

Abstract
AIM: To investigate whether high-fat-feeding is associated with increased intestinal permeability via alterations in bile acid metabolism.
METHODS: Male C57Bl/6J mice were fed on a high-fat (n = 26) or low-fat diet (n = 24) for 15 wk. Intestinal permeability was measured from duodenum, jejunum, ileum and colon in an Ussing chamber system using 4 kDa FITC-labeled dextran as an indicator. Fecal bile acids were analyzed with gas chromatography. Segments of jejunum and colon were analyzed for the expression of farnesoid X receptor (FXR) and tumor necrosis factor (TNF).
RESULTS: Intestinal permeability was significantly increased by high-fat feeding in jejunum (median 0.334 for control vs 0.393 for high-fat, P = 0.03) and colon (0.335 for control vs 0.433 for high-fat, P = 0.01), but not in duodenum or ileum. The concentration of nearly all identified bile acids was significantly increased by high-fat feeding (P < 0.001). The proportion of ursodeoxycholic acid (UDCA) in all bile acids was decreased (1.4% ± 0.1% in high-fat vs 2.8% ± 0.3% in controls, P < 0.01) and correlated inversely with intestinal permeability (r = -0.72, P = 0.01). High-fat feeding also increased jejunal FXR expression, as well as TNF expression along the intestine, especially in the colon.
CONCLUSION: High-fat-feeding increased intestinal permeability, perhaps by a mechanism related to bile acid metabolism, namely a decreased proportion of fecal UDCA and increased FXR expression.
Keywords: Bile acids, Bile salts, Diet-induced obesity, Farnesoid X-activated receptor, Intestinal permeability, Ursodeoxycholic acid
INTRODUCTION
Intestinal permeability is a term for the paracellular and transcellular translocation of large molecules foreign to the body. Paracellular permeability is mediated by tight-junction proteins, which prevent uncontrolled transport through the epithelium[1]. Deterioration of tight-junction proteins may permit translocation of bacterial lipopolysaccharides into the serum, i.e., endotoxemia, which may then cause inflammation. In obesity, low-grade inflammation is a risk factor for type 2 diabetes and cardiovascular diseases[2,3]. Impaired gut barrier function is related to several disease states such as steatohepatitis, fatty liver disease and diabetes[4-6]. Furthermore, it may form the link between obesity and its related disorders[7-12].
High dietary fat content may in part lead to barrier dysfunction, supported by cross-sectional data indicating that a diet high in energy and fat is associated with endotoxemia[13]. Dietary fat affects bile acid metabolism, because the absorption of fat requires an increase in bile flow. Consequently, a high-fat diet elevates the fecal concentration of bile acids[14,15]. In mice, an orally fed secondary bile acid deoxycholic acid (DCA) induces intestinal inflammation[16], and in vitro bile acids provoke permeability of intestinal cell monolayers[17,18]. In contrast, the most hydrophilic bile acid, ursodeoxycholic acid (UDCA), may counteract increased intestinal permeability[19].
Proteins involved in bile acid absorption are mediated by the intestinal farnesoid X receptor (FXR), also known as the bile acid receptor, which is highly expressed in intestinal and liver tissues[20]. Mice deficient in FXR show compromised barrier function and localization of neutrophils in their intestinal villi[21].
The aim of this study was to investigate whether high-fat feeding is associated with increased intestinal permeability via alterations in bile acid metabolism.
MATERIALS AND METHODS
Animals
Male C57Bl/6 mice were obtained from Charles River (Sulzfeld, Germany) and acclimatized for a week prior to the experiment. At 6 wk of age, they were randomized into two groups equal in weight: High-fat (n = 26; 60E% fat, D12492; Research Diets, New Brunswick, NJ, United States) and control (n = 24; 10E% fat, D12450B; Research Diets). Diets were paired for fiber and protein content. Mice were housed six per cage in a standard animal laboratory with a dark/light cycle of 12/12 h, with free access to food and water. After 15 wk of feeding, mice were euthanized with a gas mixture of CO2 (70%) and O2 (30%) (AGA, Riihimäki, Finland), and cervical dislocation. Animal experiments were approved by the National Animal Experiment Board of Finland.
