

Abstract
Prostaglandins (PGs) are powerful lipid mediators in many physiological and pathophysiological responses. They are produced by oxidation of arachidonic acid (AA) by cyclooxygenases (COX-1 and COX-2) followed by metabolism of endoperoxide intermediates by terminal PG synthases. PG biosynthesis is inhibited by nonsteroidal anti-inflammatory drugs (NSAIDs). Specific inhibition of COX-2 has been extensively investigated, but relatively few COX-1-selective inhibitors have been described. Recent reports of a possible contribution of COX-1 in analgesia, neuroinflammation, or carcinogenesis suggest that COX-1 is a potential therapeutic target. We designed, synthesized, and evaluated a series of (E)-2′-des-methyl-sulindac sulfide (E-DMSS) analogues for inhibition of COX-1. Several potent and selective inhibitors were discovered, and the most promising compounds were active against COX-1 in intact ovarian carcinoma cells (OVCAR-3). The compounds inhibited tumor cell proliferation but only at concentrations >100-fold higher than the concentrations that inhibit COX-1 activity. E-DMSS analogues may be useful probes of COX-1 biology in vivo and promising leads for COX-1-targeted therapeutic agents.
Introduction
Cyclooxygenases (COX-1 and COX-2) catalyze the oxygenation of arachidonic acid (AA) to form prostaglandins and thromboxane, which mediate a range of physiological and pathophysiological responses.1 The discovery in 1971 that nonsteroidal anti-inflammatory drugs (NSAIDs) inhibit prostaglandin synthesis in guinea pig lung and human platelets established COX-1 as a molecular target for this ancient class of drugs.2−4 The subsequent discovery in 1991 of COX-2, which is induced by cytokines during inflammation, suggested that this form of the enzyme represents the molecular target for the anti-inflammatory effects of NSAIDs.5−8 The differential expression of COX-1 and COX-2, and in particular the finding that COX-1 is the principal form expressed in the gastrointestinal tract, led to the search for COX-2-selective inhibitors as potential anti-inflammatory agents that might lack the gastrointestinal side effects of traditional NSAIDs.9−11 A number of highly selective inhibitors were introduced to the market including celecoxib (6), rofecoxib (7), valdecoxib, etoricoxib, and lumiracoxib (Figure 1).12−17 These compounds exhibited reduced gastrointestinal side effects in human testing but did not completely eliminate this liability.18,19 In addition, long-term placebo-controlled studies revealed cardiovascular side effects that led to the withdrawal of rofecoxib and valdecoxib from the market in the United States and Europe.20−23
Figure 1.

Upper row: the structures of sulindac (sulfoxide, 1a), its in vivo metabolites (1b and 1c), and 2′-des-methyl sulindac sulfide (2) as well as indomethacin (3, nonselective COX-1/-2 inhibitor). Lower row: N-(5-amino-2-pyridinyl)-4-(trifluoromethyl) benzamide (TFAP, 4), SC-560 (5) (selective COX-1 inhibitors), celecoxib (6), and rofecoxib (7) (selective COX-2 inhibitors).
The “COX-2” hypothesis posited that COX-1 inhibition is responsible for the gastrointestinal toxicity of NSAIDs. However, animal studies suggest that the total extent of COX inhibition is more important than the inhibition of the individual enzymes and that highly selective COX-1 inhibitors do not exhibit gastrointestinal toxicity analogous to highly selective COX-2 inhibitors.24,25 COX-1-dependent prostaglandin synthesis has been implicated in many pathophysiological processes including atherosclerosis, endothelial dysfunction, neuroinflammation, preterm labor, pain, and cancer.26,27 The role of COX-1 in neurodegenerative diseases is of particular interest. Neurodegenerative disorders that exhibit a strong inflammatory component produce prostaglandins (PGs) in neurons and glial cells.28 The extent of neurodegeneration is reduced in COX-1 knockout mice or by administration of COX-1-selective inhibitors.29−31 Very recent results indicate that COX-1 is a major source of pro-inflammatory PGs in the brains of both LPS-treated mice and mice treated with the Parkinsonism-inducing compound, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine.32 Therefore, COX-1-selective inhibitors represent a potentially useful class of drugs that has not been extensively investigated.
The COX-1 inhibitors that have been described in the literature fall into two main structural classes.26 One class comprises diarylheterocycles discovered during the COX-2 inhibitor programs that led to the development of 6 and 7 (Figure 1). Noteworthy among this class of inhibitors is SC-560 (5, Figure 1), which exhibits COX-1 selectivity greater than 700-fold and which has been extensively used as an in vitro and in vivo probe, for example, for investigating inhibition of PGE2 synthesis in rat primary spinal cord neurons or in (mouse) models of human ovarian cancer (OVCAR-3).33−36 The other class of COX-1 inhibitors comprises substituted benzamides related to N-(5-amino-2-pyridinyl)-4-(trifluoromethyl)-benzamide (TFAP, 4; Figure 1).25,37 This compound exhibits high COX-1 selectivity but has not been used extensively as a probe. TFAP and its analogues have been suggested as candidates for analgesics that do not cause stomach erosions or ulcers.25 Other compounds related to resveratrol, a noncompetitive COX inhibitor with anti-inflammatory, cardiovascular protective, and cancer chemopreventive properties, and curcumin, the active component of turmeric that possesses anti-inflammatory and anticancer activity, are reported to exhibit modest COX-1 selectivity. A recent study of 92 stilbene analogues, whose biological activities were assessed with various chemoprevention targets, revealed a handful of nitro- or amino-substituted resveratrol derivatives that inhibit COX-1 with potency roughly comparable to the parent compound but without affecting COX-2.39 Finally, valeryl salicylate, a derivative of aspirin, selectively inhibits COX-1 by covalent modification of an active site serine residue.26,38 Biological effects of valeryl salicylate have been, for example, studied in models of acute inflammation in mice.39
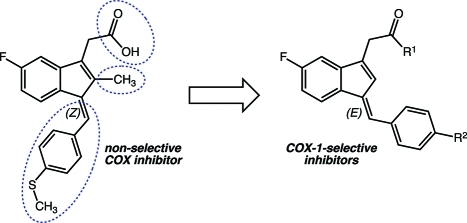
Sulindac (compound 1a, Figure 1) is an NSAID that exhibits cancer chemopreventive activity in animal models, induces polyp regression in familial polyposis patients, and inhibits polyp recurrence in patients when combined with α-difluoromethylornithine.40−46 Sulindac is a pro-drug that is reduced to the active sulfide metabolite by colonic microflora.47 Sulindac sulfide (compound 1b, Figure 1) is a slow, tight-binding inhibitor of both COX-1 and COX-2. We recently reported that the 2′-des-methyl derivative of 1b (compound 2, Figure 1) does not inhibit COX-2 but selectively inhibits COX-1 (Table 1 and Figure S5 in the Supporting Information).48 The absence of the 2′-methyl group during indene synthesis quantitatively directs the geometry of the introduced benzylidene double bond to (E) rather than (Z), which is formed when the 2′-methyl group is present;49 compound 2 is a rapid, reversible inhibitor of COX-1. The novelty of this scaffold prompted us to investigate its potential for the construction of more potent and selective COX-1 inhibitors.
Table 1. COX-1 and COX-2 Inhibition by Arylmethylidene Variants of E-DMSS*.


