

Abstract
In the title molecular salt, C26H28N4 2+·2Br−, the central benzene ring makes dihedral angles of 76.75 (11) and 82.40 (10)° with the pendant benzimidazole rings. The corresponding angle between the benzimidazole rings is 57.03 (9)°. In the crystal, the cations and anions are linked via C—H⋯Br hydrogen bonds, forming sheets lying parallel to the bc plane. The crystal structure also features weak C—H⋯π interactions.
Related literature
For background to the biological activities of benzimidazole compounds, see: Mohan et al. (2011 ▶).
Experimental
Crystal data
C26H28N4 2+·2Br−
M r = 556.34
Monoclinic,
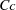
a = 9.7093 (7) Å
b = 35.796 (3) Å
c = 8.0340 (6) Å
β = 118.230 (1)°
V = 2460.1 (3) Å3
Z = 4
Mo Kα radiation
μ = 3.32 mm−1
T = 296 K
0.45 × 0.32 × 0.23 mm
Data collection
Bruker APEXII DUO CCD diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2009 ▶) T min = 0.318, T max = 0.516
16606 measured reflections
8158 independent reflections
6244 reflections with I > 2σ(I)
R int = 0.023
Refinement
R[F 2 > 2σ(F 2)] = 0.033
wR(F 2) = 0.080
S = 1.02
8158 reflections
291 parameters
2 restraints
H-atom parameters constrained
Δρmax = 0.71 e Å−3
Δρmin = −0.28 e Å−3
Absolute structure: Flack (1983 ▶), 3727 Friedel pairs
Flack parameter: 0.009 (6)
Data collection: APEX2 (Bruker, 2009 ▶); cell refinement: SAINT (Bruker, 2009 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXTL (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXTL; molecular graphics: SHELXTL; software used to prepare material for publication: SHELXTL and PLATON (Spek, 2009 ▶).
Supplementary Material
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S1600536812002802/hb6611sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536812002802/hb6611Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536812002802/hb6611Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
Cg4 is the centroid of the C11–C16 ring.
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C9—H9A⋯Br2 | 0.93 | 2.76 | 3.610 (2) | 152 |
| C10—H10A⋯Br1i | 0.97 | 2.92 | 3.884 (2) | 171 |
| C10—H10B⋯Br2 | 0.97 | 2.91 | 3.820 (2) | 156 |
| C15—H15A⋯Br2ii | 0.93 | 2.80 | 3.700 (3) | 163 |
| C17—H17B⋯Br2 | 0.97 | 2.78 | 3.696 (3) | 158 |
| C24—H24A⋯Br1 | 0.93 | 2.73 | 3.579 (2) | 152 |
| C5—H5A⋯Cg4iii | 0.93 | 2.92 | 3.630 (3) | 135 |
Symmetry codes: (i) 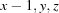 ; (ii)
; (ii) 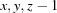 ; (iii)
; (iii) 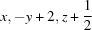 .
.
Acknowledgments
RAH thanks Universiti Sains Malaysia (USM) for the Research University grant Nos. 1001/PKIMIA/811157 and 1001/PKIMIA/823082. MAI is grateful to (IPS) USM for financial support [fellowship USM.IPS/JWT/1/19 (JLD 6)] and research attachment fund [P-KM0018/10(R)–308/AIPS/415401]. HKF and MH thank the Malaysian Government and Universiti Sains Malaysia for the Research University grant No. 1001/PFIZIK/811160. MH also thanks Universiti Sains Malaysia for a post-doctoral research fellowship.
supplementary crystallographic information
Comment
Many benzimidazole containing compounds are biologically active (Mohan et al., 2011). As part of our studies in this area, we now describe the title compound, (I).
The central benzene ring (C11–C16) makes dihedral angles of 76.75 (11) and 82.40 (10)° with the adjacent benzimidazole rings (N1,N2/C3–C9) and (N3,N4/C18–C24). The dihedral angle between the benzimidazole rings (N1,N2/C3–C9) and (N1,N2/C3–C9) is 57.03 (9)°. The benzimidazole (N1,N2/C3–C9) and (N3,N4/C18–C24) rings are approximately planar [maximum deviations of 0.020 (3) Å for atom C3 and 0.014 (2) Å for atom N3, respectively].