Intestinal permeability measurements
Fresh segments of duodenum, jejunum, ileum and proximal colon were collected in duplicate, opened along the mesenteric border, pinned onto 0.3 cm2 sliders and mounted into an EasyMount Ussing chamber system with a voltage-clamp apparatus (Physiologic Instruments, San Diego, CA, United States). Two intestinal segments were collected in duplicate from each mouse, because only four chambers were available. Tissues were surrounded by 5 mL Ringer solution (120 mmol/L NaCl, 5 mmol/L KCl, 25 mmol/L NaHCO3, 1.8 mmol/L Na2HPO4, 0.2 mmol/L NaH2PO4, 1.25 mmol/L CaCl2, 1 mmol/L MgSO4, 10 mmol/L glucose) on each side. The system was water-jacketed to 37 °C and carbonated with a carbogen (95% O2 , 5% CO2; AGA) gas flow. After an equilibration period of 10 min, solutions were replaced with fresh Ringer, and 4 kDa FITC-dextran (TdB Cons, Uppsala, Sweden) was added to the luminal side to a final concentration of 2.2 mg/mL. Resistance and short-circuit current were followed for 60 min, after which, serosal fluorescence was detected with a Wallac Victor2 1420 Multilabel counter (Perkin-Elmer, Waltham, MA, United States). Intestinal permeability was determined by comparing serosal fluorescence to luminal fluorescence as per mille of translocated dextran.
Fecal bile acid analysis
Feces were collected at 13 wk by placing mice individually in metabolic cages for 24 h. Feed and water was provided ad libitum. Feces were carefully separated from all other material and frozen at -20 °C. Upon analysis, feces of six mice from each group were dried overnight with nitrogen and pulverized. Bile acids were extracted and measured by gas-liquid chromatography according to the method by Grundy et al[22] in the laboratory of the Hospital District of Helsinki and Uusimaa. Internal standards were run for isolithocholic acid, lithocholic acid, epideoxycholic acid, DCA, chenodeoxycholic acid, cholic acid and UDCA.
Farnesoid X receptor and tumor necrosis factor expression assays
Segments of jejunum were collected from each mouse, snap frozen in liquid nitrogen, and stored at -80 °C. RNA was extracted with TRI Reagent (RT111; Molecular Research Center, Cincinnati, OH, United States) according to the manufacturer’s protocol. Tissues were homogenized with Precellus24 (6500 rpm, 2 × 15 s; Bertin Technologies, Montigny-le-Bretonneux, France). Phase separation was performed with chloroform (34 854; Sigma-Aldrich, St Louis, MO, United States), and RNA precipitation with isopropanol (P/7507/15X, Fisher Scientific, United States). RNA concentration was measured with NanoDrop 8000 (Thermo Scientific, Waltham, MA, United States), and converted to cDNA with a SuperScript VILO cDNA synthesis kit (Applied Biosystems, Carlsbad, CA, United States) according to the manufacturer’s instructions, using 1 μg RNA in a reaction volume of 20 μL. Reactions for quantitative real time polymerase chain reaction (qPCR) were run using TaqMan chemistry (Applied Biosystems) for FXR (Mm00436420_m1) and tumor necrosis factor (TNF) (Mm00443258_m1). Gene expression was normalized to beta-actin and beta-glucuronidase. Reactions were run on a CFX96 real-time PCR detection system (Bio-Rad, Hercules, CA, United States) in triplicate. Skeletal muscle was used as a non-expressing negative control. Gene expression was calculated with Bio-Rad CFX Manager software using the normalized expression ΔΔC(t) method.
Statistical analysis
Permeability results were statistically analyzed with a Mann-Whitney U test due to the uneven distribution of values around the means. In addition to calculating intestinal permeability for each segment (duodenum, jejunum, ileum and colon), a value for overall intestinal permeability was calculated for each mouse as an average of the permeability in its two measured intestinal segments. Bile acid and gene expression data were analyzed with a t test between control and high-fat groups. As there was a special focus on UDCA, the concentration of this bile acid relative to total identified bile acids was also calculated. The gene expression data had a setting of four groups, characterized by two factors possibly affecting intestinal FXR and TNF expression: intestinal segment and diet. The independent effects of these two factors were statistically analyzed with a factorial experiment. Groups were also compared with each other individually for both intestinal segments with a t test. Equality of variances was tested with Levene’s test. All statistical analyses were performed with PASW Statistics version 18.0.2 (IBM, United States). All data are expressed as mean ± SE unless otherwise stated.