Compounds were screened at 4 μM. A concentration of 5 μM of AA-substrate was employed; n.d., not determined.
Mean ± SEM of two (n) experiments.
In the present manuscript, we describe the synthesis of a series of compounds designed to probe the importance of the various functionalities in 2 with regard to selective inhibition of COX-1. We find that modification of the benzylidene group leads to improved inhibitors, but alterations in the 5′-fluoro or carboxyl group are less well tolerated. The most active compounds are potent inhibitors of COX-1 in cultured human ovarian cells but are weak inhibitors of the growth of the cells in vitro.
Results
The indene template is a widely used pharmacophore that has found many applications in medicinal chemistry, for example, in NSAIDs.50−53 Recent reports have indicated that the benzylidene-indene acetic acid derivative 1b can be structurally re-engineered to improve isoform-specific COX binding inter alia.43,54,55 In the present study, compound 2, the E-2′-des-methyl-sulindac sulfide [(E)-DMSS] analogue of 1b, served as the lead scaffold for construction of potential COX-1-selective inhibitors. Modifications were made at the carboxyl, benzylidene, and 5′-position of the indene ring (Figure 2). The synthetic scheme followed the general approaches previously described for assembly of sulindac analogues with some modifications (Scheme 1).53,56 In contrast to the standard procedures, which employ “activated” zinc and expensive α-haloalkanoyl esters, we reacted the indanone precursors, 8, with inexpensive ethyl alkanoates at low temperatures using lithium diisopropylamide in tetrahydrofuran.57 This method generated the intermediate alcohols, 9, after an acidic quench in a much shorter time (2 h). The quantities of side products detected by HPLC were less than with the lengthy Reformatsky reaction (overnight with Zn0 catalyst).58 The applied alkylation reaction tolerated different substituents at the core aromatic annulus (e.g., 6′-F, 6′-OCH3) and allowed the introduction of α-unsubstituted (9a,b), monomethylated (9c), or geminal-dimethylated (9d) carboxymethyl moieties at position 1 of the indanone ring. Compounds 9a, 9c, and 9d hold a 6′-F substituent at the 1-hydroxy-2,3-dihydro-1H-inden-1-yl partial structure, whereas 9b has a methoxy group at its 6′-position. HPLC analysis of 9a, 9b, and 9d, which contain a single asymmetric carbon, afforded a single peak, whereas compound 9c, which has two chiral centers, afforded two substantial peaks due to the presence of diastereomers. Attempts to prepare a cyclopropyl analogue using ethyl cyclopropanecarboxylate as an alkylation reagent failed due to the instability of the intermediate cycloalkyl carbanion.59,60
Figure 2.

Intended chemical modifications on 2 and 13.
Scheme 1. General Synthetic Route to Multisubstituted (E)-2′-Des-methyl Sulindac Sulfide Analogues Including Procedures for Ester Cleavage and (Re)Esterification Reactions.

Reagents and conditions: (a) For compound 2: ethyl bromoacetate, activated zinc dust, benzene/ether, reflux, 16 h. (b) 2 M LDA, THF, (substituted) ethyl alkanoate, −75 °C, 2 h. (c) TsOH·2H2O, CaCl2·2H2O, toluene, reflux, overnight. (d) 1 N NaOH, MeOH, 65–70 °C. (e) Potassium trimethylsilanoate (90% material), THF, microwave radiation, 90–120 °C, 15 min. (f) Water, 2–4 M HCl. (g) 1 M LiOH, THF, RT, 5–6 h. (h) Alkyl alcohol, H2SO4, reflux, overnight.
Dehydration of 9a and 9b produced a mixure of exo- and endo-olefins for both 10a and 10b (Table S1 in the Supporting Information).50,57 Steric hindrance from the exocyclic alkyl chain of the racemic mixture 10c favored conversion to the endo-olefin as indicated by HPLC analysis and by comparison to the UV spectrum of 10d, which cannot exist in the exo-form (Table S1 in the Supporting Information). Unseparated isomeric mixtures of ethyl esters, 10a–c, were employed in the subsequent condensation step. Knoevenagel (aldol) condensation between 10a–d and the appropriate aldehydes proceeded under basic conditions to afford a single endo-olefin in all cases. Considerable amounts of byproduct, most likely from the Cannizzaro reaction, could be detected by HPLC, MS, and NMR. Together with the recognized light instability of the E-DMSS analogues (UV-induced isomerization about the exocyclic double bond from E to Z), this complicated the purification process and decreased the isolatable yields for some compounds.48,50,61 The basic conditions of the condensation led to ester hydrolysis for compounds derived from 10a and 10b. In addition, the lipophilic nature of the final E-DMSS analogues occasionally allowed their quantitative isolation as pure sodium salts directly from the aqueous-alkaline mother liquor (Table S2 in the Supporting Information). The acids could be esterified with alcohols under acidic conditions and hydrolyzed with LiOH.55 Hydrolysis was not observed for compounds derived from 10c and 10d. The ester products either crystallized from the reaction mixture or were extracted and purified by chromatography. The stereochemistry of the exocyclic double bond of all of the E-DMSS analogues was E as anticipated by the absence of a methyl group at the 2′-position of the indenyl ring (compare 1H NMR and NOESY spectrum of 20b in Figures S2 and S3 in the Supporting Information).48 Detailed experimental conditions and spectral properties of all intermediate and final compounds are provided in the Supporting Information.
Individual compounds were preincubated with purified ovine COX-1 or murine COX-2 for 17 min followed by addition of [1-14C]-AA (Figure S4 in the Supporting Information: experimental timeline). After 3 min, the reaction was quenched, and radioactive products were extracted and quantified as described in Supporting Information. For initial screening, a single concentration of 4 μM inhibitor was employed, and the concentration of [1-14C]-AA was 5 μM. The 4 μM inhibitor concentration was chosen based on the previously determined IC50 of 1.8 μM for compound 2. The AA concentration of 5 μM represents the Km for COX-1 and COX-2, which enables rapid, reversible, and slow, tight-binding inhibitors to be detected. Compound 2 was used as a reference for all new E-DMSS analogues.
Table 1 lists the percent inhibition of COX for the initial series of arylmethylidene analogues of 2.51 We determined the effects of different steric and electronic properties upon COX-1 and COX-2 inactivation. Oxidation of the sulfide (11a and 11b) reduces activity as previously reported for sulindac sulfide analogues. Substitution with trifluoromethylsulfide (11c) maintains some COX-1 selectivity, while oxidation of the sulfur again reduces activity (11d and 11e). A range of para substituents exhibited modest COX-1 selectivity (11f–m). Substitution of carboxyl at the para position reduced the potency against COX-1 but more substantially reduced inhibition of COX-2. Conversion of the phenyl ring to a naphthyl or aza-naphthyl ring maintained COX-1 selectivity and in some cases increased it (12d and 12f). The most potent and selective COX-1 inhibitor in this series was the biphenyl analogue 13a (COX-1 IC50, 570 nM; COX-2 IC50, > 4 μM). Such hydrophobic biphenyl systems are a common structure template seen in other small molecule inhibitors of the AA pathway (e.g., flurbiprofen [NSAID] or MK-866 analogues [microsomal prostaglandin E2 synthase-1 (mPGES-1) inhibitors]).60,62 Concentration dependences were determined for the most potent compounds, which led to the determination of IC50 values for a subset of the inhibitors (Table 1 and Figure S5 and Table S4 in the Supporting Information).
To build on the discovery of 13a, a series of substituted biphenyls were synthesized by either Knoevenagel condensation or Suzuki–Miyaura coupling of brominated benzylidene precursors with (hetero)aryl boronic acids (e.g., 13f and 13k; Scheme 2 and Figure S1 in the Supporting Information).63 Evaluation of this series (Table 2) indicated that multiple substitutions were tolerated, although none dramatically increased either the potency or the selectivity of COX-1 inhibition over compound 13a. Interestingly, introduction of a 2-aza substituent into a meta-substituted biphenyl scaffold reversed the selectivity of inhibition, yielding compounds with modest COX-2 selectivity (17a–e; compare Figure S6 in the Supporting Information).
Scheme 2. Synthesis of New “Biphenyl Derivatives” of 2′-Des-methyl Sulindac Sulfide Using Suzuki–Miyaura Coupling Reactions.

