

Abstract
Prediction methods as well as experimental methods for T-cell epitope discovery have developed significantly in recent years. High-throughput experimental methods have made it possible to perform full-length protein scans for epitopes restricted to a limited number of MHC alleles. The high costs and limitations regarding the number of proteins and MHC alleles that are feasibly handled by such experimental methods have made in silico prediction models of high interest. MHC binding prediction methods are today of a very high quality and can predict MHC binding peptides with high accuracy. This is possible for a large range of MHC alleles and relevant length of binding peptides. The predictions can easily be performed for complete proteomes of any size. Prediction methods are still, however, dependent on good experimental methods for validation, and should merely be used as a guide for rational epitope discovery. We expect prediction methods as well as experimental validation methods to continue to develop and that we will soon see clinical trials of products whose development has been guided by prediction methods.
Keywords: CTL, epitope, HLA, MHC, prediction, T cell, vaccine
Background
Why epitope discovery?
The immune system, which is the ‘natural’ defense system of most multicellular organisms, can be manipulated in the fight against infections, cancer and autoimmune diseases. The immune system as a whole reacts against a large number of factors, many of which are not of protein origin. The innate immune system, in particular, reacts quickly against a number of compounds supposed to be foreign or very rare in a healthy and uninfected individual [1]. The adaptive immune system is able to very specifically react against proteins and peptides specific for pathogenic cells and foreign organisms. The immune system has for centuries primarily been a tool for prophylactic treatments in the form of vaccination against pathogens and toxins. However, most vaccines, including those that are the most successful, have been developed empirically without much utilization of specific immunological knowledge [1]. The insight obtained within the last decades, on the other hand, has revealed an intriguing potential for more rational approaches. This is not only an opportunity in the field of vaccine design, but also opens up the possibility of highly specific therapeutic interventions [2].
Epitopes were originally defined as the part of an antigen that defines the binding to an immunoglobulin [3]. For antibodies, this is an obvious and straightforward definition as the immunoglobulin generally binds to the native antigen with unambiguously and well-defined interactions without any help or interactions with other proteins. When considering the cellular arm of the adaptive immune system, the active immunoglobulin is the T-cell receptor (TCR) and the binding partner, the antigen, is generally a processed part of a protein in complex with an MHC protein. Thus, in this definition, the epitope will consist of part of a processed protein and parts of the host-expressed MHC protein. What is usually of interest, though, is which part of a protein (peptide) is responsible for an immune response. Thus, often this part is referred to as the epitope and the native protein from which the epitope originates as the antigen. In the following text, an antigen will thus refer to a protein that is able to induce an immune response in a given host context. The definition of a T-cell epitope will be a subpeptide from the antigen, which is able to complex with a host MHC and, in this context, can be recognized by antigen-specific TCRs.
Epitope discovery is obviously of major interest in traditional protective vaccine research. Here, the identification of specific epitopes can lead to the identification of important antigens and help define the most important parts of selected antigens [4]. In traditional vaccine development, such information is important in order to ensure that selected production strains and subtypes carry the relevant versions of the most important antigens. Recently, the knowledge of specific antigens and epitopes has been used to guide the design of minimal vaccines consisting of only the relevant antigens [5], which will very likely soon lead to vaccines consisting of minimal artificial polypeptides designed to contain several strong epitopes relevant for the specific disease [6]. In addition to protective vaccinology, the potential of using immune-related tools to fight established diseases such as cancer and chronic infections attracts increasing interest. Immunotherapy is the general term of two, conceptually very different, approaches. First, most mature, and increasingly used in the clinic, is the use of monoclonal antibodies against a specific disease target [7]. The second approach is to manipulate a patient’s immune system to focus on specific disease-related targets that are otherwise being ignored by the host immune system. Here, carefully selected epitopes might be used to induce a more potent and efficient immune response.
The knowledge of which strong epitopes a protein contains has further importance when considering the use of proteins and peptides as therapeutic drugs. Here, epitope identification can be used for deselection or de-immunization purposes, where therapeutic proteins, including monoclonal antibodies, that might otherwise have unwanted immunogenic effects can be deselected early in the preclinical phase or the proteins may be de-immunized by removal of identified epitopes [8]. Finally, epitope identification is of general interest in basic immunology. The studies of how and when specific epitopes will or will not lead to immunogenic responses and which signaling pathways coincide with specific epitopes is an important part of immunology research that can help in the general understanding of the behavior and evolution of the immune system.
From antigen to epitope: processing & presentation of T-cell epitopes
For the sake of the later descriptions of different prediction approaches, the key events in cellular immune responses are briefly summarized here with an emphasis on the events that have been successfully characterized by prediction methods, and other known important events that are presently not predictable by mathematical models or computational algorithms.
T-cell epitopes in complex with MHC proteins are recognized by TCRs on the surface of T cells with different functionality, T-helper (Th) cells and cytotoxic T lymphocytes (CTL). Besides having different coreceptors, CD4 and CD8, respectively, the MHC molecules presenting Th epitopes or CTL epitopes have some important differences that seem to be universal for jawed vertebrates, with one recently discovered exception in cod, as this fish seems to be lacking the genes that are specific for Th responses [9]. Furthermore, only a limited panel of animals have been investigated in depth with respect to their immune systems, thus this section will consider primarily the knowledge obtained by studying the human immune system.
The CTL response is induced when a TCR on a CD8+ T cell is able to bind to an MHC class I–peptide complex presented by a nucleated cell. Peptides bind to the MHC class I in a defined binding groove that is closed at the end so that binding peptides are limited in length, where most MHC class I molecules prefer binding to peptides of nine amino acids in length (Figure 1A). The MHC class I proteins are able to exchange bound peptides with free peptides in solution if available, but generally the peptides presented on the surface of a cell originate from longer polypeptides and proteins produced within the cell. The pathway of MHC class I peptide presentation is described in detail in a number of recent reviews [10-12], and is an extension of the general protein turnover machinery present in all eukaryotic cells. The proteasome complex digests proteins labeled with the regulatory protein ubiquitin, producing short peptides. Thus, under normal circumstances, this process is performed by the constitutive proteasome that has strong stochastic elements in cleavage preferences [13,14]. However, careful mapping of cleavage sites has led to a general description of the cleavage specificity [15]. In immune-alert cells, certain subunits in the proteasome complex that are responsible for the actual cleavage are replaced, and the cleavage becomes more specific, but still with stochastic elements. This variant of the proteasome is generally referred to as the immunoproteasome, and the change in specificity moves the length distribution of the peptide products towards longer peptides. Some of the generated peptides will be able to bind to the transporter associated with antigen processing (TAP) and transported into the endoplasmic reticulum (ER). Upon entrance to the ER, the peptide will encounter semifolded MHC class I molecules anchored at the inner side of the ER membrane. Suitable peptides will induce further folding of the MHC as the peptide fully enters the ER, further enabled by the protein tapasin that has a chaperone-like function [16,17]. Fully folded MHC–peptide complexes will be transported to the cell surface where they will be available for binding by a suitable TCR.
Figure 1. Structural surface representations of MHC class I (A) and MHC class II (B) molecules in complex with peptide shown in stick representation.
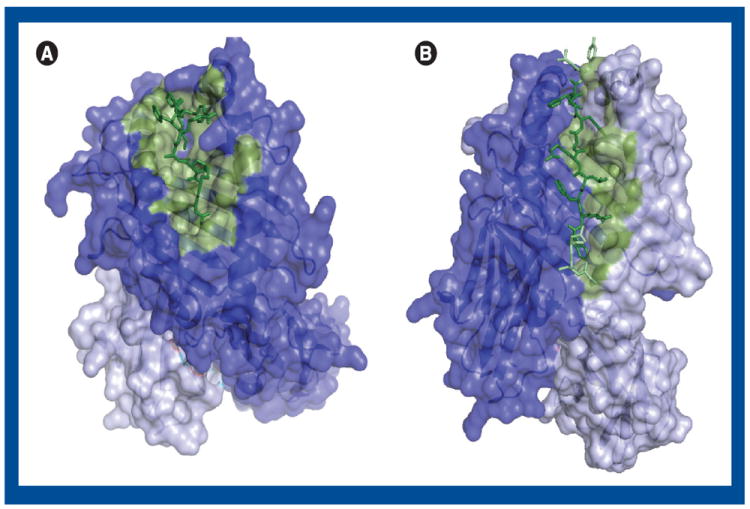
(A) The peptide is green and the residues in the α chain considered important for peptide binding [73] is colored dust green. The rest of the α chain is dark blue and β-2-microglobulin is light blue. (B) The binding core of the peptide is dark green, and the rest of the peptide is bright green. The residues in the β chain considered important for peptide binding [83] are colored dust green. The rest of the β chain is colored dark blue, and the α chain is light blue. The figure has been created using PyMol with the PDB available templates of an HLA-A*11:01-peptide complex structure, 2HN7 [105] (A) and an HLA-DR1-peptide structure, 1AQD [106] (B).