RESULTS
Intestinal permeability
The weight of the high-fat-fed mice was significantly higher compared to control mice (49.5 ± 0.59 g vs 28.6 ± 0.36 g, P < 0.001). High-fat-feeding increased intestinal permeability significantly in the jejunum (median 0.334 per mille translocated dextran for control vs 0.393 for high-fat, P = 0.03) and colon (median 0.335 for control vs 0.433 for high-fat, P = 0.01), but not in the duodenum (median 0.359 for control vs 0.360 for high-fat, P = 0.33) or ileum (median 0.351 for control vs 0.452 for high-fat, P = 0.69, Figure 1). Resistance and short-circuit current were measured during the Ussing chamber experiments to monitor tissue viability. The stability of these values indicates good viability of tissue segments throughout the experiments (Figure 2).
Figure 1.

Effect of high-fat-feeding on intestinal permeability. Intestinal permeability in duodenum (A), jejunum (B), ileum (C), and colon (D) of high-fat-fed vs control mice. Permeability was measured in an Ussing chamber. Results are shown as per mille translocated dextran. Box plots show median, upper and lower quartiles, and Tukey’s whiskers (highest and lowest values, outliers shown as black dots). aP < 0.05 between high-fat and control.
Figure 2.

Stability of Ussing chamber experiments. Short-circuit current (A) and resistance (B) in Ussing chamber experiments. Bars indicate mean and SEM. n = 9-15/group.
Fecal bile acids
Feces from high-fat fed mice contained substantially more bile acids compared to feces from control mice (2.13 ± 0.29 mg/g dry feces vs 0.37 ± 0.03 mg/g dry feces, P < 0.001; Figure 3A). This was reflected as a significantly elevated concentration of each bile acid (P < 0.01) except isolithocholic acid (Figure 3B). We were especially interested in the effects of UDCA, which is considered cytoprotective, therefore, we calculated the ratio of UDCA to total bile acids. The proportion of UDCA in feces of high-fat-fed mice was nearly halved compared to that in control mice (1.4% ± 0.1% in high-fat vs 2.8% ± 0.3% in controls, P < 0.01, Figure 3C). There was also a marked inverse correlation of overall intestinal permeability to the proportion of UDCA in total fecal bile acids (r = -0.72, P = 0.01, n = 11, Figure 4A). The correlation remained significant even after controlling for body weight (partial correlation r = -0.64, P < 0.05, n = 11). A trend for a similar association was seen in the subgroups of jejunal and colonic permeability (r = -0.88, P = 0.05, n = 5 for jejunum and r = -0.70, P = 0.12, n = 6 for colon, Figure 4B and C). In addition, we correlated permeability to the ratio of UDCA to DCA, a cytotoxic bile acid. This was done to prevent bias by the unmeasured muricholic acid, which is a primary bile acid present in murine bile[23], and is left unidentified by methods designed for clinical use. The relation of UDCA to DCA showed a similar halved concentration and inverse correlation to permeability (r = -0.70, P = 0.02).
Figure 3.

Fecal bile acids. A: Concentration of total measured bile acids; B: Concentration of measured bile acids in feces; C: Proportion of ursodeoxycholic acid (UDCA) in total measured bile acids. Bars indicate mean and SEM. n = 6/group, aP < 0.001, bP < 0.01 between high-fat and control groups.
Figure 4.

Correlation of proportion of ursodeoxycholic acid with intestinal permeability. Pearson’s correlation of fecal proportion of ursodeoxycholic acid (UDCA) percentage to overall permeability of intestine (A), jejunal permeability (B), and colonic permeability (C). Each dot represents an individual animal.
Intestinal farnesoid X receptor and tumor necrosis factor expression
The effect of high-fat-feeding on bile acid metabolism and intestinal inflammation was assayed as expression of intestinal FXR and TNF in the jejunum and colon. Jejunal FXR expression was increased 30% by high-fat feeding (0.74 ± 0.049 for high-fat vs 0.57 ± 0.051 for control, P = 0.03) and colonic TNF expression was doubled (0.82 ± 0.148 for high-fat vs 0.42 ± 0.068 for control, P = 0.02) but no significant differences were seen in jejunal tumor necrosis factor (TNF) (P = 0.30) or colonic FXR (P = 0.63, Table 1). FXR and TNF expressions did, however, correlate with each other (r = 0.41, P < 0.01, n = 50, Figure 5).
Table 1.