Reagents and conditions: Pd(PPh3)4, K+-carbonate, DME/H20, 90 °C, 14 h. Suzuki–Miyaura couplings were performed on submillimolar scale in a sealed glass tube under argon.63
Table 2. COX-1 and COX-2 Inhibition by Substituted Biphenylmethylidene Derivatives of E-DMSS*.


Compounds were screened at 4 μM. A concentration of 5 μM of AA-substrate was employed.
Mean ± SEM of two (n) experiments.
Neutralization of the carboxylic acid by conversion to an ester or amide in the benzylidene series reduced potency but maintained selectivity for COX-1, except for the biphenyl methylidene analogue 16e, which revealed potent anti-COX-1 activity but also increased COX-2 inhibition (Table 3). No significant differences were observed on changing the ester from methyl to isopropyl (16a–c).64
Table 3. COX-1 and COX-2 Inhibition by Ester and Amide Derivatives of E-DMSS*.


Compounds were screened at 4 μM. A concentration of 5 μM of AA-substrate was employed.
Mean ± SEM of two (n) experiments.
Substitution α- to the carboxyl was explored by synthesis of a series of esters and acids (Table 4). The general route outlined in Scheme 1 was employed using ethyl esters of the indene acetic acids, 10c and 10d, as building blocks. It was not possible to make a complete series of monomethyl-substituted analogues because of low yields and the generation of multiple side products (Scheme S1 in the Supporting Information). The only monomethyl analogue prepared was the ethyl ester, 18a (Table 4), which demonstrated reduced COX-1 inhibitory activity (30% inhibition at 4 μM) as compared to the ethyl ester of the unsubstituted biphenyl methylidene compound, 16e (COX-1 IC50, 1.1 μM) (Table 3 and Figure 7).65 Dimethyl analogues were prepared in high yield with minimal side products. The steric hindrance introduced by α,α-disubstitutions dramatically reduced the ease of hydrolysis to the acids. Thus, a nonaqueous hydrolytic strategy was employed in which THF solutions of individual esters were heated in a microwave with potassium trimethylsilanoate (Scheme 1).66 This afforded nearly quantitative conversion of the esters to the acids. The two initially prepared acids, 19a and 19c, were less active than the unsubstituted free acids, 2 and 13a (Table 1), so no further acids were synthesized. The ethyl esters of the α,α-disubstituted compounds, 19b and 19d–20b (Table 4) retained COX-1 selectivity but exhibited reduced potency. The most selective inhibitor was the oxazole, 19i.
Table 4. COX-1 and COX-2 Inhibition by α-Substituted E-DMSS Analogues*.


Compounds were screened at 4 μM. A concentration of 5 μM of AA-substrate was employed.
Mean ± SEM of two (n) experiments.
Figure 7.

Derivatization and COX activity data of lead compound 13a.
A series of substituted alkyl- and arylsulfonimides as carboxylic acid isosteres were synthesized as depicted in Scheme 3.60,67,68 Bioisosteric replacements represent a common approach for the rational modification of lead compounds into safer and more clinically effective agents with similar biological properties to the parent compound. Moderate COX-1 selectivity was observed for compounds 21a, 21b, and 21d–n (Table 5), but the trifluoromethylsulfonimide, 21c, with biphenylmethylidene substituent, was the most potent and selective compared of all of the analogues synthesized in the present study (COX-1 IC50, 470 nM; COX-2 IC50, 14 μM; compare Figure S6 in the Supporting Information). This meant that it exhibited a more than 4-fold improvement in activity against COX-1 as compared with our early lead and reference compound 2. Analogue 21c was approximately 30-fold more selective for COX-1 over COX-2.
Scheme 3. Preparation of Sulfonimide Derivatives of 2′-Des-methyl Sulindac Sulfide.

Syntheses were performed on a parallel synthesis apparatus using a straightforward one-pot, two-step CDI carbodiimide coupling reaction (route A).67 Alternatively, the two-step sulfonamide couplings could be accomplished with oxalyl chloride as the carboxylic acid activator, according to a disclosed procedure, as established for compound 21c (route B).65,86,87 Reagents and conditions: (Route A): (a1) CDI/DCM, 0–5 °C, 2 h; (b1) alkyl-/arylsulfonamide, DBU, RT, overnight. (Route B): (a2) oxalyl chloride/DCM, RT, overnight; (b2) alkyl-/arylsulfonamide, 1,2-DCE, pyridine, RT, overnight.
Table 5. COX-1 and COX-2 Inhibition by Sulfonimide Derivatives of E-DMSS*.


Differently substituted alkyl- and arylsulfonimides (21a–n) were prepared as bioisosteres of 2 and 13a, respectively, to alter acidity or modify lipophilicity without making significant changes to the core structure or affecting pKa (Scheme 3; see also the Experimental Section). Compounds were screened at 4 μM. A concentration of 5 μM of AA-substrate was employed.
Mean ± SEM of two (n) experiments.
Substitution of methoxy for fluoro at the indene 5′-position in analogues 15a and 15b, respectively (like in compound 3), led to a substantial loss of inhibitory potency for both the phenylmethylsulfide and the biphenyl derivatives (Figure 3 and Table S3 in the Supporting Information).69 The differential was most dramatic for the phenylmethylsulfide derivative, 15a, which was a poor COX-1 inhibitor. The 5′-methoxy derivative of the biphenyl, 15b, exhibited modest COX-1 inhibitory activity, although the presence of a plateau at higher inhibitor concentrations suggests that inhibition is readily reversible (also see Figures S7 and S8 in the Supporting Information).49
Figure 3.