Th responses will be induced if a TCR on a CD4+ T cell binds to an MHC class II–peptide complex, which is generally expressed in professional APCs [18]. The MHC class II binding groove is open at both ends allowing binding of peptides of any length (Figure 1B). Also, the MHC class II-presented peptides have usually been subject to substantial processing before presentation at the cell surface, and this process has been reviewed in detail elsewhere [19-22]. Generally, the presented peptides originate from from native or partly degraded proteins or protein complexes from the extracellular space. The proteins have entered the APC by phagocytosis and continued into the endosomic pathway, where the protein will be denatured and processed by proteases such as the cathepsins, although the specificity of these are not fully understood [23,24]. Membrane-anchored MHC class II molecules are present in the endosomes, fully folded with the invariant chain (Ii) in the binding groove. The Ii undergoes proteolysis, and the MHC class II binding groove will finally be occupied by only a part of the Ii, the MHC class II-associated invariant chain peptide (CLIP). CLIP is then exchanged with free peptides resulting from proteolysis of the phagocytosed antigens facilitated by the action of HLA-DM. Local structural properties of the source antigen have been shown to influence antigen processing and/or the accessibility of peptides to the MHC class II molecule, imposing a bias so that fragments that are exposed in the native fold of antigens are more likely to be presented compared with peptides from the antigen protein core [25]. Loaded MHC class II is next transported to the cell surface, available for potential recognition by CD4+ T cells carrying an appropriate TCR.
The above-described pathways are simplified and a number of exceptions have been described, such as alternative routes for MHC class I-presented peptides to be processed and enter the ER [26-28]. Furthermore, in recent years it has become more evident that the pathways for MHC class I and MHC class II peptide presentation are not always separated and that peptides from autophagocytosed extracellular proteins can end up being presented by MHC class I proteins. This phenomenon is generally described as cross-presentation [29,30].
MHC genes & proteins
MHC class I and II proteins share extensive overall structural similarities (Figure 1) but are constructed differently. MHC class I proteins consist of a membrane-anchored α chain and a smaller β-2-microglobulin chain. The α chain has three structural domains and is linked noncovalently to the β-2-microglobulin chain via interactions of the three domains. The binding groove is made up by the α 1 and α 2 domains and is closed at the ends, which restricts the length of the peptides that will be able to bind to the MHC. Generally, peptides 8–13 amino acids in length are considered able to bind to the MHC, with a preference of nonamer peptides. However, even longer epitopes have been reported [31]. The structure of the MHC molecule has revealed a number of binding pockets important for peptide binding specificity, and for MHC class I two major pockets exist, placed in each end of the binding groove in such a way that generally peptide amino acid number 2 and the C-terminal amino acid will bind in these pockets [32,33]. In humans the α chains for the classical MHC class I proteins are encoded by genes at the HLA-A, -B and -C loci. These genes are highly polymorphic, and thousands of HLA-A, -B and -C alleles have at present been reported [34]. Most of the polymorphic sites are placed in the binding groove and will influence which peptides will be able to bind the particular MHC (Figure 1A).
MHC class II proteins consist of two membrane-anchored polypeptide chains, an α and a β chain. The binding groove is made up of domains from both the α and the β chain, the α-1 and the β-1 domains (Figure 1B). The binding groove is open ended, which enables binding of longer peptides, and generally peptides 12–20 amino acids in length are observed [35]. However, the length of the binding groove resembles that of MHC class I and only nine consecutive amino acid residues from the binding peptide are in contact with residues in the binding groove. The MHC class II binding groove has special pockets that will fit defined amino acids of the binding peptide, and have a major influence on the binding energy [36]. In humans MHC class II chains are encoded by genes in the HLA-DR, -DQ and -DP loci. HLA-DRα chains are expressed from a single locus (HLA-DRA) and no functional variations have been reported. HLA-DRβ chains (HLA-DRB) can be expressed from several loci (HLA-DRB-1, -3, -4 and -5) where most individuals will express the gene present at the HLA-DRB1 locus. Almost 1000 HLA-DRB1 alleles have presently been identified, expressing more than 700 variant proteins, whereas just 46, eight and 16 protein variants have been identified for HLA-DRB-3, -4 and -5, respectively [34]. For HLA-DQ and -DP only one α (HLA-DQA1 and HLA-DPA1, respectively) and one β (HLA-DQB1 and HLA-DPB1, respectively) locus is active at each chromosome. However, for HLA-DP and -DQ, both the α and β chains are polymorphic with one to two dozen α variants and a little more than 100 β variants each. Furthermore, α and β chains can combine with the corresponding partner expressed from either the cis or the trans chromosome, resulting in up to four different class II proteins per locus. As for MHC class I the polymorphic sites are predominantly present in the binding groove and will thus be responsible for diversities in peptide binding specificities. Thus, in principle each allelic version of an MHC molecule will bind a specific set of peptides. As epitopes are a true subset of what are able to bind the MHCs of a given individual, the high degree of polymorphism imposes a big challenge on epitope discovery. Fortunately, not all alleles are equally (in)frequent. Almost half of the European population will have the class I HLA-A*02:01 allele according to the two major web-accessible allele frequency databases, alllelefrequencies.net [37] and dbMHC [38]. The HLA-A*02:01 allele is also the most investigated HLA allele so far, with 1287 reported CTL epitopes available from the major epitope deposit, the ImmuneEpitope database (IEDB) [39]. For other frequent european alleles such as HLA-A*01:01 (~30%) and HLA-B*07:02 (~25%), the numbers are 68 and 136, respectively. Despite this bias, a number of different alleles have actually been examined and it appears that some alleles have similarities at sequence positions coinciding with the major binding pockets, and that these alleles also show similarities in peptide binding preferences. This observation led to the suggestion that the alleles could actually be clustered into a dozen functional clusters, named supertypes [32].
Experimental approaches for T-cell epitope discovery, detection & validation
A number of assays have been developed to detect and verify T-cell epitopes, and these methods have recently been described in more detail [40-42]. Each of these methods have various benefits and pitfalls and should be carefully considered with respect to the purpose of the experiment.
Stimulation in vitro or in vivo
In classical antigen discovery, T cells were stimulated to proliferate with antigens and cytokines. Proliferated T cells were cloned and used to screen COS cells expressing both the relevant HLA and one of several potential antigens in a cDNA library setup. This approach would reveal which antigen was the parent of the stimulating epitope. Other, very tedious, steps could be included in order to reveal the actual epitope. T-cell cultures could also be stimulated by bare peptides; however, this procedure imposes the question of whether the T cells will react to cells that have processed the parent antigen [40]. Lack of recognition could be due to the fact that the native antigen might not be processed correctly, and such peptides are referred to as cryptic. Today, the concept of being cryptic is often interchanged with being subdominant. Epitope dominance is the concept that only certain peptides from a given antigen/pathogen will be epitopes in a given individual (dominant epitopes), but in cases where the dominant epitopes are absent from the antigen, other peptides will now be able to induce an immune response [43]. This phenomenon has not been pinpointed as being caused by any single or few events but is most probably a result of competition on several or all steps in the immunogenic pathway [44]. These steps includes expression level, stability of the antigen, all the different steps in the given presentation pathway and finally the TCR affinity towards the MHC–peptide complex [43,45]. Even if subdominant epitopes are not immunogenic determinants in the native antigen or pathogen context, it is often possible to detect a strong recall response against the native antigen using T-cells stimulated with the subdominant epitope either in vitro or in vivo.
Instead of in vitro stimulation of T cells, it has been shown to be possible to use peripheral blood mononuclear cells (PBMCs) consisting of T cells, B cells, NK cells, basophils and DCs. If taken from an individual (animal or human) that has been exposed to a given antigen, either by vaccination or natural infection, it is possible to detect a T-cell response when the PBMC is stimulated with the peptides containing the right epitopes. This response can be detected by the amount of released cytokines (e.g., IFN-γ) in ELISA, enzyme-linked immunosorbent spot assays or by intracellular staining. All three methods demand that antibody staining reagents have been developed for detection of the given cytokine. This has been achieved for mice and men, but for many animals the reagents are not readily available, if at all. The first two methods can easily be performed in multiwell plates, but have the disadvantage that additional extensions have to be introduced in order to be able to distinguish between responses from different cell types, such as CD4+ and CD8+ T cells. By use of intracellular staining in combination with flow cytometry it is possible to simultaneously detect the activation state and cell type of a given cell. This can in principle be done in a high-throughput (HTP) system; however, the preparation of the collected data still needs a large degree of human intervention. Using PBMCs from infected (previous or current) or vaccinated individuals will lead to the discovery of basically only dominant epitopes. In order to optimally discover subdominant epitopes by purely experimental methods, several rounds of immunizations have to be performed with new antigens in which the dominant epitopes are no longer present.
