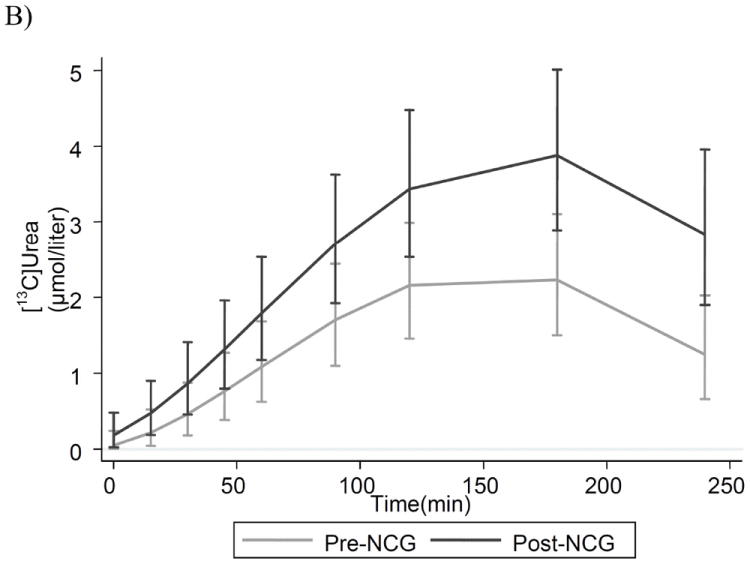
Isotopic enrichment in plasma 13CO2 (A), and [13C]urea (B) in 7 subjects with propionic acidemia who were administered 27.5 mg/kg of [1-13C]sodium-acetate. Results are model estimates +/- 95% CI. Subject 4 contributes to mean data only at time-points 0, 15, 30, 60 and 120 minutes.
Fig. 2B presents the detailed model estimates, +/- 95% CI, of the change in plasma [13C]urea over the course of the study for same participants when on and not on NCG. Pre- and post- treatment, plasma [13C]urea concentration increased rapidly from 0 to 90 minutes, demonstrating rapid incorporation of 13CO2 into urea, which peaks at approximately 180 min. When receiving NCG, patients produce significantly greater levels of [13C]urea at every time point after 15 min. and this difference widened with time. The peak [13C]urea following therapy with NCG was 3.8 μM (95% CI, 2.9-5.0), over 70% greater than the pre-NCG value of 2.2 μM (95% CI, 1.5-3.1). In comparison, the peak concentration of this parameter in 38 adult control studies was 4.01 ± 0.59 μM[8].
Ammonia, amino acids and blood urea nitrogen
Fig. 3a illustrates plasma ammonia levels for all subjects before and after NCG treatment. Each point represents the mean ammonia measurement in 9 blood samples in each subject (only 5 in subject 4) over the duration of the study. All subjects, with the exception of subject 2, show a decrease in ammonia after treatment. Subjects with a higher baseline ammonia level show the greatest decline in response to NCG. The grand mean is shown in bold, +/- 95% CI, and illustrates a decrease in ammonia from 59 to 43 μmol/l (p = 0.018).
Figure 3.
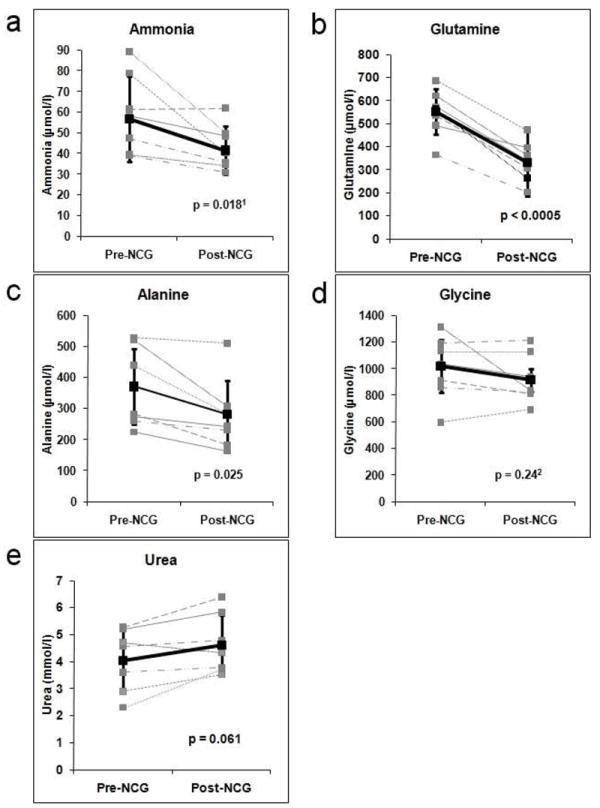
Plasma levels of ammonia (A), glutamine (B), alanine (C), glycine (D) and urea (E) in 7 patients with propionic acidemia before and following a 3-day treatment with N-carbamylglutamate. Mean values +/- 95% CI are shown in bold. 1Based on log transformation; 2Based on square root transformation
An increase in nitrogen disposal following NCG administration was also reflected in changes in amino acids. Mean plasma glutamine, alanine and glycine concentrations from each subject are shown in Fig. 3b-d, with the collective average, and mean of means, in bold. In each patient, there is a large drop in plasma glutamine, with the mean aggregate glutamine of all patients decreasing from 552 to 331 μmol/l (p<0.0005). All subjects, with the exception of subject 3, show a decrease in plasma alanine (Fig. 3c). The collective mean decreased from 370 to 282 μmol/l (p=0.025), with the greatest response again seen among patients with the highest baseline values. Glycine (Fig. 3d) was relatively unchanged (1018 vs. 914 μmol/l, p=0.24).