In the crystal (Fig. 2), the cations and anions are linked via C—H···Br (Table 1) hydrogen bonds, forming two-dimensional networks lying parallel to the the bc-plane. The crystal structure also features weak C—H···π interactions involving the centroid of the phenyl (C11–C16) ring.
Experimental
A mixture of benzimidazole (2.36 g, 20 mmol) and finely ground potassium hydroxide (2.36 g, 30 mmol) in 30 ml of DMSO was stirred at room temperature (27–28 °C) for 30 min. 1-Bromoethane (1.50 ml, 20 mmol) was added drop wise in this consistently stirring mixture and further stirred for 2 h at same temperature, poured into water (300 ml) and was extracted by chloroform (5 × 20 ml). The extract was dried by magnesium sulphate and evaporated under reduced pressure to get N-ethylbenzimidazole as a thick yellowish fluid (2.52 g, 86.30%). Furthermore, a mixture of 1 (1.46 g, 10 mmol) and 1,2-bis(bromomethyl)benzene (1.32 g, 5 mmol) in dioxane (30 ml) was refluxed at 110 °C for 18 h. Desired compound (2.2Br) appeared as beige–colored precipitates in dark brown solution. The mixture was filtered and precipitates were washed by fresh dioxane (3 × 5 ml), dried at room temperature for 24 h, and soft lumps so obtained were ground to fine powder (2.40 g, 86.33%). Saturated solution of 2.2Br in methanol (0.5 ml) was exposed to diethyl ether vapours (vapour diffusion) at room temperature overnight to get colourless blocks of (I).
Refinement
All hydrogen atoms were positioned geometrically [C–H = 0.93–0.97 Å] and were refined using a riding model, with Uiso(H) = 1.2 or 1.5 Ueq(C). A rotating group model was applied to the methyl groups. 3727 Friedel pairs were used to determine the absolute structure. One outliner, (4 0 -4), was omitted in the final refinement.
Figures
Fig. 1.

The asymmetric unit of the title compound, showing 30% probability displacement ellipsoids.
Fig. 2.

The crystal packing of the title compound.
Crystal data
| C26H28N42+·2Br− | F(000) = 1128 |
| Mr = 556.34 | Dx = 1.502 Mg m−3 |
| Monoclinic, Cc | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: C -2yc | Cell parameters from 5440 reflections |
| a = 9.7093 (7) Å | θ = 2.9–27.3° |
| b = 35.796 (3) Å | µ = 3.32 mm−1 |
| c = 8.0340 (6) Å | T = 296 K |
| β = 118.230 (1)° | Block, colourless |
| V = 2460.1 (3) Å3 | 0.45 × 0.32 × 0.23 mm |
| Z = 4 |
Data collection
| Bruker APEXII DUO CCD diffractometer | 8158 independent reflections |