However, subdominant epitopes might be important in vaccine development for several reasons. Regions containing subdominant epitopes are less influenced by selective pressure by the human immune system and therefore cover more of the pathogen strain variation, or might for other reasons turn out to be more effective for use in vaccines [44,46-48].
Peptide pools
When mapping immune responses using peptides, the binding nature of the MHC has to be considered. As described earlier, preferentially nonamer peptides will bind to MHC class I molecules, but it is possible to use longer peptides in the recall assays [41]. Often peptides 15–20 amino acids in length will be used when a full antigen, or even a full viral proteome, is to be scanned. As the binding core of both MHC class I and class II binding peptide is nine consecutive amino acids, peptides will have to overlap with at least eight amino acids in order for all nonamer binding cores to be present in at least one peptide. To lower the considerable experimental effort, the peptides are pooled in samples with up to 64 peptides per pool and prepared in a matrix system. The longer peptides often give a cytotoxic immune response even if the actual reacting peptide is usually 8–11 amino acids in length. This is most likely because the peptides will be partly digested by proteases present in the monocyte suspension, even though the origin of such protease/peptidase activity has at present not been fully identified [49]. The use of longer peptides also has the benefit that the same peptides may be used both in CTL and Th epitope mapping [41,50]. In order to identify the exact epitope, often referred to as the ‘minimal’ epitope, new shorter versions of the reactive peptide must be designed and used in additional assays, preferably without the interference of the proteolytic effects. This can, however, be a challenge, as the living cells in the assays might potentially secrete proteases, which can lead to the observation that peptides with weak binding to the relevant MHC molecules give strong CTL responses whereas peptides with more optimal binding do not. To unambiguously verify the minimal epitope, MHC–peptide binding should be tested by biochemical assays, as described in the next section. If the peptide can in fact bind to the restricting MHC protein, a strong method of verification of the minimal epitope is to create MHC–peptide multimers in order to detect T cells with a compatible TCR. The concept has recently been reviewed [51]. The MHC-multimer is prepared with synthetic peptides of interest that are able to bind the MHC and are subsequently used as markers in flow cytometric assays. MHC multimers are ideal to verify whether the T cells of interest recognize the particular MHC–peptide complex, and to assess the amount of specific T cells. MHC-multimers are most commonly used in the original form, as tetramers [52]; however, several variants have been made with higher number MHC–peptide complexes per entity (e.g., as pentamers or dextramers [53,54], or improved functionality in the form of streptamers [55]). In the end, however, it can be hard to estimate whether a given epitope is actually presented on infected or malign cells. For this purpose, it should be possible to clone TCRs generated by artificial stimulation and tested for specificities using MHC multimers. The TCRs can then be used as markers in flow cytometric assays in order to detect cells presenting the peptide.
MHC class I–peptide binding assays
The MHC–peptide binding is the most restrictive step in the peptide presentation pathway [43]. Thus, the affinity of peptides to a given MHC is a strong indication of whether the peptide is a potential epitope or not. For this reason, a number of biochemical assays have been developed in order to determine the MHC–peptide affinity. A classic method is a competition assay where a usually radioactively labeled reference peptide is bound to the MHC. The labeled peptide is then competed out with increasing concentrations of the peptide in order to estimate the IC50 value, which, depending on the reference peptide affinity, is considered to be close to the constant of dissociation (Kd). Several HTP affinity assays have been developed, spanning from spin column-based gel filtration, over ELISA, to highly automated proximity assays [56]. The automatization of the measurement of MHC–peptide binding has been an important factor leading to the fact that now more than 100,000 different MHC–peptide affinities have been measured and are available from the IEDB.
Predictions of T-cell epitopes
The above-described experimental methods are all relatively resource intensive even using the newest HTP methods, especially if several HLA types must be considered. The cost of synthetic peptides alone are still in the order of US$10–100 per peptide dependent on purity, and with a standard protein length of 200–300 amino acid residues, the price of a complete protein scan is still significant and a full scan of all potential antigens from more complex organisms such as bacteria and large viruses is not presently feasible, even for a single HLA type.
Prediction of peptide binding to MHC class I
Because of the amount of resources demanded for complete epitope scans, in silico predictions of T-cell epitopes is of major interest in epitope discovery. Here, it is worth mentioning that not only is it a necessity for a peptide to be able to bind an MHC to become an epitope, it is also the most restrictive step in the pathway from native protein to an immune response. On average, only one in 200 of the possible peptides that can be generated from a native protein will be able to bind a given MHC class I protein [43]. Even though these numbers are more uncertain regarding MHC class II, in both cases, specific positions in the binding peptide are responsible for the majority of the binding affinity, making MHC–peptide binding an interesting subject for development of prediction methods [14,35,57,58]. Any prediction method is dependent on data, at least for evaluation, although some approaches, such as structure-based methods using knowledge of docking and molecular interactions [59-61], have been developed using only very few measurements of MHC–peptide binding. The most accurate prediction methods, however, are data-driven and depend on large amounts of binding data in order to be able to develop various prediction models. For these methods, it has been a clear observation that the more data, the more accurate the predictions. Pre-experimental use of, and filtering by, successful prediction methods naturally greatly improve the success rate in experimental assays as more of the tested peptides will turn out to be binders. However, as the information that a peptide is able to bind a given MHC is much more valuable in prediction development than a nonbinder, this increase in binder identification has further improved the prediction methods. Thus, very beneficial scientific collaborations generating iterative assay–prediction–assay loops have been practiced between experimentalists, assay developers and bioinformatics prediction developers, which have further sped up the process of developing accurate prediction methods [62]. As the price of synthetic peptides is not insignificant, only peptides predicted to bind to an MHC will be synthesized and tested. This iterative process has an inherent risk of developing predictors that are only able to predict a limited part of the potential MHC binders. In the next iteration of training the newly measured data will be used and only in the cases were the predictions are false will the system be given new information in the next iteration. False predictions will then always be a predicted binder that turned out to be a nonbinder. The complementary situation will not occur as predicted nonbinders will never be tested. In order to at least partly circumvent this problem, methods have been developed to assess the confidence of the predictions. These measures have then been used to select peptides believed to be in the periphery of the known space. The affinity of such peptides is then measured and fed back into the loop [62]. This method will not guarantee that submotifs very different from those already estimated will not be missed. Thus, full peptide scanning of various antigens of interest is still of major importance in order to get unbiased inputs into the development cycle. Full antigen scans will also be important in order to get an unbiased validation of the prediction methods.
As described previously in the text, the HLA molecules are highly polymorphic and the issue of identifying which peptides bind any given HLA is important in order to understand how different individuals and populations react to certain stimuli. Thus, in principle, the whole peptide identification, binding measurement and prediction cycle should start from scratch for each new MHC allele, of which thousands of different alleles are known at present [34]. Furthermore, MHC–peptide binding assays have been broadened to include most of the more common HLA types in human as well as MHCs from the most used strains of inbred mice. As described, a HLA supertype consists of a number of HLA-encoded proteins binding basically the same peptides. However, how to cluster the HLAs and how to score the overlap in peptide binding space is not a trivial task, indicated by the fact that a number of different methods have been used for the purpose [32,63-66]. Naturally, the smaller the sequence distance between two proteins, the bigger the chance that they will bind the same peptides, but many exceptions have been observed, and even for two HLAs that have a high degree of homology, a significant number of the peptides that will bind to one protein might not bind to the other. This can be exemplified with the two very similar HLAs, HLA-A*30:01 and HLA-A*30:02. According to the definitions of Sidney et al. these could both belong to the A1 supertype [32]. However, HLA-A*30:01 are also assigned to the A3 supertype, which is confirmed by the work of Lamberth et al. [67]. Using the web accessible tool, MHC Motif Viewer [68], it is also apparent that the two HLAs have considerable differences in their peptide binding motif (Figure 2). These distinct binding patterns have been verified using peptides parented by a Mycobacterium tuberculosis antigen [69].