Comparison of the inhibition of ovine COX-1 by 15a (black circle), 15b (red triangle), and 2 (blue square).
The ability of the various des-methylsulindac sulfide analogues to inhibit COX-1 activity in intact cells was evaluated using the human ovarian cancer cell line, NIH-OVCAR-3.35,36 This cell line expresses high levels of COX-1 but no detectable COX-2 as demonstrated by the Western blot in Figure 4a.36 Individual compounds were incubated with the cells for 30 min followed by the addition of [1-14C]-AA and another 30 min incubation. Compound 2, 13a, and 21c inhibited COX-1-dependent [1-14C]-AA oxidation with potencies comparable to that of the known COX-1 inhibitor, SC-560 (compound 5).33 Minimal inhibition of COX-1 in OVCAR-3 cells was detected with celecoxib (compound 6) (Figure 4a). When parallel experiments were conducted in the COX-2 expressing human head and neck cancer cell line, 1483, no inhibition of [1-14C]-AA oxygenation was observed by 2 and 13a, whereas 6 potently inhibited oxygenation (IC50 = 54 nM) (Figure 4b).
Figure 4.

(a) Inhibition of COX-1 in ovarian cancer cells. The OVCAR-3 human ovarian epithelial cancer cell line expresses high levels of catalytically active COX-1 but no COX-2 (see Western blot above).36 Whole-cell lysates from the OVCAR-3 human ovarian epithelial cancer cell lines were fractionated on a 10% PAGE and probed with polyclonal antibodies specific for COX-1 and COX-2. The control lane for the COX-1 blot is a standard solution of 0.3 μg/μL purified oCOX-1 in M-PER and Laemmli sample buffer, whereas the control lane for the COX-2 blot is a standard solution of 0.3 μg/μL purified mCOX-2 in M-PER and Laemmli sample buffer. Lower graph: Inhibition of hCOX-1 in OVCAR-3 cells (8 μM 14C-AA, 30 min at 37 °C) by E-DMSS analogues 2 (green circle; IC50, 116 nM), 13a (red triangle; IC50, 300 nM), and 21c (blue square; IC50, 495 nM) in comparison with literature reference compounds 5 (brown diamond; positive control; IC50, 160 nM) and 6 (○; negative control 16% inhibition at 5 μM). (b) Inhibition of hCOX-2 in HNSCC 1483 cells by E-DMSS analogues 2 (blue circle, n.i. at 5 μM) and 13a (red square, n.i. at 5 μM) in comparison with literature reference compound 6 (○; positive control; IC50, 54 nM).
Sulindac (compound 1a) and sulindac sulfide (compound 1b) inhibit the growth of tumor cells in culture, and there has been some controversy about the role of COX inhibition (especially COX-2 inhibition) in this antiproliferative activity.43,70−75 Therefore, we evaluated the effect of 2, 13a, 1b, and 1a on the viability of OVCAR-3 cells (Figure 5 and Supporting Information).76 Following a 2 day treatment, comparable EC50 values for inhibition of cell growth were observed with 2 (223 μM), 13a (132 μM), and 1a (210 μM), but no inhibition was observed with 1b up to 250 μM. Comparison of Figures 4a and 5 reveals that inhibition of cell growth requires concentrations of 2 and 13a that are more than 100-fold higher than the concentrations required for inhibition of COX-1 activity in the OVCAR-3 cells. Therefore, it appears that the antiproliferative effects of this series of compounds on OVCAR-3 cells grown in culture are independent of their ability to inhibit COX-1 activity. Evaluation of sulindac sulfide and compounds 13a and 21b in the breast cancer cell line, MDA-MB231, yielded EC50 values of 209, 141, and 215 μM (Figure S11 in the Supporting Information).
Figure 5.

OVCAR-3 cell growth inhibition by 1a (purple triangle, n.i. at 250 μM), 1b (blue diamond; EC50, 210 μM), 2 (green circle; EC50, 223 μM), and 13a (red square; EC50, 132 μM).
Discussion
Figure 6 summarizes the structure–activity relationships (SARs) for the series of compounds evaluated in the present study. Increasing the size and hydrophobicity of the aryl group from methylsulfanylbenzylidene to biphenylmethylidene significantly improved the potency and selectivity of COX-1 inhibition, so a number of analogues were made (Figure 7). Substitutions on the biphenyl ring (e.g., fluoro, trifluoromethyl) did not increase potency. Introduction of nitrogen into either biphenyl ring to increase hydrophilicity decreased potency, and the presence of multiple nitrogens in one ring reduced it further. Fusing the biphenyl (fluorene) retained potency against COX-1 but introduced a higher activity against COX-2. Alkyl substitution α- to the carboxyl group reduced activity in all cases. Conversion of the carboxylic acid to an ester or amide derivative significantly reduced potency, but introduction of the sulfonimide isostere in place of the carboxylate generated the most potent and selective COX-1 inhibitor in the series. Substitution on the sulfonyl sulfur was tolerated from methyl to substituted aryl. Perhaps the most surprising result was the finding of significantly reduced activity on substitution of a 5′-methoxy for the 5′-fluoro.
Figure 6.