| Radiation source: fine-focus sealed tube | 6244 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.023 |
| φ and ω scans | θmax = 32.5°, θmin = 2.5° |
| Absorption correction: multi-scan (SADABS; Bruker, 2009) | h = −14→14 |
| Tmin = 0.318, Tmax = 0.516 | k = −54→42 |
| 16606 measured reflections | l = −12→12 |
Refinement
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.033 | H-atom parameters constrained |
| wR(F2) = 0.080 | w = 1/[σ2(Fo2) + (0.P)2 + 0.1437P] where P = (Fo2 + 2Fc2)/3 |
| S = 1.02 | (Δ/σ)max = 0.003 |
| 8158 reflections | Δρmax = 0.71 e Å−3 |
| 291 parameters | Δρmin = −0.28 e Å−3 |
| 2 restraints | Absolute structure: Flack (1983), 3727 Friedel pairs |
| Primary atom site location: structure-invariant direct methods | Flack parameter: 0.009 (6) |
Special details
| Geometry. All s.u.'s (except the s.u. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell s.u.'s are taken into account individually in the estimation of s.u.'s in distances, angles and torsion angles; correlations between s.u.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell s.u.'s is used for estimating s.u.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > 2σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| Br1 | 0.68944 (3) | 0.918762 (6) | 0.48259 (3) | 0.05182 (7) | |
| Br2 | 0.38351 (4) | 0.822199 (8) | 0.90664 (4) | 0.06457 (9) | |
| N1 | 0.4060 (2) | 0.95348 (6) | 0.8359 (3) | 0.0441 (4) | |
| N2 | 0.2014 (2) | 0.92236 (5) | 0.6352 (3) | 0.0378 (4) | |
| N3 | 0.4142 (2) | 0.82699 (5) | 0.4234 (3) | 0.0386 (4) | |
| N4 | 0.5754 (2) | 0.79004 (5) | 0.3842 (3) | 0.0416 (4) | |
| C1 | 0.6769 (3) | 0.97225 (8) | 0.9292 (4) | 0.0569 (7) | |
| H1A | 0.7799 | 0.9729 | 1.0354 | 0.085* | |
| H1B | 0.6509 | 0.9964 | 0.8706 | 0.085* | |
| H1C | 0.6739 | 0.9541 | 0.8395 | 0.085* | |
| C2 | 0.5611 (3) | 0.96170 (10) | 0.9949 (4) | 0.0615 (7) | |
| H2A | 0.5990 | 0.9399 | 1.0759 | 0.074* | |
| H2B | 0.5519 | 0.9820 | 1.0690 | 0.074* | |
| C3 | 0.3020 (3) | 0.97982 (6) | 0.7130 (3) | 0.0405 (4) | |
| C4 | 0.3092 (3) | 1.01828 (7) | 0.7066 (4) | 0.0503 (6) | |
| H4A | 0.3959 | 1.0316 | 0.7930 | 0.060* | |
| C5 | 0.1818 (4) | 1.03575 (7) | 0.5662 (5) | 0.0578 (7) | |
| H5A | 0.1826 | 1.0616 | 0.5568 | 0.069* | |
| C6 | 0.0502 (3) | 1.01593 (7) | 0.4361 (4) | 0.0542 (6) | |
| H6A | −0.0333 | 1.0290 | 0.3424 | 0.065* | |
| C7 | 0.0416 (3) | 0.97753 (7) | 0.4438 (4) | 0.0453 (5) | |