Figure 2. The Kullback–Leibler binding peptide sequence logo for the HLA-A*30:01 (A) and HLA-A*30:02 (B) alleles is generated using the MHC Motif Viewer website.
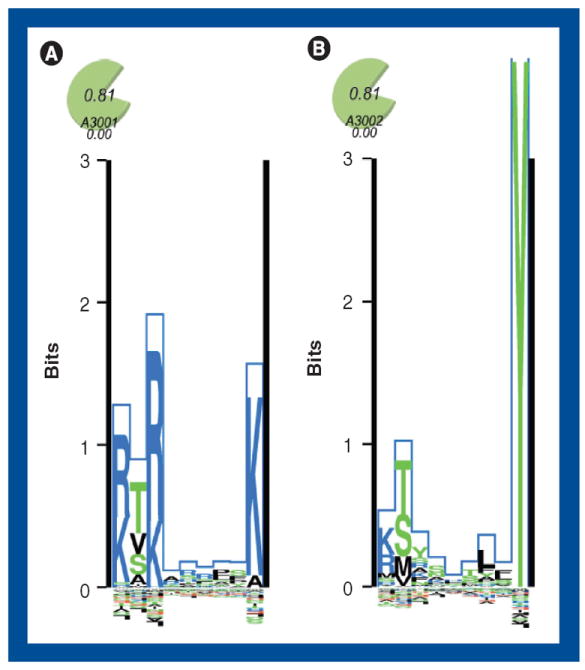
The Kullback–Leibler information content is plotted along the nonamer. Amino acids with positive influence on binding are plotted on the positive y-axis, and amino acids with a negative influence on binding are plotted on the negative y-axis. The height of each amino acid is determined by their relative contribution to the binding specificity.
One of the most complicated parts in MHC–peptide binding assays is to get folded or refoldable MHC proteins expressed from a large number of different alleles, and one of the more expensive parts is the synthesis of peptides. However, it has been a relatively inexpensive and uncomplicated task to test peptides already synthesized, for their ability to bind to several of the MHC proteins available. This synergy has enhanced the quantity of available affinity data for each HLA allele significantly as peptides synthesized with the assumption of being able to bind a specific HLA allele have been measured by testing them against a large number of different MHC proteins. These kinds of data have been of great value in developing prediction systems because peptides not necessarily predicted to bind to a given MHC nevertheless sometimes turn out to be able do so. The data are important for the iteration loops because they limit the risk of only being able to predict a part of the possible motif space. Furthermore, having peptide binding data for a large diversity of different peptides to a high diversity of MHCs has made it possible to create prediction systems that are able to predict not only whether new peptides will bind to previously characterized MHCs, but also predict the affinity of new peptides to new MHCs for which no peptide affinity data are yet available [70,71]. Where the single allele predictors are trained to deduce a binding motif from a large number of peptides known to bind to a given MHC, the most successful pan-allele predictors have been trained to predict the affinity of a given MHC–peptide combination. The amino acid sequence information from the full binding peptide is combined with the amino acid information regarding the residues in the MHC sequence known to influence binding so as to be used as input for the training (Figure 1) [58,72,73]. With these systems, accurate predictions can now be made for basically all HLA class I proteins and for several animal class I MHCs as well, including those of pigs, mice and nonhuman primates. Predictions of peptide binding to most HLA class I and chimpanzee MHC class I variants are at present very accurate. The errors between measured and predicted affinities are generally very close to the difference in the measured affinity using two slightly different binding assays [57,70,74]. The pan-specific method often outcompetes the single allele trained methods, and a combination of the two is generally even better. For this reason, a new web server has been developed that will automatically decide the optimal combination of predictions to use [75]. Concerns have been raised that available MHC–peptide binding prediction methods can only be used to predict binding of peptides eight, nine and maybe ten amino acid residues in length [31,76]. However, methods have been developed that are able to predict longer peptides [77] and these methods have been included in a number of different prediction schemes [74,78,79]. The length approximation method has currently been benchmarked to perform well on peptides ten and 11 amino acids in length, which are the lengths that have enough available binding data to carry out statistically robust validations. However, the method can, in principle, also be used for longer epitopes. Both proteasomal cleavage and TAP binding can be predicted, and integrated prediction systems have been developed (for more information, see [15,58,80]). However, even though the improvement of prediction accuracy regarding verified MHC ligands has been statistically significant, the impact seems to be marginal compared with MHC binding predictions alone [79]. This fact is probably linked to the coevolutionary effect on connected steps in the pathway [81,82].
Prediction of Th epitopes
For MHC class II binding, it is inherently harder to go from peptide binding data to a defined motif of the binding core as this is a continuous stretch of nine amino acid residues placed somewhere in a larger peptide usually in the range of 12–20 residues in length (Figure 1B). Furthermore, MHC class II proteins consists of two chains and both chains participates in peptide binding. Also, in the case of HLA-DQ and -DP, both chains contains polymorphic residues. Whereas the issue regarding defining the binding core is mainly a theoretical problem that can in principle be deduced from large sets of binding data, MHC class II is also an experimental challenge as it has turned out to be much harder to do in vitro folding of recombinantly expressed MHC class II. Thus, at present, we do not have the same degree of HTP biochemical assays for MHC class II proteins as for MHC class I, and far fewer different MHC class II proteins have been investigated in relation to peptide binding. As mentioned before, placing the binding core correctly is a challenge; thus, not only do we have far less binding data available for MHC class II protein–peptide binding, but we do not get the same improvement in prediction accuracy per data point because of this higher degree of uncertainty. Since the first prediction methods on MHC class II binding, the fields have developed slowly compared with MHC class I. However, at present, validated accurate predictions of peptide binding to several HLA-DR alleles, and some HLA-DQ and -DP alleles, are available [83].
Moreover, for MHC class II peptide binding, pan-MHC prediction methods have been developed, enabling predictions for basically any mammalian MHC class II protein. However, the predictions of nonhuman mammal MHCs have only been validated for a very limited number of alleles owing to lack of data [84]. Even though it is possible to create class II multimers, it has turned out to be technically more complicated to produce refoldable MHC class II recombinantly [85,86].
Accuracy of in silico methods
The HLA class I binding predictions for some alleles have for some time been very accurate, as exemplified with a test on predicted HLA binding peptides from the SARS proteome. Here, 86–93% of the tested peptides turned out to be binders for alleles for which a mature prediction system aleady existed [87]. Some of the methods have recently been compared in an open competition [88], where it was clearly shown that basically all newer prediction methods are better than the two pioneer methods BIMAS [89] and SYFPEITHI [90], which, despite this fact, are still widely used in epitope discovery. Hopefully, these kinds of competitions will continue and develop to concern not only MHC class I binding but also class II binding and actual epitope discovery success rates. Based on accuracy, speed and consistent availability, we generally recommend the tools listed in Table 1.
Table 1.
Recommended MHC binding prediction tools†.
In terms of the accuracy of epitope prediction, the validation is more complicated, as what is immunogenic in one might not be in another, for various reasons. Thus, the more individuals that are tested, the higher the chance of getting a positive response, provided the peptide is immunogenic at all. In an experiment where highly conserved influenza peptides were tested in individuals supposed to have had an infection within the last 3–7 years we found that 10% of the binding peptides could elicit a CTL recall response [91]. However, the time span from the immunogenic event to detection has a major influence on the success rate, as exemplified by the attempt to detect CTL responses more than 30 years post-vaccinia virus vaccination [92]. Here, only 6% of the predicted binders were able to induce a recall response. The state of an infection is also important, as in acute infections the hit rate has been shown to be much higher. That prediction methods are strong prefiltering tools has been shown in an attempt to discover all possible epitopes in vaccinia in mice after vaccination [93]. In this scenario, only 1% of peptides were selected based on MHC class I binding predictions, and 95% of the immunogenicity could be explained by 50 peptides within this selection.
Prediction of peptide binding to animal MHCs
Whereas we have extensive knowledge of the HLAs and MHCs of the most animal models used in research (i.e., mice and chimpanzees), we lack fundamental knowledge of many other animal species. Some genetic variations are known for important animals in food production, but the variation and differences in these genes between mammals are extensive. Rhesus monkeys, for example, have several MHC class I loci that individually are less variable than HLAs, but turn out not to all be expressed in a given individual. Also, MHC class II genes show species differences, as in cattle and goat it seems that they are analogs to the human HLA-DRB3 genes that display the most extensive polymorphism. Furthermore, MHC binding assays and epitope discovery tools are still only developed for very few species, including pig and cattle. However, despite the limited possibility of thorough benchmarking, it has been shown that MHC class II predictions can to some extent be applied to cattle [94], and pan predictors have been able to predict peptide binding to pig MHC class I [95].