SAR for selective COX-1 inhibition.
The in vitro screen utilized ovine COX-1 and mouse COX-2. Ovine COX-1 is the closest in sequence to human COX-1 among several mammalian species and is commonly used as a surrogate for human COX-1.77−79 The human enzyme has proven difficult to routinely express and purify with high activity. To verify the in vitro results, we tested the most active compounds against human COX-1 in cultured human ovarian cancer cells, OVCAR-3, and against human COX-2 in cultured head and neck cancer cells, 1483. All of the compounds inhibited COX-1 in the OVCAR cells to an extent that was comparable to that of the established COX-1 inhibitor, 5. None of the compounds inhibited COX-2 in the 1483 cells. In contrast, 6 potently inhibited COX-2 in the 1483 cells but had no effect on COX-1 in the OVCAR-3 cells. Thus, the lead compounds identified in the in vitro screen were effective inhibitors of human COX-1 in intact cells.
There was a dramatic differential in the ability of the lead compounds to inhibit PG synthesis by COX-1 in the OVCAR-3 cells and their ability to reduce proliferation in the same cell type. Whereas complete COX-1 inhibition was observed below 1 μM, none of the E-DMSS analogues reduced proliferation below 50 μM, and the EC50 values were on the order of 130–220 μM (Figures S9a,b and S10 in the Supporting Information). Thus, it appears that the ability of compound 1b and the analogues described herein to inhibit proliferation is independent of their ability to inhibit COX-1. Because these experiments were performed in cell culture, one cannot conclude that COX-1 inhibition would not inhibit tumor growth in vivo.74,80 Tumor growth and metastasis are complex processes involving multiple steps, such as angiogenesis, motility, invasion etc., which are not components of growth in vitro.36,81 Indeed, literature compound 5 inhibits tumor growth in different mouse models of ovarian carcinoma.35,82 Thus, E-DMSS analogues may have antitumor activity as well.76
Compound 3 (Figure 1) is a structurally related COX inhibitor to 1b (and 2) and was used for modeling of COX-1 inhibitor interactions.25 In 3, the p-chlorobenzoyl group at the indole nitrogen can rotate around the connecting σ-bond. This allows the molecule to take up an S-trans-conformation, besides its preferred and lower energy S-cis-conformation, to interact with COX. Whereas the cis conformer of 3 had been found to bind to both COX forms, the trans conformer was only present in cocrystal complexes with COX-1.25 This trans-conformation of 3 emulates the molecule geometry of the rigid E-2′-DMSS derivatives described in the present study (see Figure 1).
The nearly identical 3D structures of the two COX isoforms constitute a substantial obstacle for the design of selective COX-1 inhibitors.78,83 This is particularly true because in COX-1, the cavity of the selectivity pocket is less accessible due to the presence of Ile523. The same cavity is more spacious in COX-2, having a Val at the same amino acid position.1 It is assumed that compound 2 and its analogues described herein exert an atypical binding mode with COX, as the orientation of the p-methylsulfanylphenyliden group is predicted to cause steric clashes when the compounds are modeled into a similar configuration as 1b or 3 in the active site.84 Unfortunately, the attempt to cocrystallize one of the more potent E-DMSS analogues (2 or 13a) together with oCOX-1 has not succeeded thus far.83
There has been a report proposing an optional binding mode of COX-1-selective inhibitors derived from 3.84 Moreover, a recent docking study on the COX binding of 2-methyl-3-indolylacetic acid derivatives that preferentially inactivate COX-1 suggested that the binding specificity of the reported compounds resulted from individual plasticities of the 3D protein structure close to the binding site (“selective induced fit”).85 These data further point out the formation of a new lipophilic interaction site by COX-1 structural rearrangements triggered by the ligand that favors a boost in inhibitory activity for accurately fitting compounds. The less flexible COX-2 protein does not allow such conformational changes to take place and thus confines binding affinity.85 This hypothesis could be taken into consideration to rationalize the benefit of the rather bulky and lipophilic biphenyl substituent as part of our optimized COX-1 selective E-DMSS analogues (e.g., 13a or 21c).
COX-1 has emerged as a potential pharmacological target for the treatment of several clinical conditions, and preclinical and clinical data suggest that selective COX-1 inhibitors may not exhibit significant gastrointestinal and cardiovascular side effects.24,25,56 Thus, the E-DMSS analogues described in the present report may be useful leads for novel COX-1 inhibitors or useful probes for the involvement of COX-1 in physiological or pathophysiological processes. However, some of the inhibitors may have off-target effects that would limit their utility in vivo. For example, compound 13a is a potent activator of peroxisome proliferator-activated receptor (PPAR)γ in cell culture.51 Interestingly, the SAR for activation of PPARγ indicates that a free carboxyl group is absolutely required for activity. Thus, analogues such as the trifluoromethylsulfonimide, 21c, may be more useful as probes for COX-1 inhibition in vivo.
Experimental Section
General
Reagents and solvents were of commercial quality and were used without further purification. All synthesized intermediates and final compounds were structurally characterized using 1H NMR (optionally 19F NMR, 13C NMR) and electrospray ionization mass spectrometry. The purity of the test compounds was determined using HPLC [UV detection at 220 and 254 nm (optionally 288 and 347 nm) along with ELSD detection] and was generally ≥95%, if not denoted otherwise. The general synthetic procedures A–J are specified in the Supporting Information.
Preparation and Analytical Characterization of Exemplified Test Compounds
(E)-2-(5-Fluoro-1-((2-fluorobiphenyl-4-yl)methylene)-1H-inden-3-yl)acetic Acid (13d)