Blood urea nitrogen (BUN) is shown in Fig. 3e. Although all but one subject showed an increase in mean BUN, this change does not reach significance (4.04 vs. 4.61 mmol/l, p=0.061).
Discussion
We present herein the first clinical trial evidence for the salutary effect of NCG on plasma ammonia and ureagenesis in patients with propionic acidemia. Prior studies of NCG efficacy[8, 13, 21-23], with the exception of our report [8], were non-controlled experiments during acute illness. Under such circumstances, the concurrent application of other standard treatment modalities may confound the therapeutic contribution of NCG. In the current study, seven patients were investigated prospectively while metabolically stable. Thus, each subject could serve as his/her own control, and both experimental and control studies were performed in nearly identical circumstances to best discriminate the effect of NCG.
NCG is an allosteric activator of CPSI[5] that has a lower affinity than NAG (the natural CPSI co-factor) for the enzyme in vitro[5]. However, NCG is the preferred pharmacological agent, since its oral bioavalibility is superior to that of NAG, which is hydrolyzed before reaching the liver. Thus, our interest in NCG stems from its potential therapeutic benefit in conditions in which NAG is deficient. Even when NAG is not deficient, such as in unaffected controls, NCG supplementation can enhance the rate of ureagenesis[12]. Therefore, we hypothesized that NCG might be effective in the treatment of hyperammonemia in PA in which a decrease in NAG concentration develops either from the competitive inhibition of NAG synthase by propionyl-CoA or diminution of the hepatic mitochondrial pool of acetyl-CoA or free coenzyme-A[17-19].
In this trial, we demonstrated that oral NCG treatment accelerates ureagenesis in patients with PA, presumably by increasing flux through the CPS I reaction, the rate-limiting step of the urea cycle. In our cohort, augmentation of ureagenesis was reflected in an increased concentration of plasma [13C]urea, although this parameter was 6% lower than that of adult controls. Pediatric control data are unavailable, since human protection guidelines preclude performing the isotopic study in healthy youngsters.
Some subjects (5/7), manifested enhanced post-treatment nitrogen disposal with concurrent reduction in plasma ammonia, glutamine and alanine, even though the baseline value was not elevated in each instance. A plausible inference is that there occurred an overall contraction of the nitrogen pool. This finding might be clinically relevant during acute illness in PA, when tissue protein catabolism and impaired ureagenesis result in hyperammonemia. In such circumstances, the salutary effect of NCG might be even more appreciable.
Our cohort represents a relatively severe form of PA, as indicated by a marked increase of the baseline glycine level (1018 μM) and a tendency toward early clinical onset of symptoms. Overall, we found no statistically significant decline of plasma glycine following treatment with NCG (1018 vs. 914 μM), probably because NCG did not alter the hepatic concentration of propionyl-CoA, which inhibits the glycine-cleavage system[24].
We did not observe a significant post-treatment increase of plasma urea (4.04 vs. 4.61 mmol/l), perhaps because of greater renal clearance in response to enhanced ureagenesis.
We employed a stable isotope method to assess ureagenesis in vivo. Stable isotopes are safe, and the analysis of isotopic enrichment in body fluids by ion-ratio mass-spectrometry is highly sensitive and reproducible [25, 26]. Our method reliably documented the in vivo conversion of an single oral bolus of [1-13C]acetate to 13C-bicarbonate (measured as blood 13CO2), and the subsequent incorporation of this label into urea. In all 14 studies (7 pre- and 7 post-NCG), we observed an initial increase of the [13C]urea concentration, which approximated a plateau. The augmentation in ureagenesis following NCG therapy is reflected both in the initial slope and the peak urea enrichment. This study illustrates the robustness and reproducibility of this isotopic approach, and its utility in assessing changes in ureagenesis in response to NCG.