Expert commentary
In this article we have described a number of methods used for T-cell epitope discovery. At present, HTP ex vivo assays do not have the sensitivity of the more elaborate assay types, and it is not feasible to make a full peptide proteome scan for an extensive number of HLA alleles, even for relatively simple pathogens. Furthermore, by use of pure assay-based methods it is a challenge to identify the subdominant epitopes that might be crucial in prevention or treatment of certain diseases. On the other hand, in silico models can help identify potential subdominant epitopes. Using in silico methods in the primary selection of peptides can successfully be combined with other selection criteria, such as conservation state of the potential epitope. In the previously mentioned example of discovery of CTL epitopes induced by influenza A H1N1 infections, this led to the discovery of 13 epitopes, of which 11 were fully conserved in the H5N1 avian strain that has been responsible for several fatal infections in humans [91] and all were conserved in the recent H1N1 influenza pandemic. The selection schemes have been further developed to include strain variability, and also to benefit from predictions of all alleles of a given population and include the allele frequencies obtained from databases. In this way, a pool of peptides has been predicted that should optimally cover all HIV strains and most of the human population. In such experiments, we see that up to 70% of the selected peptides will elicit a response in at least one individual [96,97]. This approach will take over from the previously described supertype approach as it will be more sensitive and accurate, and is better suited for taking different populations into consideration.
Epitope prediction systems will probably never be able to detect all the possible epitopes in a given setup. However, some experimentalists seem to neglect that experimental assays are also only accurate to a certain sensitivity and specificity, which is in fact the general condition for all natural sciences. It has been argued that prediction systems are in general not sensitive enough and that resources are better spent on thorough HTP scans [76,98]. The fact that there are no valid data that go against the generally reported high accuracy of MHC–peptide binding predictions has been argued elsewhere [33]. Even though new methods have been developed that are able to predict binding of longer peptides, the Burrows paper [31] is still put forward as an argument against prediction methods [76]. It is our strong belief that it is possible to successfully predict a large majority, if not all of the 25 longer epitopes mentioned in the paper by Burrows et al. Furthermore, Wu et al. argue that some newly identified epitopes cannot be predicted based on the claim that they do not contain the correct motif [76]. It is, however, possible to test for peptides 8–11 amino acids in length using existing tools such as HLArestrictor [82] and NetMHCcons [101]. We strongly belief that the use of these tools will show that a majority of the epitopes claimed not to be predictable will turn out to be so.
In the end, it is our opinion that it is not a choice between two scenarios where, in one scenario, prediction methods lead to the end of experimentally based assay procedures, or, in another scenario, of experimental puritanism in which every step in an epitope discovery effort has to be based on positive results in experimentally based assays alone, thereby excluding the involvement of theoretical-based selection. It is our strong belief that a concerted effort in a nonhostile arms race of the development of assay procedures and computational modeling will lead to mutual benefits where each side will strongly benefit on the progress of the other.
Five-year view
As described in the previous sections, the field of T-cell epitope prediction is expected to develop significantly within the next few years. The amount of data for MHC class II–peptide binding will most likely increase significantly, leading to better prediction systems for MHC binding. Also very likely is that we will see accurate predictions not only for all HLA-DR alleles, but also for HLA-DQ and -DP alleles, and probably for MHC class II proteins from several other mammals as well. A new possibility for HTP assays has been developed in recent years that will potentially have a great impact on the amount of available MHC class II–peptide affinity data. Peptides spotted or synthesized in microarrays can be used to measure a very high number of different peptides in one go. The micro-array peptide chip technology is maturing now and will be very suitable for such multipeptide assays in the future, especially for MHC class II because of the open ends of the peptide binding groove enabling the MHC to bind even though the peptide is immobilized in one end.
The newly discovered involvement of MHC class I pathway elements in presentation of MHC class II restricted peptides will certainly be investigated and might improve significantly on Th epitope predictions [12]. The MHC class I–peptide binding prediction systems will be highly accurate for most mammals, and it is very likely that usable prediction systems will be available for several bird alleles as well. The development of direct epitope prediction systems will move slower because of the resources needed in generating the data and the complexity involved in discriminating immunogenic MHC binding from nonimmunogenic MHC binders. The effect of MHC–peptide stability measurements and predictions of this will be an important integration in prediction of T-cell epitopes. New and improved knowledge of TCRs, positive and negative selection in the development of TCR specificity, and the interactions between the TCR and the MHC–peptide complex will also be obtained as a result of recent discoveries, combined with the new and emerging, fast and cheap sequencing methods. Such knowledge will undoubtedly have a significant impact on the modeling of usable true Th and CTL immune response models already at the end of the coming 5-year period. As data increase and prediction methods develop, a much greater understanding will emerge on what defines a dominant T-cell epitope. We believe that we will observe a preliminary HLA hierarchy that will explain some of the dominance effects that we see. The understanding of dominance will probably further increase when we gain knowledge of how specific TCR germline genes influence the preferences of the TCR for specific MHC–peptide complexes. Likewise, evidence is emerging that suggests that the integration of signals from other immune system receptors, such as KIR, has a strong impact on our T-cell immunity and epitope dominance [99]. Recently, a new accurate MHC–peptide stability HTP assay has been described [100], and it will be interesting to follow in the coming years whether stability will be an even better indicator of peptide immunity than affinity.
The first field where selected epitopes will have a clinical impact is probably in cancer treatments based on specific selected T-cell epitopes. Several trials are already initiated for various cancer types (for more information see [102-104]). These epitopes are often identified using one or more in silico methods combined with more traditional assay-driven discovery methods. Within traditional vaccine development, it is our impression that several developers, especially smaller startup companies, are using prediction methods as a major step in epitope discovery to design vaccines with specially selected epitopes in order to secure and improve population coverage. The first designed vaccines on the market are expected to be an extension of the subunit vaccine approach extended with selected T-cell epitopes either inserted into the selected subunits or as individual peptides. Since these are still in the early development phase we will probably not see this kind of vaccines for human use on the market until 7–10 years from now at the earliest. However, for veterinary vaccines, the rational vaccine design approach might go much faster for the species in which detection protocols are established and the MHCs are known. We may even see vaccines on the market already at the end of the next 5-year period.
The development of computational methods in combination with general obtained biological knowledge will lead to the possibility of highly personalized treatments. Such treatments might be seen in trials against otherwise untreatable serious diseases within a 5-year period and most certainly within the next 10 years.
Key issues.
Full genome sequences of pathogens and somatic cells are available in ever-increasing amounts.
Traditional T-cell epitope discovery methods are resource intensive despite development of intelligent assay methods and high-throughput systems.
A number of T-cell epitope discovery methods are dependent on biological materials (e.g., patient blood samples), which are usually available in limiting amounts.
High polymorphy of MHCs and length variation of reacting peptides further increase the number of experiments needed for traditional T-cell discovery.
Accurate MHC binding prediction methods exists for all HLA class I and several mammal MHC class I proteins, for all HLA-DR proteins and several other MHC class II proteins.
MHC class I binding predictions can be obtained even for peptides with an unusual epitope length.
The use of MHC peptide binding predictions highly improve the success rates in epitope discovery and are easily integrated with, for example, sequence variation analysis.
The combination of in silico, in vitro and ex vivo methods will, in the near future, be the optimal setup in epitope discovery.
Predictions and mathematical models will not fully replace experimental methods.
Acknowledgments
The authors are supported by NIH/NIAID, contract # HHSN2662004000 06C and contract #HHSN266200400025C.