According to general procedure C, the title compound was obtained from the isomer mixture 10a (0.05 g, 0.23 mmol) and 2-fluorobiphenyl-4-carbaldehyde (0.05 g, 0.25 mmol) after heating to 65 °C for 3 h and subsequent stirring at ambient temperature overnight. After acidification of the reaction mixture and repeated extraction with dichloromethane, the organic layers were combined and concentrated in vacuo. The remaining yellow solid material revealed good analytical quality and required no further purification. Yield, 63 mg (74%): C24H16F2O2, Mr = 374.39. 1H NMR (400 MHz, DMSO-d6) δ: 3.70 (s, 2H), 7.08 (dtd, J = 1.2/2.4/8.4 Hz, 1H), 7.16–7.19 (m, 2H), 7.41–7.45 (m, 1H), 7.49–7.53 (m, 2H), 7.58–7.67 (m, 6H), 7.84 (dd, J = 5.2/8.0 Hz, 1H). HPLC (method 1) tR: 11.30 min (>99%, UV347). ESI/MS: calcd, 373; found, 373.13 ([M – 1]+, 22%), 329.27 ([M – 1]+–CO2, 100%).
(E)-2-(5-Fluoro-1-((4-methyl-2-phenylpyrimidin-5-yl)methylene)-1H-inden-3-yl)acetic Acid (13j)
According to general procedure C, the title compound was obtained from the isomer mixture 10a (0.05 g, 0.23 mmol) and 4-methyl-2-phenylpyrimidine-5-carbaldehyde (0.050 g, 0.25 mmol) after heating to 70 °C for 3 h and subsequent stirring at ambient temperature overnight. The crude product was purified by preparative thin-layer chromatography (SiO2, EtOAc/hexane, 0.5% AcOH) to afford 43 mg (51%) of 13h: C23H17FN2O2, Mr = 372.40. 1H NMR (400 MHz, DMSO-d6) δ: 2.69 (s, 3H), 3.67 (s, 2H), 6.87 (s, 1H), 7.11 (td, J = 2.4/8.4 Hz, 1H), 7.18 (dd, J = 2.4/9.2 Hz, 1H), 7.54–7.56 (m, 3H), 7.75 (s, 1H), 7.98 (dd, J = 5.2/8.4 Hz, 1H), 8.44–8.47 (m, 2H), 8.82 (s, 1H). HPLC (method 1) tR: 10.16 min (97%, UV347). LCMS (ESI) (method 2) tR: 2.95 min (97%, ELSD); m/z: 373.2 [M + 1]+.
(E)-2-(5-Fluoro-1-(4-(6-fluoropyridin-3-yl)benzylidene)-1H-inden-3-yl)acetic Acid (13k)
According to general procedure E, the title compound was obtained from (E)-2-(1-(4-bromobenzylidene)-5-fluoro-1H-inden-3-yl)acetic acid 11i (0.50 g, 0.14 mmol) and 2-fluoro-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine (0.037 g, 0.17 mmol) after 14 h at 90 °C. The crude product was purified by recrystallization from MeOH/hexanes. Yield, 25 mg (48%), bright yellow solid: C23H15F2NO2, Mr = 375.37. 1H NMR (400 MHz, DMSO-d6) δ: 3.69 (s, 2H), 7.07 (td, J = (1.2)2.4/8.8 Hz, 1H), 7.15–7.18 (m, 2H), 7.31 (dd, J = 2.8/8.8 Hz, 1H), 7.69 (s, 1H), 7.79–7.88 (m, 5H), 8.36 (td, J = 2.8/8.2 Hz, 1H), 8.63 (d, J = 2.8 Hz, 1H). 19F NMR (282 MHz, DMSO-d6) δ: −112.96 (d, 1F, C5′-F), −68.78 (d, 1F, Pyr-F). HPLC (method 1) tR: 9.75 min. LCMS (ESI) (method 2) tR: 2.70 min (≥95%, ELSD); m/z: 376.0 [M + 1]+.
Sodium (E)-2-(1-([1,1′-Biphenyl]-4-ylmethylene)-5-methoxy-1H-inden-3-yl)acetate [15b(Na)]
The title compound was prepared by the aldol condensation reaction specified in general procedure D (step 1) from the isomer mixture 10b (0.050 g, 0.22 mmol) and commercially available [1,1′-biphenyl]-4-carbaldehyde (0.043 g, 0.24 mmol). After stirring for 72 h at room temperature, the accumulated product precipitate was filtered and washed first with a little ice-cold methanol and then with diethylether. Compound 15b was obtained as a yellow solid (Na-salt) in 37% yield (31.5 mg): C25H19NaO3, Mr = 390.41. 1H NMR (300 MHz, DMSO-d6) δ: 3.23 (s, 2H), 3.79 (s, 3H), 6.74 (dd, J = 2.22/8.22 Hz, 1H), 6.89 (s, 1H), 6.99 (d, J = 2.22 Hz, 1H), 7.34–7.41 (m, 2H), 7.47–7.52 (m, 2H), 7.65 (d, J=8.22 Hz, 1H), 7.73–7.79 (m, 6H).
(E)-2-(1-([1,1′-Biphenyl]-4-ylmethylene)-5-methoxy-1H-inden-3-yl)acetic Acid (15b)
The title compound was prepared according to general procedure D (step 2) using 200 μL of 1.5 N HCl, starting from 15b(Na) (0.016 g, 0.041 mmol) in 1 mL of H2O. Yield, 13 mg (86%): C25H20O3, Mr = 368.42. 1H NMR (400 MHz, DMSO-d6) δ: 3.64 (s, 2H), 3.80 (s, 3H), 6.81 (dd, J = 2.4/8.2 Hz, 1H), 6.92 (d, J = 2.0 Hz, 1H), 7.05 (s, 1H), 7.39 (tt, J = 0.8/2.0/7.2 Hz, 1H), 7.47–7.51 (m, 3H), 7.72–7.79 (m, 7H). LCMS (ESI) (method 2) tR: 3.14 min (≥95%, ELSD); m/z: 369.2 [M + 1]+.
(E)-Isopropyl 2-(5-fluoro-1-(4-(methylthio)benzylidene)-1H-inden-3-yl)acetate (16c)
According to general procedure F, (E)-2-(5-fluoro-1-(4-(methylthio)benzylidene)-1H-inden-3-yl)acetic acid 2 (100 mg, 0.31 mmol) was subjected to reaction with 2-propanol (5 mL) to afford 16c as a fluffy yellow solid. Yield, 118 mg (100%). C22H21FO2S, Mr = 368.46. 1H NMR (400 MHz, DMSO-d6) δ: 400 MHz: 1.19 (d, J = 6.4 Hz, 6H), 2.52 (s, 3H), 3.72 (s, 2H), 4.94 (sept, J = 6.4 Hz, 1H), 7.05 (td, J = 8.9/2.4 (1.2) Hz, 1H), 7.13–7.15 (m, 2H), 7.35 (d, J = 8.4 Hz, 2H), 7.59–7.64 (m, 3H), 7.83 (dd, J = 8.4/5.2 Hz, 1H). 19F NMR (282 MHz, DMSO-d6) δ: −113.39 (1F, C5′-F). LCMS (ESI) 3.60 min (>99%, ELSD); m/z: 369.2 [M + 1]+.
(E)-2-(5-Fluoro-1-(4-(methylthio)benzylidene)-1H-inden-3-yl)-2-methylpropanoic Acid (19a)
Ester cleavage as described in general procedure G_variant2 was performed on 19b (15 mg, 0.039 mmol). The crude product was purified by flash chromatography (ethyl acetate/hexane 1:1, 0.5% HOAc, gradient) to produce 19a. Yield, 12.5 mg (90%). C21H19FO2S, Mr = 354.44. 1H NMR (400 MHz, DMSO-d6) δ: 1.53 (s, 6H), 2.53 (s, 3H), 6.98 (dd, J = 2.4/10.0 Hz, 1H), 7.02–7.07 (m, 2H), 7.36 (d, J = 8.8 Hz, 2H), 7.60 (s, 1H), 7.66 (d, J = 8.4 Hz, 2H), 7.86 (dd, J = 5.4/8.2 Hz, 1H). 19F NMR (282 MHz, DMSO-d6) δ: −113.31 (q, 1F, C5′-F). LCMS (ESI) (method 2) tR: 3.12 min (98%, ELSD); m/z: 355.2 [M + 1]+.
(E)-Ethyl 2-(1-([1,1′-biphenyl]-4-ylmethylene)-5-fluoro-1H-inden-3-yl)-2-methylpropanoate (19d)
The title compound was prepared by the aldol condensation reaction specified in general procedure D (step 1) from 10d (0.10 g, 0.40 mmol) and commercially available [1,1′-biphenyl]-4-carbaldehyde (0.081 g, 0.44 mmol). After stirring for 72 h at room temperature, the accumulated product precipitate was filtered and carefully washed with water. Compound 19d was obtained as a yellow solid (ethyl ester) in 98% yield (163 mg): C28H25FO2, Mr = 412.50. 1H NMR (400 MHz, DMSO-d6) δ: 1.07 (t, J = 7.2 Hz, 3H), 1.57 (s, 6H), 4.09 (q, J = 7.2 Hz, 2H), 6.90 (dd, J = 2.4/9.6 Hz, 1H), 7.05–7.10 (m, 2H), 7.40 (tt, J = 7.2 Hz, 1H), 7.50 (t, J = 7.2 Hz, 1H), 7.72–7.76 (m, 3H), 7.81 (s, 4H), 7.91 (dd, J = 5.2/8.4 Hz, 1H). LCMS (ESI) (method 2) tR: 3.89 min (>99%, ELSD); m/z: 413.2 [M + 1]+.