Gene expression of farnesoid X receptor and tumor necrosis factor (mean ± SEM)
|
Control |
High-fat |
P value | |||
| n | expression | n | expression | ||
| FXR | |||||
| Jejunum | 14 | 0.57 ± 0.051 | 12 | 0.74 ± 0.049 | 0.03 |
| Colon | 10 | 1.36 ± 0.234 | 14 | 1.49 ± 0.159 | 0.63 |
| TNF | |||||
| Jejunum | 14 | 0.21 ± 0.041 | 12 | 0.27 ± 0.040 | 0.30 |
| Colon | 10 | 0.42 ± 0.068 | 14 | 0.82 ± 0.148 | 0.02 |
FXR: Farnesoid X receptor; TNF: Tumor necrosis factor.
Figure 5.
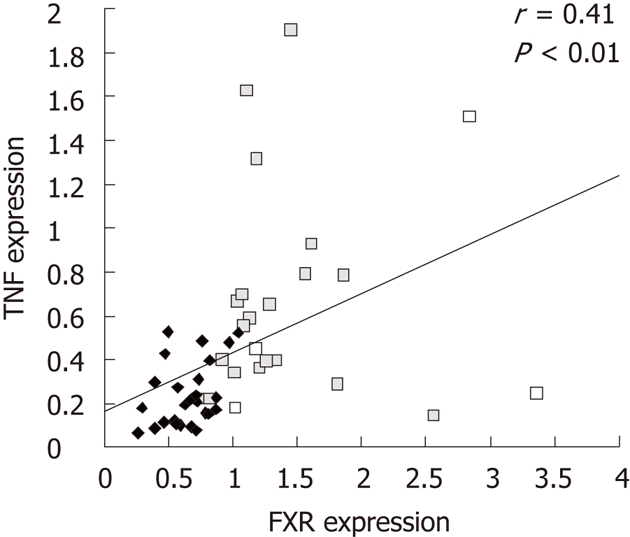
Correlation of tumor necrosis factor and farnesoid X receptor expression. Pearson’s correlation of tumor necrosis factor (TNF) and farnesoid X receptor (FXR) expression. Jejunal values are shown as black diamonds and colonic values as grey squares. Each dot represents an individual animal.
A factorial experiment on the independent effects of diet and intestinal segment on gene expression revealed that overall intestinal TNF expression was elevated by high-fat-feeding (P = 0.02). Moreover, both TNF and FXR expressions were higher in colon compared to jejunum (P < 0.001 for both).
DISCUSSION
The objective of this study was to see whether intestinal permeability was increased by high-fat-feeding via a mechanism related to bile acid metabolism. These data showed that permeability of jejunum and colon in high-fat-fed mice was increased. The animals were clearly obese solely due to dietary fat content (60E%), and not by genetic modification. Our results on increased permeability are supported by a similar previous study, which investigated the effects of obesity and a high-fat diet on rat intestinal permeability[12]. Also a carbohydrate-free diet containing an extremely high amount of fat (72E%) increased intestinal permeability in mice[9]. Our 60E% diet may be considered more physiologically relevant compared to carbohydrate-free diets. We also took care to pair the diets in protein and fiber content. Therefore, the barrier dysfunction in high-fat-feeding was not biased by any fiber-mediated effect. There is no conclusion on whether gut permeability may be affected by obesity alone, without modifications in diet composition[7,12].
In the present study, we found that, in addition to increased fecal bile acid content, high-fat feeding also modulated fecal bile acid profile. Bile acids and bile juice are known to impair barrier function in enterocyte monolayers in vitro[12,17,18], as well as recovery from tissue damage ex vivo[24], and are elevated in serum of high-fat-fed rats[12]. Our data indicate that high-fat-feeding modifies gut permeability by a mechanism related to bile acids, as previously hypothesized[12].
This is, to the best of our knowledge, the first study to show that high-fat-induced intestinal barrier dysfunction is related to increased intestinal FXR expression. Inagaki et al[21] have shown in a FXR knockout mouse model that FXR is involved in tight junction integrity. It is unclear, however, what role FXR plays in the pathogenesis of high-fat-induced barrier dysfunction.