Footnotes
Financial & competing interests disclosure
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
-
•
of interest
-
••
of considerable interest
- 1.Pulendran B, Ahmed R. Immunological mechanisms of vaccination. Nat Immunol. 2011;12:509–517. doi: 10.1038/ni.2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Flinsenberg TW, Compeer EB, Boelens JJ, Boes M. Antigen cross-presentation: extending recent laboratory findings to therapeutic intervention. Clin Exp Immunol. 2011;165:8–18. doi: 10.1111/j.1365-2249.2011.04411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang J, Honda W. CED: a conformational epitope database. BMC Immunol. 2006;7:7. doi: 10.1186/1471-2172-7-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sette A, Peters B. Immune epitope mapping in the post-genomic era: lessons for vaccine development. Curr Opin Immunol. 2007;19:106–110. doi: 10.1016/j.coi.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 5.Ottenhoff TH, Doherty TM, van Dissel JT, et al. First in humans: a new molecularly defined vaccine shows excellent safety and strong induction of long-lived Mycobacterium tuberculosis-specific Th1-cell like responses. Hum Vaccin. 2010;6:1007–1015. doi: 10.4161/hv.6.12.13143. [DOI] [PubMed] [Google Scholar]
- 6.Söllner J, Heinzel A, Summer G, et al. Concept and application of a computational vaccinology workflow. Immunome Res. 2010;6(Suppl 2):S7. doi: 10.1186/1745-7580-6-S2-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hansel TT, Kropshofer H, Singer T, Mitchell JA, George AJ. The safety and side effects of monoclonal antibodies. Nat Rev Drug Discov. 2010;9:325–338. doi: 10.1038/nrd3003. [DOI] [PubMed] [Google Scholar]
- 8.Cantor JR, Yoo TH, Dixit A, Iverson BL, Forsthuber TG, Georgiou G. Therapeutic enzyme deimmunization by combinatorial T-cell epitope removal using neutral drift. Proc Natl Acad Sci USA. 2011;108:1272–1277. doi: 10.1073/pnas.1014739108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Star B, Nederbragt AJ, Jentoft S, et al. The genome sequence of Atlantic cod reveals a unique immune system. Nature. 2011;477:207–210. doi: 10.1038/nature10342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Del Val M, Iborra S, Ramos M, Lázaro S. Generation of MHC class 1 ligands in the secretory and vesicular pathways. Cell Mol Life Sci. 2011;68:1543–1552. doi: 10.1007/s00018-011-0661-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mester G, Hoffmann V, Stevanovi S. Insights into MHC class 1 antigen processing gained from large-scale analysis of class 1 ligands. Cell Mol Life Sci. 2011;68:1521–1532. doi: 10.1007/s00018-011-0659-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dragovic SM, Hill T, Christianson GJ, et al. Proteasomes, TAP, and endoplasmic reticulum-associated aminopeptidase associated with antigen processing control CD4+ Th cell responses by regulating indirect presentation of MHC class 2-restricted cytoplasmic antigens. J Immunol. 2011;186(12):6683–6692. doi: 10.4049/jimmunol.1100525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sijts EJ, Kloetzel PM. The role of the proteasome in the generation of MHC class 1 ligands and immune responses. Cell Mol Life Sci. 2011;68:1491–1502. doi: 10.1007/s00018-011-0657-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lundegaard C, Lund O, Kesmir C, Brunak S, Nielsen M. Modeling the adaptive immune system: predictions and simulations. Bioinformatics. 2007;23:3265–3275. doi: 10.1093/bioinformatics/btm471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Diez-Rivero CM, Lafuente EM, Reche PA. Computational analysis and modeling of cleavage by the immunoproteasome and the constitutive proteasome. BMC Bioinformatics. 2010;11:479. doi: 10.1186/1471-2105-11-479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roder G, Geironson L, Darabi A, et al. The outermost N-terminal region of tapasin facilitates folding of major histocompatibility complex class 1. Eur J Immunol. 2009;39:2682–2694. doi: 10.1002/eji.200939364. [DOI] [PubMed] [Google Scholar]
- 17.Roder G, Geironson L, Rasmussen M, Harndahl M, Buus S, Paulsson KM. Tapasin discriminates peptide-HLA-A*02:01 complexes formed with natural ligands. J Biol Chem. 2011 doi: 10.1074/jbc M111.230151. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van den Hoorn T, Paul P, Jongsma ML, Neefjes J. Routes to manipulate MHC class 2 antigen presentation. Curr Opin Immunol. 2011;23:88–95. doi: 10.1016/j.coi.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 19.Call MJ. Small molecule modulators of MHC class 2 antigen presentation: mechanistic insights and implications for therapeutic application. Mol Immunol. 2011;48:1735–1743. doi: 10.1016/j.molimm.2011.05.022. [DOI] [PubMed] [Google Scholar]
- 20.Watts C. The endosome–lysosome pathway and information generation in the immune system. Biochim Biophys Acta. 2011 doi: 10.1016/j.bbapap.2011.07.006. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Landsverk OJ, Bakke O, Gregers TF. MHC II and the endocytic pathway: regulation by invariant chain. Scand J Immunol. 2009;70:184–193. doi: 10.1111/j.1365-3083.2009.02301.x. [DOI] [PubMed] [Google Scholar]
- 22.Münz C. Enhancing immunity through autophagy. Ann Rev Immunol. 2009;27:423–449. doi: 10.1146/annurev.immunol.021908.132537. [DOI] [PubMed] [Google Scholar]
- 23.Zavasnik-Bergant T, Turk B. Cysteine proteases: destruction ability versus immunomodulation capacity in immune cells. Biol Chem. 2007;388:1141–1149. doi: 10.1515/BC.2007.144. [DOI] [PubMed] [Google Scholar]
- 24.Conus S, Simon HU. Cathepsins and their involvement in immune responses. Swiss Med Wkly. 2010;140:w13042. doi: 10.4414/smw.2010.13042. [DOI] [PubMed] [Google Scholar]
- 25.Jørgensen KW, Buus S, Nielsen M. Structural properties of MHC class 2 ligands, implications for the prediction of MHC class 2 epitopes. PLoS One. 2010;5:e15877. doi: 10.1371/journal.pone.0015877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Endert P. Post-proteasomal and proteasome-independent generation of MHC class 1 ligands. Cell Mol Life Sci. 2011;68:1553–1567. doi: 10.1007/s00018-011-0662-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chemali M, Radtke K, Desjardins M, English L. Alternative pathways for MHC class 1 presentation: a new function for autophagy. Cell Mol Life Sci. 2011;68:1533–1541. doi: 10.1007/s00018-011-0660-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Larsen MV, Nielsen M, Weinzierl A, Lund O. TAP-independent MHC class 1 presentation. Curr Immunol Rev. 2006;2:233–245. [Google Scholar]
- 29.Segura E, Villadangos JA. A modular and combinatorial view of the antigen cross-presentation pathway in dendritic cells. Traffic. 2011;12(12):1677–1685. doi: 10.1111/j.1600-0854.2011.01254.x. [DOI] [PubMed] [Google Scholar]
- 30.Amigorena S, Savina A. Intracellular mechanisms of antigen cross presentation in dendritic cells. Curr Opin Immunol. 2010;22:109–117. doi: 10.1016/j.coi.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 31.Burrows SR, Rossjohn J, McCluskey J. Have we cut ourselves too short in mapping CTL epitopes? Trends Immunol. 2006;27:11–16. doi: 10.1016/j.it.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 32.Sidney J, Peters B, Frahm N, Brander C, Sette A. HLA class 1 supertypes: a revised and updated classification. BMC Immunol. 2008;9:1471–2172. doi: 10.1186/1471-2172-9-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lundegaard C, Lund O, Buus S, Nielsen M. Major histocompatibility complex class 1 binding predictions as a tool in epitope discovery. Immunology. 2010;130:309–318. doi: 10.1111/j.1365-2567.2010.03300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robinson J, Mistry K, McWilliam H, Lopez R, Parham P, Marsh SG. The IMGT/HLA database. Nucleic Acids Res. 2011;39:D1171–D1176. doi: 10.1093/nar/gkq998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nielsen M, Lund O, Buus S, Lundegaard C. MHC class 2 epitope predictive algorithms. Immunology. 2010;130:319–328. doi: 10.1111/j.1365-2567.2010.03268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bordner AJ. Towards universal structure-based prediction of class 2 MHC epitopes for diverse allotypes. PLoS One. 2010;5:e14383. doi: 10.1371/journal.pone.0014383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gonzalez-Galarza FF, Christmas S, Middleton D, Jones AR. Allele frequency net: a database and online repository for immune gene frequencies in worldwide populations. Nucleic Acids Res. 2011;39:D913–D919. doi: 10.1093/nar/gkq1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sayers EW, Barrett T, Benson DA, et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2011;39:D38–D51. doi: 10.1093/nar/gkq1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vita R, Zarebski L, Greenbaum JA, et al. The immune epitope database 2.0. Nucleic Acids Res. 2010;38:D854–D862. doi: 10.1093/nar/gkp1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40•.Scharnagl NC, Klade CS. Experimental discovery of T-cell epitopes: combining the best of classical and contemporary approaches. Exp Review Vaccin. 2007;6(4):605–615. doi: 10.1586/14760584.6.4.605. Very comprehensive review of experimental T-cell epitope dscovery. [DOI] [PubMed] [Google Scholar]