Conclusion
We describe the first clinical trial evidence of the beneficial effects of N-carbamylglutamate on ureagenesis in patients with propionic acidemia. We also revalidate our robust stable isotope approach to evaluating the effect of N-cabamylglutamate on ureagenesis in metabolic disorders associated with hyperammonemia. Future directions include a long-term trial of NCG in patients with propionic acidemia, as well as investigating the utility of NCG therapy in other conditions which cause hyperammonemia.
Acknowledgments
This work was supported by public health service grants R01HD058567 R01DK47870 R01DK64913, U54RR019453, P01HD26979, NS054900, RR024134 from the National Institutes of Health
We thank the General Clinical Research Center staff at Children’s National Medical Center and Dr. Steven Lossef for their help in performing the study. We also thank Drs. Kyrieckos Aleck, Karla Komersová, J. Cilliano, Dorothy Grange, Jose Abdenur, Stephen Cederbaum, Rani Singh, Barbara Burton, and Rebecca Mardach for their patient referrals to this study
Abbreviations
- CI
confidence intervals
- CPSI
carbamyl-phosphate synthetase I
- MMA
methylmalonic acidemia
- NAG
N-acetylglutamate
- NAGS
NAG synthase
- NCG
N-carbamylglutamate
- PA
propionic acidemia
- PCCB
propionyl-CoA carboxylase B
References
- 1.Tuchman M, Lee B, Lichter-Konecki U, Summar ML, Yudkoff M, Cederbaum SD, et al. Cross-sectional multicenter study of patients with urea cycle disorders in the United States. Mol Genet Metab. 2008;94(4):397–402. doi: 10.1016/j.ymgme.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brusilow SW, Maestri NE. Urea cycle disorders: diagnosis, pathophysiology, and therapy. Adv Pediatr. 1996;43:127–70. [PubMed] [Google Scholar]
- 3.Batshaw ML, Brusilow S, Waber L, Blom W, Brubakk AM, Burton BK, et al. Treatment of inborn errors of urea synthesis: activation of alternative pathways of waste nitrogen synthesis and excretion. N Engl J Med. 1982;306(23):1387–92. doi: 10.1056/NEJM198206103062303. [DOI] [PubMed] [Google Scholar]
- 4.Batshaw ML, Brusilow SW. Treatment of hyperammonemic coma caused by inborn errors of urea synthesis. J Pediatr. 1980;97(6):893–900. doi: 10.1016/s0022-3476(80)80416-1. [DOI] [PubMed] [Google Scholar]
- 5.Hall LM, Metzenberg RL, Cohen PP. Isolation and characterization of a naturally occurring cofactor of carbamyl phosphate biosynthesis. J Biol Chem. 1958;230(2):1013–21. [PubMed] [Google Scholar]
- 6.Bachmann C, Krahenbuhl S, Colombo JP, Schubiger G, Jaggi KH, Tonz O. N-acetylglutamate synthetase deficiency: a disorder of ammonia detoxication. N Engl J Med. 1981;304(9):543. doi: 10.1056/NEJM198102263040918. [DOI] [PubMed] [Google Scholar]
- 7.Morris AA, Richmond SW, Oddie SJ, Pourfarzam M, Worthington V, Leonard JV. N-acetylglutamate synthetase deficiency: favourable experience with carbamylglutamate. J Inherit Metab Dis. 1998;21(8):867–8. doi: 10.1023/a:1005478904186. [DOI] [PubMed] [Google Scholar]
- 8.Tuchman M, Caldovic L, Daikhin Y, Horyn O, Nissim I, Korson M, et al. N-carbamylglutamate markedly enhances ureagenesis in N-acetylglutamate deficiency and propionic acidemia as measured by isotopic incorporation and blood biomarkers. Pediatr Res. 2008;64(2):213–7. doi: 10.1203/PDR.0b013e318179454b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guffon N, Vianey-Saban C, Bourgeois J, Rabier D, Colombo JP, Guibaud P. A new neonatal case of N-acetylglutamate synthase deficiency treated by carbamylglutamate. J Inherit Metab Dis. 1995;18(1):61–5. doi: 10.1007/BF00711374. [DOI] [PubMed] [Google Scholar]
- 10.Hinnie J, Colombo JP, Wermuth B, Dryburgh FJ. N-Acetylglutamate synthetase deficiency responding to carbamylglutamate. J Inherit Metab Dis. 1997;20(6):839–40. doi: 10.1023/a:1005344507536. [DOI] [PubMed] [Google Scholar]