| H7A | −0.0461 | 0.9643 | 0.3592 | 0.054* | |
| C8 | 0.1711 (2) | 0.95996 (6) | 0.5847 (3) | 0.0372 (4) | |
| C9 | 0.3416 (3) | 0.92020 (6) | 0.7867 (3) | 0.0433 (5) | |
| H9A | 0.3878 | 0.8981 | 0.8494 | 0.052* | |
| C10 | 0.0966 (2) | 0.89051 (6) | 0.5423 (3) | 0.0388 (4) | |
| H10A | −0.0035 | 0.8952 | 0.5378 | 0.047* | |
| H10B | 0.1408 | 0.8682 | 0.6174 | 0.047* | |
| C11 | 0.0711 (3) | 0.88369 (5) | 0.3442 (3) | 0.0372 (4) | |
| C12 | −0.0774 (3) | 0.89068 (6) | 0.1938 (4) | 0.0485 (5) | |
| H12A | −0.1560 | 0.8994 | 0.2187 | 0.058* | |
| C13 | −0.1095 (3) | 0.88490 (7) | 0.0100 (4) | 0.0537 (6) | |
| H13A | −0.2082 | 0.8903 | −0.0886 | 0.064* | |
| C14 | 0.0046 (4) | 0.87111 (7) | −0.0274 (4) | 0.0537 (6) | |
| H14A | −0.0173 | 0.8670 | −0.1518 | 0.064* | |
| C15 | 0.1523 (3) | 0.86321 (6) | 0.1182 (4) | 0.0478 (5) | |
| H15A | 0.2289 | 0.8538 | 0.0912 | 0.057* | |
| C16 | 0.1865 (3) | 0.86927 (5) | 0.3048 (3) | 0.0368 (4) | |
| C17 | 0.3497 (3) | 0.86073 (6) | 0.4620 (4) | 0.0411 (5) | |
| H17A | 0.4179 | 0.8817 | 0.4767 | 0.049* | |
| H17B | 0.3461 | 0.8576 | 0.5798 | 0.049* | |
| C18 | 0.3557 (3) | 0.79088 (6) | 0.4066 (3) | 0.0406 (5) | |
| C19 | 0.2223 (3) | 0.77759 (8) | 0.4100 (4) | 0.0525 (6) | |
| H19A | 0.1533 | 0.7933 | 0.4260 | 0.063* | |
| C20 | 0.1996 (4) | 0.73945 (9) | 0.3877 (5) | 0.0635 (8) | |
| H20A | 0.1112 | 0.7292 | 0.3865 | 0.076* | |
| C21 | 0.3028 (4) | 0.71617 (7) | 0.3674 (5) | 0.0665 (8) | |
| H21A | 0.2825 | 0.6906 | 0.3547 | 0.080* | |
| C22 | 0.4359 (4) | 0.72922 (7) | 0.3652 (4) | 0.0581 (7) | |
| H22A | 0.5060 | 0.7133 | 0.3523 | 0.070* | |
| C23 | 0.4582 (3) | 0.76776 (6) | 0.3837 (3) | 0.0404 (4) | |
| C24 | 0.5441 (3) | 0.82501 (6) | 0.4069 (3) | 0.0406 (5) | |
| H24A | 0.6047 | 0.8455 | 0.4108 | 0.049* | |
| C25 | 0.7016 (3) | 0.77625 (9) | 0.3487 (5) | 0.0621 (7) | |
| H25A | 0.6559 | 0.7646 | 0.2251 | 0.074* | |
| H25B | 0.7602 | 0.7573 | 0.4418 | 0.074* | |
| C26 | 0.8105 (5) | 0.80600 (13) | 0.3563 (8) | 0.0893 (12) | |
| H26A | 0.8782 | 0.7964 | 0.3099 | 0.134* | |
| H26B | 0.8720 | 0.8142 | 0.4845 | 0.134* | |
| H26C | 0.7518 | 0.8267 | 0.2795 | 0.134* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Br1 | 0.05117 (12) | 0.04447 (12) | 0.05944 (14) | 0.00068 (11) | 0.02585 (11) | 0.00191 (11) |
| Br2 | 0.0914 (2) | 0.05391 (15) | 0.06103 (16) | 0.03112 (14) | 0.04636 (15) | 0.01796 (12) |
| N1 | 0.0401 (9) | 0.0531 (11) | 0.0364 (9) | 0.0054 (8) | 0.0158 (8) | 0.0004 (8) |