- 41.LiPira G, Ivaldi F, Moretti P, Manca F. High throughput T epitope mapping and vaccine development. J Biomed Biotechnol. 2010;2010:325720. doi: 10.1155/2010/325720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hoppes R, Ekkebus R, Schumacher TN, Ovaa H. Technologies for MHC class 1 immunoproteomics. J Proteom. 2010;73:1945–1953. doi: 10.1016/j.jprot.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 43.Yewdell JW. Confronting complexity: real-world immunodominance in antiviral CD8+ T cell responses. Immunity. 2006;25:533–543. doi: 10.1016/j.immuni.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 44.Riedl P, Wieland A, Lamberth K, et al. Elimination of immunodominant epitopes from multispecific DNA-based vaccines allows induction of CD8 T cells that have a striking antiviral potential. J Immunol. 2009;183:370–380. doi: 10.4049/jimmunol.0900505. [DOI] [PubMed] [Google Scholar]
- 45.Baumgartner CK, Malherbe LP. Antigendriven T-cell repertoire selection during adaptive immune responses. Immunol Cell Biol. 2011;89:54–59. doi: 10.1038/icb.2010.117. [DOI] [PubMed] [Google Scholar]
- 46.Dominguez MR, Silveira EL, de Vasconcelos JR, et al. Subdominant/cryptic CD8 T cell epitopes contribute to resistance against experimental infection with a human protozoan parasite. PLoS One. 2011;6:e22011. doi: 10.1371/journal.pone.0022011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Im EJ, Hong JP, Roshorm Y, et al. Protective efficacy of serially up-ranked subdominant CD8+ T cell epitopes against virus challenges. PLoS Pathog. 2011;7:e1002041. doi: 10.1371/journal.ppat.1002041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ruckwardt TJ, Luongo C, Malloy AM, et al. Responses against a subdominant CD8+ T cell epitope protect against immunopathology caused by a dominant epitope. J Immunol. 2010;185:4673–4680. doi: 10.4049/jimmunol.1001606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kiecker F, Streitz M, Ay B, et al. Analysis of antigen-specific T-cell responses with synthetic peptides –what kind of peptide for which purpose? Hum Immunol. 2004;65:523–536. doi: 10.1016/j.humimm.2004.02.017. [DOI] [PubMed] [Google Scholar]
- 50.Zandvliet ML, van Liempt E, Jedema I, et al. Simultaneous isolation of CD8(+) and CD4(+) T cells specific for multiple viruses for broad antiviral immune reconstitution after allogeneic stem cell transplantation. J Immunother. 2011;34:307–319. doi: 10.1097/CJI.0b013e318213cb90. [DOI] [PubMed] [Google Scholar]
- 51.Casalegno-Garduño R, Schmitt A, Yao J, et al. Multimer technologies for detection and adoptive transfer of antigen-specific T cells. Cancer Immunol Immunother. 2010;59:195–202. doi: 10.1007/s00262-009-0778-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52•.Leisner C, Loeth N, Lamberth K, et al. One-pot, mix-and-read peptide–MHC tetramers. PLoS One. 2008;3:e1678. doi: 10.1371/journal.pone.0001678. Description of easy and reproducible production of MHC–peptide tetramers. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Burrows JM, Wynn KK, Tynan FE, et al. The impact of HLA-B micropolymorphism outside primary peptide anchor pockets on the CTL response to CMV. Eur J Immunol. 2007;37:946–953. doi: 10.1002/eji.200636588. [DOI] [PubMed] [Google Scholar]
- 54.Schøller J, Singh M, Bergmeier L, et al. A recombinant human HLA-class 1 antigen linked to dextran elicits innate and adaptive immune responses. J Immunol Methods. 2010;360:1–9. doi: 10.1016/j.jim.2010.05.008. [DOI] [PubMed] [Google Scholar]
- 55.Neudorfer J, Schmidt B, Huster KM, et al. Reversible HLA multimers (streptamers) for the isolation of human cytotoxic T lymphocytes functionally active against tumor- and virus-derived antigens. J Immunol Methods. 2007;320:119–131. doi: 10.1016/j.jim.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 56.Harndahl M, Justesen S, Lamberth K, Røder G, Nielsen M, Buus S. Peptide binding to HLA class 1 molecules: homogenous, high-throughput screening, and affinity assays. J Biomol Screen. 2009;14:173–180. doi: 10.1177/1087057108329453. [DOI] [PubMed] [Google Scholar]
- 57.Peters B, Bui HH, Frankild S, et al. A community resource benchmarking predictions of peptide binding to MHC-I molecules. PLoS Comput Biol. 2006;2:e65. doi: 10.1371/journal.pcbi.0020065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lundegaard C, Hoof I, Lund O, Nielsen M. State of the art and challenges in sequence based T-cell epitope prediction. Immunome Res. 2010;6(Suppl 2):S3. doi: 10.1186/1745-7580-6-S2-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang P, Sidney J, Dow C, Mothé B, Sette A, Peters B. A systematic assessment of MHC class 2 peptide binding predictions and evaluation of a consensus approach. PLoS Comput Biol. 2008;4:e1000048. doi: 10.1371/journal.pcbi.1000048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bordner AJ, Abagyan R. Ab initio prediction of peptide–MHC binding geometry for diverse class 1 MHC allotypes. Proteins. 2006;63:512–526. doi: 10.1002/prot.20831. [DOI] [PubMed] [Google Scholar]
- 61.Zhang H, Wang P, Papangelopoulos N, et al. Limitations of Ab initio predictions of peptide binding to MHC class 2 molecules. PLoS One. 2010;5:e9272. doi: 10.1371/journal.pone.0009272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lundegaard C, Lund O, Nielsen M. Prediction of epitopes using neural network based methods. J Immunol Methods. 2010;374(1-2):26–34. doi: 10.1016/j.jim.2010.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Reche PA, Reinherz EL. Definition of MHC supertypes through clustering of MHC peptide-binding repertoires. Methods Mol Biol. 2007;409:163–173. doi: 10.1007/978-1-60327-118-9_11. [DOI] [PubMed] [Google Scholar]
- 64.Doytchinova IA, Guan P, Flower DR. Identifiying human MHC supertypes using bioinformatic methods. J Immunol. 2004;172:4314–4323. doi: 10.4049/jimmunol.172.7.4314. [DOI] [PubMed] [Google Scholar]
- 65.Heckerman D, Kadie C, Listgarten J. Leveraging information across HLA alleles/supertypes improves epitope prediction. J Comput Biol. 2007;14:736–746. doi: 10.1089/cmb.2007.R013. [DOI] [PubMed] [Google Scholar]
- 66.Hertz T, Yanover C. Identifying HLA supertypes by learning distance functions. Bioinformatics. 2007;23:e148–e155. doi: 10.1093/Bioinformatics/btl324. [DOI] [PubMed] [Google Scholar]
- 67.Lamberth K, Røder G, Harndahl M, et al. The peptide-binding specificity of HLA-A*3001 demonstrates membership of the HLA-A3 supertype. Immunogenetics. 2008;60:633–643. doi: 10.1007/s00251-008-0317-z. [DOI] [PubMed] [Google Scholar]
- 68.Rapin N, Hoof I, Lund O, Nielsen M. MHC motif viewer. Immunogenetics. 2008;60:759–765. doi: 10.1007/s00251-008-0330-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Axelsson-Robertson R, Ahmed RK, Weichold FF, et al. Human leukocyte antigens A*3001 and A*3002 show distinct peptide-binding patterns of the Mycobacterium tuberculosis protein TB10.4: consequences for immune recognition. Clin Vaccin Immunol. 2011;18:125–134. doi: 10.1128/CVI.00302-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang H, Lundegaard C, Nielsen M. Pan-specific MHC class 1 predictors: a benchmark of HLA class 1 pan-specific prediction methods. Bioinformatics. 2009;25:83–89. doi: 10.1093/bioinformatics/btn579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang L, Udaka K, Mamitsuka H, Zhu S. Toward more accurate pan-specific MHC–peptide binding prediction: a review of current methods and tools. Brief Bioinform. 2011 doi: 10.1093/bib/bbr060. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 72.Hoof I, Peters B, Sidney J, et al. NetMHCpan, a method for MHC class 1 binding prediction beyond humans. Immunogenetics. 2009;61:1–13. doi: 10.1007/s00251-008-0341-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nielsen M, Lundegaard C, Blicher T, et al. NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PLoS One. 2007;2:e796. doi: 10.1371/journal.pone.0000796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hoof I, Pérez CL, Buggert M, et al. Interdisciplinary analysis of HIV-specific CD8+ T cell responses against variant epitopes reveals restricted TCR promiscuity. J Immunol. 2010;184(9):5383–5391. doi: 10.4049/jimmunol.0903516. [DOI] [PubMed] [Google Scholar]