(E)-Ethyl 2-(5-fluoro-1-((6-(4-fluorophenyl)pyridin-2-yl)methylene)-1H-inden-3-yl)-2-methyl-propanoate (20b)
The title compound was prepared by the aldol condensation reaction specified in general procedure D (step 1) from 10d (0.050 g, 0.20 mmol) and commercially available 6-(4-fluorophenyl)picolin-aldehyde (0.045 g, 0.22 mmol). After stirring for 72 h at room temperature, the accumulated product precipitate was filtered and carefully washed with water. The title compound was obtained as a yellow solid (Na salt) in 83% yield (72.5 mg): C27H23F2NO2, Mr = 431.47. 1H NMR (400 MHz, DMSO-d6) δ: 1.06 (t, J = 7.2 Hz, 3H), 1.59 (s, 6H), 4.08 (q, J = 7.2 Hz, 2H), 6.91 (dd, J = 2.0/9.6 Hz, 1H), 7.07 (td, J = 2.4/8.8 Hz, 1H), 7.38 (pseudo t, J = 8.8 Hz, 2H), 7.61–7.63 (m, 2H), 7.87–8.00 (m, 3H), 8.07 (s, 1H), 8.20–8.24 (dd, J = 5.6/8.8 Hz, 2H). 19F NMR (282 MHz, DMSO-d6) δ: −111.98 (s, 1F), −110.74 (s, 1F). LCMS (ESI) (method 2) tR: 3.79 min (>99%, ELSD); m/z: 432.2 [M + 1]+.
(E)-2-(1-([1,1′-Biphenyl]-4-ylmethylene)-5-fluoro-1H-inden-3-yl)-N-((trifluoromethyl)sulfonyl)-acetamide (21c)
According to general procedure J, (E)-2-(1-([1,1′-biphenyl]-4-ylmethylene)-5-fluoro-1H-inden-3-yl)acetyl chloride (20 mg, 0.053 mmol) was subjected to reaction with trifluoromethanesulfonamide (11.9 mg, 0.080 mmol). The crude product was purified by flash chromatography (SiO2, EtOAc + hexane: 1 + 1 (0.5% AcOH, gradient) to afford 13.5 mg (52%) of 21c as bright yellow solid. C25H17F4NO3S, Mr = 487.47. 1H NMR (400 MHz, DMSO-d6) δ: 3.47 (s, 2H), 7.02 (td, J = 2.4/8.8 Hz, 1H), 7.09 (s, 1H), 7.15 (dd, J = 2.4/9.4 Hz, 1H), 7.39 (tt, J = 1.6/7.2 Hz, 1H), 7.49 (t, J = 7.2 Hz, 2H), 7.59 (s, 1H), 7.74 (dd, J = 1.6/7.2 Hz, 2H), 7.78 (s, 4H), 7.82 (dd, J = 5.2/8.4 Hz, 1H). 19F NMR (282 MHz, DMSO-d6) δ: −115.25 (s, 1F, C5′-F), −77.31 (s, 3F, −CF3). LCMS (ESI) (method 2) tR: 3.62 min (>99%, UV254, ELSD); m/z: 488.0 [M + 1]+.
Acknowledgments
This work was supported by the NIH (National Cancer Institute, CA89450, and the National Foundation for Cancer Research). A.J.L. gratefully acknowledges funding from the German Research Foundation (DFG-Forschungsstipendium, GZ: LI2019/1-1, Einzelprojekt). We are grateful to Dr. Donald Stec for assistance with NMR analysis and to Dr. Carol Rouzer for proofreading.
Glossary
Abbreviations Used
- NSAID
nonsteroidal anti-inflammatory drug
- PPAR
peroxisome proliferator-activated receptor
- COX
cyclooxygenase
- SAR
structure–activity relationship
- PG
prostaglandin
- AA
arachidonic acid
- RPMI
Roswell Park Memorial Institute medium
- HBSS
Hank's balanced salt solution
- FBS
fetal bovine serum
- mPGES-1
microsomal prostaglandin E2 synthase-1
- PBS
phosphate-buffered saline
- M-PER
mammalian protein extraction reagent
Supporting Information Available
Synthetic procedures, routine spectroscopic and spectrometric data, HPLC data, exemplified NMR spectra, and biochemical and biological testing methods. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Blobaum A. L.; Marnett L. J. Structural and Functional Basis of Cyclooxygenase Inhibition. J. Med. Chem. 2007, 50, 1425–1441. [DOI] [PubMed] [Google Scholar]
- Vane J. R. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nature (London), New Biol. 1971, 231, 232–235. [DOI] [PubMed] [Google Scholar]
- Smith J. B.; Willis A. L. Aspirin selectively inhibits prostaglandin production in human platelets. Nature (London), New Biol. 1971, 231, 235–237. [DOI] [PubMed] [Google Scholar]
- Ferreira S. H.; Moncada S.; Vane J. R. Indomethacin and aspirin abolish prostaglandin release from the spleen. Nature (London), New Biol. 1971, 231, 237–239. [DOI] [PubMed] [Google Scholar]
- Xie W.; Chipman J. G.; Robertson D. L.; Erikson R. L.; Simmons D. L. Expression of a mitogen-responsive gene encoding prostaglandin synthase is regulated by mRNA splicing. Proc. Natl. Acad. Sci. U.S.A. 1991, 88, 2692–2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J. Y.; Masferrer J. L.; Seibert K.; Raz A.; Needleman P. The induction and suppression of prostaglandin H2 synthase (cyclooxygenase) in human monocytes. J. Biol. Chem. 1990, 265, 16737–16740. [PubMed] [Google Scholar]
- Kujubu D. A.; Fletcher B. S.; Varnum B. C.; Lim R. W.; Herschman H. R. TIS10, a phorbol ester tumor promoter-inducible mRNA from Swiss 3T3 cells, encodes a novel prostaglandin synthase/cyclooxygenase homologue. J. Biol. Chem. 1991, 266, 12866–12872. [PubMed] [Google Scholar]
- Patel C. K.; Sen D. J. COX-1 and COX-2 inhibitors: Current status and future prospects over COX-3 inhibitors. Int. J. Drug Dev. Res. 2009, 1, 136–145. [Google Scholar]
- Kawai S.; Kojima F.; Kusunoki N. Recent advances in nonsteroidal anti-inflammatory drugs. Allergol. Int. 2005, 54, 209–215. [Google Scholar]
- Al-Hourani B. J.; Sharma S. K.; Mane J. Y.; Tuszynski J.; Baracos V.; Kniess T.; Suresh M.; Pietzsch J.; Wuest F. Synthesis and evaluation of 1,5-diaryl-substituted tetrazoles as novel selective cyclooxygenase-2 (COX-2) inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 1823–1826. [DOI] [PubMed] [Google Scholar]
- Pairet M.; Churchill L.; Trummlitz G.; Engelhardt G. Differential inhibition of cyclooxygenase-1 (COX-1) and −2 (COX-2) by NSAIDS: Consequences on anti-inflammatory activity versus gastric and renal safety. Inflammopharmacology 1996, 4, 61–70. [Google Scholar]
- Prasit P.; Riendeau D. Selective cyclooxygenase-2 inhibitors. Annu. Rep. Med. Chem. 1997, 32, 211–220. [Google Scholar]
- Talley J. J. Selective inhibitors of cyclooxygenase-2 (COX-2). Prog. Med. Chem. 1999, 36, 201–234. [DOI] [PubMed] [Google Scholar]
- Talley J. J.; Bertenshaw S. R.; Brown D. L.; Carter J. S.; Graneto M. J.; Koboldt C. M.; Masferrer J. L.; Norman B. H.; Rogier D. J. Jr.; Zweifel B. S.; Seibert K. 4,5-Diaryloxazole inhibitors of cyclooxygenase-2 (COX-2). Med. Res. Rev. 1999, 19, 199–208. [DOI] [PubMed] [Google Scholar]