Both fecal bile acids and intestinal TNF expression were elevated by high-fat-feeding in the present study. Bile acids are linked to inflammatory pathways, because DCA is able to stimulate the nuclear factor-κB route[25], which regulates TNF expression. A correlation between bile acids and TNF was not observed in our small set of fecal samples. Intestinal FXR and TNF were, however, correlated, which may reflect a link between intestinal bile acid concentration and inflammation. Incubation of jejunum in DCA (0.3 mmol/L) increased prostaglandin E2 in the supernatant, thus indicating a possible role for inflammation in increasing intestinal permeability (data not shown). Orally fed DCA induces intestinal inflammation in mice[16], which further supports the hypothesis that inflammation is involved in the pathogenesis of gut barrier dysfunction. In this study, we observed increased TNF expression in the intestine. TNF is known to increase intestinal permeability in vitro[26-29], and anti-TNF antibodies restore barrier function in vivo[30,31]. However, these data do not permit us to draw conclusions on whether increased TNF expression, in this study, was a cause or a secondary event in increased intestinal permeability.
We observed a halved proportion of UDCA in fecal bile acids of high-fat-fed mice. Moreover, this proportion of UDCA correlated with increased permeability, even when controlled for body weight. These data suggest a barrier protective effect for UDCA in our study. Our results are in agreement with previous reports showing that, as a hydrophilic bile acid, UDCA does not increase epithelial permeability in vitro in comparison to other bile acids[18]. On the contrary, it is capable of counteracting intestinal barrier dysfunction[19], protecting against chemically induced colitis[19,32,33] and colitis-associated adenocarcinoma[34] in rodents. Its role may be especially relevant in comparison to DCA, because UDCA protects mitochondria against DCA-induced reactive oxygen species production[35]. It may thus be suggested that, in addition to total bile acid concentration, the bile acid profile is relevant regarding bile-acid-related functions in the intestine.
Intestinal permeability was measured ex vivo with the Ussing chamber as the translocation of large dextrans. This direct method is more representative of intestinal permeability than the often used transepithelial resistance and tight-junction protein analysis. Furthermore, the Ussing chamber allows measurement of permeability from selected tissue segments, unlike direct in vivo methods.
The 4 kDa FITC-dextrans used here are generally believed to translocate through the paracellular spaces, although there are no reports to confirm this. Permeability to FITC-dextrans does, however, correlate with a decrease in tight-junction protein expression[10]. It has also been proposed that translocation of lipopolysaccharides occurs transcellularly via chylomicrons[36]. The methods used in the present study do not distinguish between these two pathways. Their importance in intestinal permeability needs further elucidation.
We evaluated modifications of bile acid metabolism by analyzing fecal bile acids. It must be noted that fecal bile acids only reflect alterations of bile acid metabolism, and this method does not allow us to draw conclusions about liver bile acid metabolism or bile composition. It is, however, an estimate of colonic bile acid concentrations.
In conclusion, high-fat feeding increases permeability in the jejunum and colon, elevates fecal bile acid concentration, and induces intestinal inflammation in mice. Alterations in bile acid homeostasis, namely UDCA synthesis and intestinal FXR expression, may relate to increased intestinal permeability. Our results show that alterations in bile acid metabolism may be associated with intestinal permeability and should be studied as a possible target in affecting the onset of barrier dysfunction.
ACKNOWLEDGMENTS
We are grateful to Hanna Laurikainen for help with the Ussing chamber experiments. We are grateful to Professor Eero Mervaala, MD, and Professor (emeritus) Heikki Vapaatalo, MD, for critical discussion. We are grateful to Leena Kaipiainen for analysis of bile acids. We are grateful to statistician Tuija Poussa for statistical advice.
COMMENTS
Background
Intestinal permeability has recently been linked to type 2 diabetes, steatosis and steatohepatitis. One proposed cause for increased permeability is a diet high in fat. A high-fat-diet increases excretion of bile. Secondary bile acids are known to increase permeability of epithelial monolayers in vitro. However, physiological concentrations of bile juice obtained from healthy rats have failed to increase epithelial monolayer permeability. This may be due to the fact that bile consists of a profile of several bile acids, which differ in cytotoxicity. It has not yet been addressed whether high-fat feeding alters the profile of fecal bile acids, and whether this profile plays a role in the onset of barrier dysfunction.
Research frontiers
Proposed mechanisms for the dietary induction of increased intestinal permeability have mostly been related to alterations in gut microbiota. Only a few publications have mentioned other luminal factors affecting permeability. The research hotspot in this field is how diet modifies bile acid metabolism and leads to increased intestinal permeability.