| N2 | 0.0395 (9) | 0.0323 (8) | 0.0399 (9) | 0.0074 (7) | 0.0174 (8) | 0.0014 (7) |
| N3 | 0.0384 (9) | 0.0314 (8) | 0.0517 (10) | 0.0024 (7) | 0.0261 (9) | 0.0025 (7) |
| N4 | 0.0413 (10) | 0.0384 (9) | 0.0485 (10) | 0.0098 (7) | 0.0240 (8) | 0.0037 (8) |
| C1 | 0.0458 (13) | 0.0531 (14) | 0.0583 (16) | 0.0057 (11) | 0.0136 (13) | 0.0064 (11) |
| C2 | 0.0454 (13) | 0.085 (2) | 0.0391 (13) | 0.0005 (14) | 0.0076 (11) | −0.0018 (12) |
| C3 | 0.0437 (11) | 0.0399 (10) | 0.0417 (11) | 0.0040 (9) | 0.0234 (9) | −0.0029 (8) |
| C4 | 0.0533 (13) | 0.0392 (11) | 0.0636 (15) | −0.0033 (10) | 0.0319 (12) | −0.0091 (10) |
| C5 | 0.0717 (17) | 0.0332 (11) | 0.0821 (19) | 0.0063 (11) | 0.0475 (16) | 0.0009 (11) |
| C6 | 0.0530 (13) | 0.0404 (12) | 0.0704 (17) | 0.0135 (11) | 0.0301 (13) | 0.0089 (11) |
| C7 | 0.0398 (11) | 0.0415 (11) | 0.0506 (13) | 0.0100 (9) | 0.0182 (10) | 0.0021 (9) |
| C8 | 0.0398 (10) | 0.0326 (9) | 0.0416 (11) | 0.0065 (8) | 0.0213 (9) | 0.0005 (8) |
| C9 | 0.0447 (11) | 0.0423 (11) | 0.0413 (11) | 0.0091 (9) | 0.0192 (10) | 0.0043 (8) |
| C10 | 0.0378 (10) | 0.0335 (10) | 0.0506 (12) | 0.0034 (8) | 0.0253 (10) | 0.0000 (8) |
| C11 | 0.0411 (10) | 0.0255 (8) | 0.0473 (11) | 0.0003 (7) | 0.0229 (9) | −0.0004 (7) |
| C12 | 0.0438 (12) | 0.0378 (11) | 0.0579 (14) | 0.0073 (9) | 0.0191 (11) | −0.0024 (10) |
| C13 | 0.0534 (14) | 0.0394 (11) | 0.0517 (14) | 0.0041 (11) | 0.0112 (12) | −0.0014 (10) |
| C14 | 0.0760 (18) | 0.0378 (11) | 0.0438 (13) | −0.0009 (12) | 0.0253 (13) | −0.0006 (9) |
| C15 | 0.0660 (15) | 0.0352 (10) | 0.0530 (13) | 0.0019 (10) | 0.0370 (12) | −0.0005 (9) |
| C16 | 0.0444 (11) | 0.0245 (8) | 0.0466 (11) | 0.0015 (8) | 0.0257 (9) | 0.0028 (7) |
| C17 | 0.0425 (11) | 0.0352 (10) | 0.0523 (13) | 0.0060 (9) | 0.0279 (10) | −0.0028 (9) |
| C18 | 0.0440 (11) | 0.0339 (10) | 0.0459 (11) | −0.0011 (8) | 0.0229 (10) | 0.0035 (8) |
| C19 | 0.0524 (14) | 0.0476 (13) | 0.0659 (16) | −0.0074 (11) | 0.0348 (13) | 0.0019 (11) |
| C20 | 0.0695 (18) | 0.0507 (15) | 0.0742 (18) | −0.0191 (14) | 0.0372 (15) | 0.0068 (13) |
| C21 | 0.088 (2) | 0.0359 (12) | 0.0665 (17) | −0.0097 (14) | 0.0293 (17) | 0.0035 (12) |
| C22 | 0.0764 (19) | 0.0329 (11) | 0.0570 (15) | 0.0096 (12) | 0.0249 (14) | 0.0031 (10) |
| C23 | 0.0451 (11) | 0.0337 (9) | 0.0393 (11) | 0.0056 (9) | 0.0174 (9) | 0.0034 (8) |
| C24 | 0.0373 (10) | 0.0367 (10) | 0.0523 (13) | 0.0014 (8) | 0.0249 (10) | 0.0022 (9) |