- 75.Karosiene E, Lundegaard C, Lund O, Nielsen M. NetMHCcons: a consensus method for the major histocompatibility complex class I predictions. Immunogenetics. 2011 doi: 10.1007/s00251-011-0579-8. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 76.Wu C, Zanker D, Valkenburg S, et al. Systematic identification of immunodominant CD8+ T-cell responses to influenza A virus in HLA-A2 individuals. Proc Natl Acad Sci USA. 2011 doi: 10.1073/pnas.1105624108. doi:0.1073/pnas.1105624108. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lundegaard C, Lund O, Nielsen M. Accurate approximation method for prediction of class 1 MHC affinities for peptides of length 8, 10 and 11 using prediction tools trained on 9mers. Bioinformatics. 2008;24:1397–1398. doi: 10.1093/bioinformatics/btn128. [DOI] [PubMed] [Google Scholar]
- 78.Erup Larsen M, Kloverpris H, Stryhn A, et al. HLArestrictor – a tool for patient-specific predictions of HLA restriction elements and optimal epitopes within peptides. Immunogenetics. 2011;63:43–55. doi: 10.1007/s00251-010-0493-5. [DOI] [PubMed] [Google Scholar]
- 79.Stranzl T, Larsen MV, Lundegaard C, Nielsen M. NetCTLpan: pan-specific MHC class 1 pathway epitope predictions. Immunogenetics. 2010;62:357–368. doi: 10.1007/s00251-010-0441-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Diez-Rivero CM, Chenlo B, Zuluaga P, Reche PA. Quantitative modeling of peptide binding to TAP using support vector machine. Proteins. 2010;78:63–72. doi: 10.1002/prot.22535. [DOI] [PubMed] [Google Scholar]
- 81.Schmid BV, Kesmir C, de Boer RJ. The specificity and polymorphism of the MHC class 1 prevents the global adaptation of HIV-1 to the monomorphic proteasome and TAP. PLoS One. 2008;3:e3525. doi: 10.1371/journal.pone.0003525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nielsen M, Lundegaard C, Lund O, Kesmir C. The role of the proteasome in generating cytotoxic T-cell epitopes: insights obtained from improved predictions of proteasomal cleavage. Immunogenetics. 2005;57:33–41. doi: 10.1007/s00251-005-0781-7. [DOI] [PubMed] [Google Scholar]
- 83.Wang P, Sidney J, Kim Y, et al. Peptide binding predictions for HLA DR, DP and DQ molecules. BMC Bioinform. 2010;11:568. doi: 10.1186/1471-2105-11-568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nielsen M, Justesen S, Lund O, Lundegaard C, Buus S. NetMHCIIpan-2.0 – improved pan-specific HLA-DR predictions using a novel concurrent alignment and weight optimization training procedure. Immunome Res. 2010;6:9. doi: 10.1186/1745-7580-6-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vollers SS, Stern LJ. Class 2 major histocompatibility complex tetramer staining: progress, problems, and prospects. Immunology. 2008;123:305–313. doi: 10.1111/j.1365-2567.2007.02801.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.James EA, LaFond R, Durinovic-Bello I, Kwok W. Visualizing antigen specific CD4+ T cells using MHC class 2 tetramers. J Vis Exp. 2009;6(25):1167. doi: 10.3791/1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sylvester-Hvid C, Nielsen M, Lamberth K, et al. SARS CTL vaccine candidates; HLA supertype-, genome-wide scanning and biochemical validation. Tis Antigens. 2004;63:395–400. doi: 10.1111/j.0001-2815.2004.00221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88••.Zhang GL, Ansari HR, Bradley P, et al. Machine learning competition in immunology – prediction of HLA class 1 molecules. J Immunol Methods. 2011;374(1-2):1–4. doi: 10.1016/j.jim.2011.09.010. First attempt at independent assessment of MHC class I binding prediction methods. [DOI] [PubMed] [Google Scholar]
- 89.Parker KC, Bednarek MA, Coligan JE. Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. J Immunol. 1994;152:163–175. [PubMed] [Google Scholar]
- 90.Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanovic S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics. 1999;50:213–219. doi: 10.1007/s002510050595. [DOI] [PubMed] [Google Scholar]
- 91.Wang M, Lamberth K, Harndahl M, et al. CTL epitopes for influenza A including the H5N1 bird flu; genome-, pathogen-, and HLA-wide screening. Vaccine. 2007;25:2823–2831. doi: 10.1016/j.vaccine.2006.12.038. [DOI] [PubMed] [Google Scholar]
- 92.Wang M, Tang ST, Lund O, Dziegiel MH, Buus S, Claesson MH. High-affinity human leucocyte antigen class 1 binding variola-derived peptides induce CD4+ T cell responses more than 30 years post-vaccinia virus vaccination. Clin Exp Immunol. 2009;155:441–446. doi: 10.1111/j.1365-2249.2008.03856.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93••.Moutaftsi M, Peters B, Pasquetto V, et al. A consensus epitope prediction approach identifies the breadth of murine T(CD8+)-cell responses to Vaccinia virus. Nat Biotechnol. 2006;24:817–819. doi: 10.1038/nbt1215. Shows the strength of using in silico methods in comprehensive epitope mapping. [DOI] [PubMed] [Google Scholar]
- 94.Vordermeier M, Whelan AO, Hewinson RG. Recognition of mycobacterial epitopes by T cells across mammalian species and use of a program that predicts human HLA-DR binding peptides to predict bovine epitopes. Infect Immun. 2003;71:1980–1987. doi: 10.1128/IAI.71.4.1980-1987.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pedersen LE, Harndahl M, Rasmussen M, et al. Porcine major histocompatibility complex (MHC) class 1 molecules and analysis of their peptide-binding specificities. Immunogenetics. 2011;63(12):821–834. doi: 10.1007/s00251-011-0555-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Buggert M, Norstrom M, Lundegaard C, Lund O, Nielsen M, Karlsson AC. Interdisciplinary evaluation of broadly-reactive HLA class II restricted epitopes eliciting hiv-specific CD4+T cell responses. AIDS Res Hum Retroviruses. 2011 In Press. [Google Scholar]
- 97.Perez CL, Larsen MV, Gustafsson R, et al. Broadly immunogenic HLA class 1 supertype-restricted elite CTL epitopes recognized in a diverse population infected with different HIV-1 subtypes. J Immunol. 2008;180:5092–5100. doi: 10.4049/jimmunol.180.7.5092. [DOI] [PubMed] [Google Scholar]
- 98.Gowthaman U, Agrewala JN. In silico methods for predicting T-cell epitopes: Dr Jekyll or Mr Hyde? Expert Rev Proteom. 2009;6(5):527–537. doi: 10.1586/epr.09.71. [DOI] [PubMed] [Google Scholar]
- 99.Colantonio AD, Bimber BN, Neidermyer WJ, et al. KIR polymorphisms modulate peptide-dependent binding to an MHC class 1 ligand with a Bw6 motif. PLoS Pathog. 2011;7:e1001316. doi: 10.1371/journal.ppat.1001316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100•.Harndahl M, Rasmussen M, Roder G, Buus S. Real-time, high-throughput measurements of peptide–MHC-I dissociation using a scintillation proximity assay. J Immunol Methods. 2010;374(1-2):5–12. doi: 10.1016/j.jim.2010.10.012. First high-throughput MHC–peptide stability assay. [DOI] [PMC free article] [PubMed] [Google Scholar]
Websites
- 101.NetMHCcons. www.cbs.dtu.dk/services/NetMHCcons/
- 102.Safety Study of Cancer Specific Epitope Peptides Cocktail for Cervical, GI, and Lung Tumors (peptidevac) http://clinicaltrials.gov/ct2/show/ NCT00676949.
- 103.HLA-A*0201 Restricted Peptide Vaccine Therapy With Gemcitabine With Gemcitabine in Patient Pancreatic Cancer (Phase 1) http://clinicaltrials.gov/ct2/show/NCT01266720.
- 104.Vaccine Therapy in Treating Patients With Breast Cancer. http://clinicaltrials.gov/ct2/show/NCT00524277.
- 105.PDB HLA-A*11:01 structure, 2HN7. http://dx.doi.org/10.2210/pdb2hn7/pdb.
- 106.PDB HLA-DR1 structure, 1AQD. http://dx.doi.org/10.2210/pdb1aqd/pdb.
- 107.Immune epitope database analysis tool. http://tools.immuneepitope.org.
- 108.CBS prediction servers. www.cbs.dtu.dk/services.