Innovations and breakthroughs
Although bile is considered detrimental to the gut epithelium, physiological concentrations have not increased epithelial monolayer permeability. We show that not only was bile excretion increased by high-fat feeding, but the profile of fecal bile acids had become more cytotoxic than in healthy control mice. The proportion of a hydrophilic bile acid, ursodeoxycholic acid (UDCA), was decreased by high-fat feeding and correlated inversely with intestinal permeability.
Applications
The prevention of barrier dysfunction may decrease the risk of its associated diseases. The present results suggest that bile acid metabolism is a potential target for the prevention of a barrier dysfunction.
Terminology
Intestinal permeability refers to how large molecules, such as inflammatory bacterial components, translocate through the intestinal epithelium into the circulation. Translocation may be paracellular, through tight-junction proteins, or transcellular, via chylomicrons. UDCA is a hydrophilic bile acid. Deoxycholic acid is a secondary bile acid produced from cholic acid by intestinal microbes.
Peer review
The authors tested intestinal permeability in an ex vivo model (Ussing chambers; mucosal to serosal flux of FITC-labeled dextran) in mice on a high-fat diet; moreover, analysis of fecal bile acids and expression of mucosal tumor necrosis factor and farnesoid X receptor was performed.
Footnotes
Supported by The Foundation for Nutrition Research and the Finnish Funding Agency of Technology and Innovation
Peer reviewer: Walter Fries, MD, Department Medicina Interna and Terapia Medica, UOS Malattie Intestinali Croniche, Policlinico Messina, 98125 Messina, Italy
S- Editor Shi ZF L- Editor Kerr C E- Editor Xiong L
References
- 1.Baumgart DC, Dignass AU. Intestinal barrier function. Curr Opin Clin Nutr Metab Care. 2002;5:685–694. doi: 10.1097/00075197-200211000-00012. [DOI] [PubMed] [Google Scholar]
- 2.Dehghan A, van Hoek M, Sijbrands EJ, Stijnen T, Hofman A, Witteman JC. Risk of type 2 diabetes attributable to C-reactive protein and other risk factors. Diabetes Care. 2007;30:2695–2699. doi: 10.2337/dc07-0348. [DOI] [PubMed] [Google Scholar]
- 3.Engström G, Hedblad B, Tydén P, Lindgärde F. Inflammation-sensitive plasma proteins are associated with increased incidence of heart failure: a population-based cohort study. Atherosclerosis. 2009;202:617–622. doi: 10.1016/j.atherosclerosis.2008.05.038. [DOI] [PubMed] [Google Scholar]
- 4.Arrieta MC, Bistritz L, Meddings JB. Alterations in intestinal permeability. Gut. 2006;55:1512–1520. doi: 10.1136/gut.2005.085373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miele L, Valenza V, La Torre G, Montalto M, Cammarota G, Ricci R, Mascianà R, Forgione A, Gabrieli ML, Perotti G, et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology. 2009;49:1877–1887. doi: 10.1002/hep.22848. [DOI] [PubMed] [Google Scholar]
- 6.Valenti L, Fracanzani AL, Fargion S. The immunopathogenesis of alcoholic and nonalcoholic steatohepatitis: two triggers for one disease? Semin Immunopathol. 2009;31:359–369. doi: 10.1007/s00281-009-0152-9. [DOI] [PubMed] [Google Scholar]
- 7.Brun P, Castagliuolo I, Di Leo V, Buda A, Pinzani M, Palù G, Martines D. Increased intestinal permeability in obese mice: new evidence in the pathogenesis of nonalcoholic steatohepatitis. Am J Physiol Gastrointest Liver Physiol. 2007;292:G518–G525. doi: 10.1152/ajpgi.00024.2006. [DOI] [PubMed] [Google Scholar]