| C25 | 0.0527 (14) | 0.0677 (17) | 0.0768 (19) | 0.0179 (13) | 0.0396 (14) | −0.0038 (14) |
| C26 | 0.065 (2) | 0.100 (3) | 0.133 (3) | −0.0016 (19) | 0.071 (2) | −0.011 (3) |
Geometric parameters (Å, º)
| N1—C9 | 1.316 (3) | C10—H10B | 0.9700 |
| N1—C3 | 1.393 (3) | C11—C12 | 1.397 (3) |
| N1—C2 | 1.472 (3) | C11—C16 | 1.399 (3) |
| N2—C9 | 1.332 (3) | C12—C13 | 1.373 (4) |
| N2—C8 | 1.396 (3) | C12—H12A | 0.9300 |
| N2—C10 | 1.474 (3) | C13—C14 | 1.369 (5) |
| N3—C24 | 1.331 (3) | C13—H13A | 0.9300 |
| N3—C18 | 1.393 (3) | C14—C15 | 1.385 (4) |
| N3—C17 | 1.460 (3) | C14—H14A | 0.9300 |
| N4—C24 | 1.321 (3) | C15—C16 | 1.391 (3) |
| N4—C23 | 1.388 (3) | C15—H15A | 0.9300 |
| N4—C25 | 1.469 (3) | C16—C17 | 1.519 (3) |
| C1—C2 | 1.499 (5) | C17—H17A | 0.9700 |
| C1—H1A | 0.9600 | C17—H17B | 0.9700 |
| C1—H1B | 0.9600 | C18—C23 | 1.372 (3) |
| C1—H1C | 0.9600 | C18—C19 | 1.392 (4) |
| C2—H2A | 0.9700 | C19—C20 | 1.381 (4) |
| C2—H2B | 0.9700 | C19—H19A | 0.9300 |
| C3—C4 | 1.381 (3) | C20—C21 | 1.371 (5) |
| C3—C8 | 1.394 (3) | C20—H20A | 0.9300 |
| C4—C5 | 1.368 (4) | C21—C22 | 1.382 (5) |
| C4—H4A | 0.9300 | C21—H21A | 0.9300 |
| C5—C6 | 1.402 (4) | C22—C23 | 1.393 (3) |
| C5—H5A | 0.9300 | C22—H22A | 0.9300 |
| C6—C7 | 1.380 (4) | C24—H24A | 0.9300 |
| C6—H6A | 0.9300 | C25—C26 | 1.481 (5) |
| C7—C8 | 1.383 (3) | C25—H25A | 0.9700 |
| C7—H7A | 0.9300 | C25—H25B | 0.9700 |
| C9—H9A | 0.9300 | C26—H26A | 0.9600 |
| C10—C11 | 1.510 (3) | C26—H26B | 0.9600 |
| C10—H10A | 0.9700 | C26—H26C | 0.9600 |
| C9—N1—C3 | 108.49 (19) | C13—C12—C11 | 121.3 (3) |
| C9—N1—C2 | 126.0 (2) | C13—C12—H12A | 119.3 |
| C3—N1—C2 | 125.5 (2) | C11—C12—H12A | 119.3 |
| C9—N2—C8 | 107.76 (18) | C14—C13—C12 | 119.6 (2) |
| C9—N2—C10 | 125.46 (17) | C14—C13—H13A | 120.2 |
| C8—N2—C10 | 126.77 (18) | C12—C13—H13A | 120.2 |
| C24—N3—C18 | 107.65 (19) | C13—C14—C15 | 120.7 (3) |
| C24—N3—C17 | 125.96 (19) | C13—C14—H14A | 119.6 |
| C18—N3—C17 | 126.3 (2) | C15—C14—H14A | 119.6 |
| C24—N4—C23 | 107.7 (2) | C14—C15—C16 | 120.1 (2) |
| C24—N4—C25 | 127.8 (2) | C14—C15—H15A | 119.9 |
| C23—N4—C25 | 124.4 (2) | C16—C15—H15A | 119.9 |
| C2—C1—H1A | 109.5 | C15—C16—C11 | 119.6 (2) |
| C2—C1—H1B | 109.5 | C15—C16—C17 | 119.2 (2) |
| H1A—C1—H1B | 109.5 | C11—C16—C17 | 121.2 (2) |
| C2—C1—H1C | 109.5 | N3—C17—C16 | 111.95 (19) |
| H1A—C1—H1C | 109.5 | N3—C17—H17A | 109.2 |