- 11.Caldovic L, Morizono H, Daikhin Y, Nissim I, McCarter RJ, Yudkoff M, et al. Restoration of ureagenesis in N-acetylglutamate synthase deficiency by N-carbamylglutamate. J Pediatr. 2004;145(4):552–4. doi: 10.1016/j.jpeds.2004.06.047. [DOI] [PubMed] [Google Scholar]
- 12.Mew NA, Payan I, Daikhin Y, Nissim I, Tuchman M, Yudkoff M. Effects of a single dose of N-carbamylglutamate on the rate of ureagenesis. Mol Genet Metab. 2009 doi: 10.1016/j.ymgme.2009.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gebhardt B, Dittrich S, Parbel S, Vlaho S, Matsika O, Bohles H. N-carbamylglutamate protects patients with decompensated propionic aciduria from hyperammonaemia. J Inherit Metab Dis. 2005;28(2):241–4. doi: 10.1007/s10545-005-5260-7. [DOI] [PubMed] [Google Scholar]
- 14.Gebhardt B, Vlaho S, Fischer D, Sewell A, Bohles H. N-carbamylglutamate enhances ammonia detoxification in a patient with decompensated methylmalonic aciduria. Mol Genet Metab. 2003;79(4):303–4. doi: 10.1016/s1096-7192(03)00095-7. [DOI] [PubMed] [Google Scholar]
- 15.Williams CA, Tiefenbach S, McReynolds JW. Valproic acid-induced hyperammonemia in mentally retarded adults. Neurology. 1984;34(4):550–3. doi: 10.1212/wnl.34.4.550. [DOI] [PubMed] [Google Scholar]
- 16.Stanley CA. Hyperinsulinism/hyperammonemia syndrome: insights into the regulatory role of glutamate dehydrogenase in ammonia metabolism. Mol Genet Metab. 2004;81(Suppl 1):S45–51. doi: 10.1016/j.ymgme.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 17.Coude FX, Sweetman L, Nyhan WL. Inhibition by propionyl-coenzyme A of N-acetylglutamate synthetase in rat liver mitochondria. A possible explanation for hyperammonemia in propionic and methylmalonic acidemia. J Clin Invest. 1979;64(6):1544–51. doi: 10.1172/JCI109614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glasgow AM, Chase HP. Effect of propionic acid on fatty acid oxidation and ureagenesis. Pediatr Res. 1976;10(7):683–6. doi: 10.1203/00006450-197607000-00010. [DOI] [PubMed] [Google Scholar]
- 19.Stewart PM, Walser M. Failure of the normal ureagenic response to amino acids in organic acid-loaded rats. Proposed mechanism for the hyperammonemia of propionic and methylmalonic acidemia. J Clin Invest. 1980;66(3):484–92. doi: 10.1172/JCI109879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gravel RA, Akerman BR, Lamhonwah AM, Loyer M, Leon-del-Rio A, Italiano I. Mutations participating in interallelic complementation in propionic acidemia. Am J Hum Genet. 1994;55(1):51–8. [PMC free article] [PubMed] [Google Scholar]
- 21.Jones S, Reed CA, Vijay S, Walter JH, Morris AA. N-Carbamylglutamate for neonatal hyperammonaemia in propionic acidaemia. J Inherit Metab Dis. 2008 doi: 10.1007/s10545-008-0777-1. [DOI] [PubMed] [Google Scholar]
- 22.Schwahn BC, Pieterse L, Bisset WM, Galloway PG, Robinson PH. Biochemical efficacy of N-carbamylglutamate in neonatal severe hyperammonaemia due to propionic acidaemia. Eur J Pediatr. 2009 doi: 10.1007/s00431-009-1036-7. [DOI] [PubMed] [Google Scholar]
- 23.Filippi L, Gozzini E, Fiorini P, Malvagia S, la Marca G, Donati MA. N-Carbamylglutamate in Emergency Management of Hyperammonemia in Neonatal Acute Onset Propionic and Methylmalonic Aciduria. Neonatology. 2009;97(3):286–290. doi: 10.1159/000255168. [DOI] [PubMed] [Google Scholar]
- 24.Hayasaka K, Narisawa K, Satoh T, Tateda H, Metoki K, Tada K, et al. Glycine cleavage system in ketotic hyperglycinemia: a reduction of H-protein activity. Pediatr Res. 1982;16(1):5–7. doi: 10.1203/00006450-198201001-00002. [DOI] [PubMed] [Google Scholar]
- 25.de Meer K, Roef MJ, Kulik W, Jakobs C. In vivo research with stable isotopes in biochemistry, nutrition and clinical medicine: an overview. Isotopes Environ Health Stud. 1999;35(1-2):19–37. doi: 10.1080/10256019908234077. [DOI] [PubMed] [Google Scholar]
- 26.Kalhan SC, Oliven A, King KC, Lucero C. Role of glucose in the regulation of endogenous glucose production in the human newborn. Pediatr Res. 1986;20(1):49–52. doi: 10.1203/00006450-198601000-00013. [DOI] [PubMed] [Google Scholar]