- 8.Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56:1761–1772. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- 9.Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57:1470–1481. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- 10.Cani PD, Possemiers S, Van de Wiele T, Guiot Y, Everard A, Rottier O, Geurts L, Naslain D, Neyrinck A, Lambert DM, et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut. 2009;58:1091–1103. doi: 10.1136/gut.2008.165886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun L, Yu Z, Ye X, Zou S, Li H, Yu D, Wu H, Chen Y, Dore J, Clément K, et al. A marker of endotoxemia is associated with obesity and related metabolic disorders in apparently healthy Chinese. Diabetes Care. 2010;33:1925–1932. doi: 10.2337/dc10-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suzuki T, Hara H. Dietary fat and bile juice, but not obesity, are responsible for the increase in small intestinal permeability induced through the suppression of tight junction protein expression in LETO and OLETF rats. Nutr Metab (Lond) 2010;7:19. doi: 10.1186/1743-7075-7-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amar J, Burcelin R, Ruidavets JB, Cani PD, Fauvel J, Alessi MC, Chamontin B, Ferriéres J. Energy intake is associated with endotoxemia in apparently healthy men. Am J Clin Nutr. 2008;87:1219–1223. doi: 10.1093/ajcn/87.5.1219. [DOI] [PubMed] [Google Scholar]
- 14.Cummings JH, Wiggins HS, Jenkins DJ, Houston H, Jivraj T, Drasar BS, Hill MJ. Influence of diets high and low in animal fat on bowel habit, gastrointestinal transit time, fecal microflora, bile acid, and fat excretion. J Clin Invest. 1978;61:953–963. doi: 10.1172/JCI109020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bianchini F, Caderni G, Dolara P, Fantetti L, Kriebel D. Effect of dietary fat, starch and cellulose on fecal bile acids in mice. J Nutr. 1989;119:1617–1624. doi: 10.1093/jn/119.11.1617. [DOI] [PubMed] [Google Scholar]
- 16.Bernstein H, Holubec H, Bernstein C, Ignatenko N, Gerner E, Dvorak K, Besselsen D, Ramsey L, Dall’Agnol M, Blohm-Mangone KA, et al. Unique dietary-related mouse model of colitis. Inflamm Bowel Dis. 2006;12:278–293. doi: 10.1097/01.MIB.0000209789.14114.63. [DOI] [PubMed] [Google Scholar]
- 17.Hughes R, Kurth MJ, McGilligan V, McGlynn H, Rowland I. Effect of colonic bacterial metabolites on Caco-2 cell paracellular permeability in vitro. Nutr Cancer. 2008;60:259–266. doi: 10.1080/01635580701649644. [DOI] [PubMed] [Google Scholar]
- 18.Raimondi F, Santoro P, Barone MV, Pappacoda S, Barretta ML, Nanayakkara M, Apicella C, Capasso L, Paludetto R. Bile acids modulate tight junction structure and barrier function of Caco-2 monolayers via EGFR activation. Am J Physiol Gastrointest Liver Physiol. 2008;294:G906–G913. doi: 10.1152/ajpgi.00043.2007. [DOI] [PubMed] [Google Scholar]
- 19.Bernardes-Silva CF, Damião AO, Sipahi AM, Laurindo FR, Iriya K, Lopasso FP, Buchpiguel CA, Lordello ML, Agostinho CL, Laudanna AA. Ursodeoxycholic acid ameliorates experimental ileitis counteracting intestinal barrier dysfunction and oxidative stress. Dig Dis Sci. 2004;49:1569–1574. doi: 10.1023/b:ddas.0000043365.39251.6e. [DOI] [PubMed] [Google Scholar]
- 20.Lefebvre P, Cariou B, Lien F, Kuipers F, Staels B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev. 2009;89:147–191. doi: 10.1152/physrev.00010.2008. [DOI] [PubMed] [Google Scholar]
- 21.Inagaki T, Moschetta A, Lee YK, Peng L, Zhao G, Downes M, Yu RT, Shelton JM, Richardson JA, Repa JJ, et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci USA. 2006;103:3920–3925. doi: 10.1073/pnas.0509592103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grundy SM, Ahrens EH, Miettinen TA. Quantitative isolation and gas--liquid chromatographic analysis of total fecal bile acids. J Lipid Res. 1965;6:397–410. [PubMed] [Google Scholar]