| H1B—C1—H1C | 109.5 | C16—C17—H17A | 109.2 |
| N1—C2—C1 | 112.0 (2) | N3—C17—H17B | 109.2 |
| N1—C2—H2A | 109.2 | C16—C17—H17B | 109.2 |
| C1—C2—H2A | 109.2 | H17A—C17—H17B | 107.9 |
| N1—C2—H2B | 109.2 | C23—C18—C19 | 122.5 (2) |
| C1—C2—H2B | 109.2 | C23—C18—N3 | 106.5 (2) |
| H2A—C2—H2B | 107.9 | C19—C18—N3 | 131.0 (2) |
| C4—C3—N1 | 132.0 (2) | C20—C19—C18 | 115.4 (3) |
| C4—C3—C8 | 121.7 (2) | C20—C19—H19A | 122.3 |
| N1—C3—C8 | 106.29 (19) | C18—C19—H19A | 122.3 |
| C5—C4—C3 | 116.4 (2) | C21—C20—C19 | 122.4 (3) |
| C5—C4—H4A | 121.8 | C21—C20—H20A | 118.8 |
| C3—C4—H4A | 121.8 | C19—C20—H20A | 118.8 |
| C4—C5—C6 | 122.2 (2) | C20—C21—C22 | 122.4 (2) |
| C4—C5—H5A | 118.9 | C20—C21—H21A | 118.8 |
| C6—C5—H5A | 118.9 | C22—C21—H21A | 118.8 |
| C7—C6—C5 | 121.6 (2) | C21—C22—C23 | 115.6 (3) |
| C7—C6—H6A | 119.2 | C21—C22—H22A | 122.2 |
| C5—C6—H6A | 119.2 | C23—C22—H22A | 122.2 |
| C6—C7—C8 | 116.0 (2) | C18—C23—N4 | 107.35 (18) |
| C6—C7—H7A | 122.0 | C18—C23—C22 | 121.7 (3) |
| C8—C7—H7A | 122.0 | N4—C23—C22 | 130.9 (2) |
| C7—C8—C3 | 122.1 (2) | N4—C24—N3 | 110.8 (2) |
| C7—C8—N2 | 131.4 (2) | N4—C24—H24A | 124.6 |
| C3—C8—N2 | 106.51 (18) | N3—C24—H24A | 124.6 |
| N1—C9—N2 | 110.93 (19) | N4—C25—C26 | 113.3 (3) |
| N1—C9—H9A | 124.5 | N4—C25—H25A | 108.9 |
| N2—C9—H9A | 124.5 | C26—C25—H25A | 108.9 |
| N2—C10—C11 | 112.86 (18) | N4—C25—H25B | 108.9 |
| N2—C10—H10A | 109.0 | C26—C25—H25B | 108.9 |
| C11—C10—H10A | 109.0 | H25A—C25—H25B | 107.7 |
| N2—C10—H10B | 109.0 | C25—C26—H26A | 109.5 |
| C11—C10—H10B | 109.0 | C25—C26—H26B | 109.5 |
| H10A—C10—H10B | 107.8 | H26A—C26—H26B | 109.5 |
| C12—C11—C16 | 118.6 (2) | C25—C26—H26C | 109.5 |
| C12—C11—C10 | 118.1 (2) | H26A—C26—H26C | 109.5 |
| C16—C11—C10 | 123.2 (2) | H26B—C26—H26C | 109.5 |
| C9—N1—C2—C1 | 108.0 (3) | C14—C15—C16—C17 | −179.4 (2) |
| C3—N1—C2—C1 | −72.7 (3) | C12—C11—C16—C15 | 1.7 (3) |
| C9—N1—C3—C4 | 177.2 (3) | C10—C11—C16—C15 | 178.9 (2) |
| C2—N1—C3—C4 | −2.3 (4) | C12—C11—C16—C17 | −179.5 (2) |
| C9—N1—C3—C8 | −0.8 (3) | C10—C11—C16—C17 | −2.3 (3) |
| C2—N1—C3—C8 | 179.8 (2) | C24—N3—C17—C16 | 119.9 (3) |
| N1—C3—C4—C5 | −178.5 (3) | C18—N3—C17—C16 | −63.6 (3) |
| C8—C3—C4—C5 | −0.8 (4) | C15—C16—C17—N3 | −38.6 (3) |
| C3—C4—C5—C6 | 0.4 (4) | C11—C16—C17—N3 | 142.5 (2) |
| C4—C5—C6—C7 | 0.6 (5) | C24—N3—C18—C23 | 1.2 (3) |
| C5—C6—C7—C8 | −1.1 (4) | C17—N3—C18—C23 | −175.9 (2) |
| C6—C7—C8—C3 | 0.8 (4) | C24—N3—C18—C19 | −178.7 (3) |