- 23.Alnouti Y, Csanaky IL, Klaassen CD. Quantitative-profiling of bile acids and their conjugates in mouse liver, bile, plasma, and urine using LC-MS/MS. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;873:209–217. doi: 10.1016/j.jchromb.2008.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Campbell NB, Ruaux CG, Shifflett DE, Steiner JM, Williams DA, Blikslager AT. Physiological concentrations of bile salts inhibit recovery of ischemic-injured porcine ileum. Am J Physiol Gastrointest Liver Physiol. 2004;287:G399–G407. doi: 10.1152/ajpgi.00310.2003. [DOI] [PubMed] [Google Scholar]
- 25.Baeuerle PA, Baltimore D. I kappa B: a specific inhibitor of the NF-kappa B transcription factor. Science. 1988;242:540–546. doi: 10.1126/science.3140380. [DOI] [PubMed] [Google Scholar]
- 26.Schmitz H, Fromm M, Bentzel CJ, Scholz P, Detjen K, Mankertz J, Bode H, Epple HJ, Riecken EO, Schulzke JD. Tumor necrosis factor-alpha (TNFalpha) regulates the epithelial barrier in the human intestinal cell line HT-29/B6. J Cell Sci. 1999;112(Pt 1):137–146. doi: 10.1242/jcs.112.1.137. [DOI] [PubMed] [Google Scholar]
- 27.Gitter AH, Bendfeldt K, Schmitz H, Schulzke JD, Bentzel CJ, Fromm M. Epithelial barrier defects in HT-29/B6 colonic cell monolayers induced by tumor necrosis factor-alpha. Ann N Y Acad Sci. 2000;915:193–203. doi: 10.1111/j.1749-6632.2000.tb05242.x. [DOI] [PubMed] [Google Scholar]
- 28.Ma TY, Iwamoto GK, Hoa NT, Akotia V, Pedram A, Boivin MA, Said HM. TNF-alpha-induced increase in intestinal epithelial tight junction permeability requires NF-kappa B activation. Am J Physiol Gastrointest Liver Physiol. 2004;286:G367–G376. doi: 10.1152/ajpgi.00173.2003. [DOI] [PubMed] [Google Scholar]
- 29.Ma TY, Boivin MA, Ye D, Pedram A, Said HM. Mechanism of TNF-{alpha} modulation of Caco-2 intestinal epithelial tight junction barrier: role of myosin light-chain kinase protein expression. Am J Physiol Gastrointest Liver Physiol. 2005;288:G422–G430. doi: 10.1152/ajpgi.00412.2004. [DOI] [PubMed] [Google Scholar]
- 30.Suenaert P, Bulteel V, Lemmens L, Noman M, Geypens B, Van Assche G, Geboes K, Ceuppens JL, Rutgeerts P. Anti-tumor necrosis factor treatment restores the gut barrier in Crohn’s disease. Am J Gastroenterol. 2002;97:2000–2004. doi: 10.1111/j.1572-0241.2002.05914.x. [DOI] [PubMed] [Google Scholar]
- 31.Fries W, Muja C, Crisafulli C, Cuzzocrea S, Mazzon E. Dynamics of enterocyte tight junctions: effect of experimental colitis and two different anti-TNF strategies. Am J Physiol Gastrointest Liver Physiol. 2008;294:G938–G947. doi: 10.1152/ajpgi.00469.2007. [DOI] [PubMed] [Google Scholar]
- 32.Kullmann F, Arndt H, Gross V, Rüschoff J, Schölmerich J. Beneficial effect of ursodeoxycholic acid on mucosal damage in trinitrobenzene sulphonic acid-induced colitis. Eur J Gastroenterol Hepatol. 1997;9:1205–1211. [PubMed] [Google Scholar]
- 33.Kullmann F, Gross V, Rüschoff J, Arndt H, Benda W, Winkler von Mohrenfels A, Schölmerich J. Effect of ursodeoxycholic acid on the inflammatory activity of indomethacin-induced intestinal inflammation in rats. Z Gastroenterol. 1997;35:171–178. [PubMed] [Google Scholar]
- 34.Loddenkemper C, Keller S, Hanski ML, Cao M, Jahreis G, Stein H, Zeitz M, Hanski C. Prevention of colitis-associated carcinogenesis in a mouse model by diet supplementation with ursodeoxycholic acid. Int J Cancer. 2006;118:2750–2757. doi: 10.1002/ijc.21729. [DOI] [PubMed] [Google Scholar]
- 35.Rodrigues CM, Fan G, Wong PY, Kren BT, Steer CJ. Ursodeoxycholic acid may inhibit deoxycholic acid-induced apoptosis by modulating mitochondrial transmembrane potential and reactive oxygen species production. Mol Med. 1998;4:165–178. [PMC free article] [PubMed] [Google Scholar]
- 36.Ghoshal S, Witta J, Zhong J, de Villiers W, Eckhardt E. Chylomicrons promote intestinal absorption of lipopolysaccharides. J Lipid Res. 2009;50:90–97. doi: 10.1194/jlr.M800156-JLR200. [DOI] [PubMed] [Google Scholar]