| C6—C7—C8—N2 | 178.7 (3) | C17—N3—C18—C19 | 4.2 (4) |
| C4—C3—C8—C7 | 0.2 (4) | C23—C18—C19—C20 | −0.3 (4) |
| N1—C3—C8—C7 | 178.4 (2) | N3—C18—C19—C20 | 179.6 (3) |
| C4—C3—C8—N2 | −178.2 (2) | C18—C19—C20—C21 | 1.2 (5) |
| N1—C3—C8—N2 | 0.0 (3) | C19—C20—C21—C22 | −0.9 (5) |
| C9—N2—C8—C7 | −177.4 (3) | C20—C21—C22—C23 | −0.4 (5) |
| C10—N2—C8—C7 | 1.1 (4) | C19—C18—C23—N4 | 179.1 (2) |
| C9—N2—C8—C3 | 0.8 (3) | N3—C18—C23—N4 | −0.8 (3) |
| C10—N2—C8—C3 | 179.3 (2) | C19—C18—C23—C22 | −1.0 (4) |
| C3—N1—C9—N2 | 1.4 (3) | N3—C18—C23—C22 | 179.0 (2) |
| C2—N1—C9—N2 | −179.2 (2) | C24—N4—C23—C18 | 0.1 (3) |
| C8—N2—C9—N1 | −1.4 (3) | C25—N4—C23—C18 | −175.1 (2) |
| C10—N2—C9—N1 | −179.9 (2) | C24—N4—C23—C22 | −179.7 (3) |
| C9—N2—C10—C11 | −114.9 (2) | C25—N4—C23—C22 | 5.0 (4) |
| C8—N2—C10—C11 | 66.8 (3) | C21—C22—C23—C18 | 1.4 (4) |
| N2—C10—C11—C12 | −111.6 (2) | C21—C22—C23—N4 | −178.8 (3) |
| N2—C10—C11—C16 | 71.3 (2) | C23—N4—C24—N3 | 0.7 (3) |
| C16—C11—C12—C13 | −2.3 (3) | C25—N4—C24—N3 | 175.7 (3) |
| C10—C11—C12—C13 | −179.6 (2) | C18—N3—C24—N4 | −1.2 (3) |
| C11—C12—C13—C14 | 1.7 (4) | C17—N3—C24—N4 | 175.9 (2) |
| C12—C13—C14—C15 | −0.5 (4) | C24—N4—C25—C26 | 5.3 (5) |
| C13—C14—C15—C16 | 0.0 (4) | C23—N4—C25—C26 | 179.6 (3) |
| C14—C15—C16—C11 | −0.6 (3) |
Hydrogen-bond geometry (Å, º)
Cg4 is the centroid of the C11–C16 ring.
| D—H···A | D—H | H···A | D···A | D—H···A |
| C9—H9A···Br2 | 0.93 | 2.76 | 3.610 (2) | 152 |
| C10—H10A···Br1i | 0.97 | 2.92 | 3.884 (2) | 171 |
| C10—H10B···Br2 | 0.97 | 2.91 | 3.820 (2) | 156 |
| C15—H15A···Br2ii | 0.93 | 2.80 | 3.700 (3) | 163 |
| C17—H17B···Br2 | 0.97 | 2.78 | 3.696 (3) | 158 |
| C24—H24A···Br1 | 0.93 | 2.73 | 3.579 (2) | 152 |
| C5—H5A···Cg4iii | 0.93 | 2.92 | 3.630 (3) | 135 |
Symmetry codes: (i) x−1, y, z; (ii) x, y, z−1; (iii) x, −y+2, z+1/2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: HB6611).
References
- Bruker (2009). APEX2, SAINT and SADABS Bruker AXS Inc., Madison, Wisconsin, USA.
- Flack, H. D. (1983). Acta Cryst. A39, 876–881.
- Mohan, V. G., Sreenivasulu, N., Rao, A. S. & Chigiri, S. (2011). Der Pharma Chem. 3, 446–452.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S1600536812002802/hb6611sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536812002802/hb6611Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536812002802/hb6611Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report