- Chan C. C.; Boyce S.; Brideau C.; Charleson S.; Cromlish W.; Ethier D.; Evans J.; Ford-Hutchinson A. W.; Forrest M. J.; Gauthier J. Y.; Gordon R.; Gresser M.; Guay J.; Kargman S.; Kennedy B.; Leblanc Y.; Leger S.; Mancini J.; O'Neill G. P.; Ouellet M.; Patrick D.; Percival M. D.; Perrier H.; Prasit P.; Rodger I.; Tagari P.; Therien M.; Vickers P.; Visco D.; Wang Z.; Webb J.; Wong E.; Xu L. J.; Young R. N.; Zamboni R.; Riendeau D. Rofecoxib [Vioxx, MK-0966; 4-(4′-methylsulfonylphenyl)-3-phenyl-2-(5H)-furanone]: A potent and orally active cyclooxygenase-2 inhibitor. Pharmacological and biochemical profiles. J. Pharmacol. Exp. Ther. 1999, 290, 551–560. [PubMed] [Google Scholar]
- Esser R.; Berry C.; Du Z.; Dawson J.; Fox A.; Fujimoto R. A.; Haston W.; Kimble E. F.; Koehler J.; Peppard J.; Quadros E.; Quintavalla J.; Toscano K.; Urban L.; van Duzer J.; Zhang X.; Zhou S.; Marshall P. J. Preclinical pharmacology of lumiracoxib: A novel selective inhibitor of cyclooxygenase-2. Br. J. Pharmacol. 2005, 144, 538–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marnett L. J. The COXIB experience: A look in the rearview mirror. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 265–290. [DOI] [PubMed] [Google Scholar]
- Rodriguez L. A. G.; Tolosa L. B. Risk of upper gastrointestinal complications among users of traditional NSAIDs and COXIBs in the general population. Gastroenterology 2007, 132, 498–506. [DOI] [PubMed] [Google Scholar]
- Hippisley-Cox J.; Coupland C.; Logan R. Risk of adverse gastrointestinal outcomes in patients taking cyclo-oxygenase-2 inhibitors or conventional non-steroidal anti-inflammatory drugs: Population based nested case-control analysis. BMJ [Br. Med. J.] 2005, 331, 1310–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott E.; Nussmeier N. A.; Duke P. C.; Feneck R. O.; Alston R. P.; Snabes M. C.; Hubbard R. C.; Hsu P. H.; Saidman L. J.; Mangano D. T. Efficacy and safety of the cyclooxygenase 2 inhibitors parecoxib and valdecoxib in patients undergoing coronary artery bypass surgery. J. Thorac. Cardiovasc. Surg. 2003, 125, 1481–1492. [DOI] [PubMed] [Google Scholar]
- Nussmeier N. A.; Whelton A. A.; Brown M. T.; Langford R. M.; Hoeft A.; Parlow J. L.; Boyce S. W.; Verburg K. M. Complications of the COX-2 inhibitors parecoxib and valdecoxib after cardiac surgery. N. Engl. J. Med. 2005, 352, 1081–1091. [DOI] [PubMed] [Google Scholar]
- Cannon C. P.; Curtis S. P.; FitzGerald G. A.; Krum H.; Kaur A.; Bolognese J. A.; Reicin A. S.; Bombardier C.; Weinblatt M. E.; van der Heijde D.; Erdmann E.; Laine L. Cardiovascular outcomes with etoricoxib and diclofenac in patients with osteoarthritis and rheumatoid arthritis in the multinational etoricoxib and diclofenac arthritis long-term (MEDAL) programme: A randomized comparison. Lancet 2006, 368, 1771–1781. [DOI] [PubMed] [Google Scholar]
- Blobaum A. L.; Marnett L. J. NSAID action and the foundations for cardiovascular toxicity. Cardiotoxicity Non-Cardiovascular Drugs 2010, 257–285. [Google Scholar]
- Wallace J. L.; McKnight W.; Reuter B. K.; Vergnolle N. NSAID-induced gastric damage in rats: requirement for inhibition of both cyclooxygenase 1 and 2. Gastroenterology 2000, 119, 706–714. [DOI] [PubMed] [Google Scholar]
- Kakuta H.; Zheng X.; Oda H.; Harada S.; Sugimoto Y.; Sasaki K.; Tai A. Cyclooxygenase-1-Selective Inhibitors Are Attractive Candidates for Analgesics That Do Not Cause Gastric Damage. Design and in Vitro/in Vivo Evaluation of a Benzamide-Type Cyclooxygenase-1 Selective Inhibitor. J. Med. Chem. 2008, 51, 2400–2411. [DOI] [PubMed] [Google Scholar]
- Perrone M. G.; Scilimati A.; Simone L.; Vitale P. Selective COX-1 inhibition: A therapeutic target to be reconsidered. Curr. Med. Chem. 2010, 17, 3769–3805. [DOI] [PubMed] [Google Scholar]
- Aid S.; Bosetti F. Targeting cyclooxygenases-1 and -2 in neuroinflammation: Therapeutic implications. Biochimie 2011, 93, 46–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons D. L.; Botting R. M.; Hla T. Cyclooxygenase isozymes: The biology of prostaglandin synthesis and inhibition. Pharmacol. Rev. 2004, 56, 387–437. [DOI] [PubMed] [Google Scholar]
- Teismann P.; Tieu K.; Choi D.-K.; Wu D.-C.; Naini A.; Hunot S.; Vila M.; Jackson-Lewis V.; Przedborski S. Cyclooxygenase-2 is instrumental in Parkinson's disease neurodegeneration. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 5473–5478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S.-H.; Bosetti F. Cyclooxygenase-1 null mice show reduced neuroinflammation in response to β-amyloid. Aging 2009, 1, 234–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee A. C.; Carreras I.; Hossain L.; Ryu H.; Klein W. L.; Oddo S.; LaFerla F. M.; Jenkins B. G.; Kowall N. W.; Dedeoglu A. Ibuprofen reduces Aβ, hyperphosphorylated tau and memory deficits in Alzheimer mice. Brain Res. 2008, 1207, 225–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura D. K.; Morrison B. E.; Blankman J. L.; Long J. Z.; Kinsey S. G.; Marcondes M. C. G.; Ward A. M.; Hahn Y. K.; Lichtman A. H.; Conti B.; Cravatt B. F. Endocannabinoid Hydrolysis Generates Brain Prostaglandins That Promote Neuroinflammation. Science (Washington, DC, U.S.) 2011, 334, 809–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C. J.; Zhang Y.; Koboldt C. M.; Muhammad J.; Zweifel B. S.; Shaffer A.; Talley J. J.; Masferrer J. L.; Seibert K.; Isakson P. C. Pharmacological analysis of cyclooxygenase-1 in inflammation. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 13313–13318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenneis C.; Maier T. J.; Schmidt R.; Hofacker A.; Zulauf L.; Jakobsson P.-J.; Scholich K.; Geisslinger G. Inhibition of prostaglandin E2 synthesis by SC-560 is independent of cyclooxygenase 1 inhibition. FASEB J. 2006, 20, 1352–1360. [DOI] [PubMed] [Google Scholar]
- Daikoku T.; Tranguch S.; Trofimova I. N.; Dinulescu D. M.; Jacks T.; Nikitin A. Y.; Connolly D. C.; Dey S. K. Cyclooxygenase-1 Is Overexpressed in Multiple Genetically Engineered Mouse Models of Epithelial Ovarian Cancer. Cancer Res. 2006, 66, 2527–2531. [DOI] [PubMed] [Google Scholar]
- Gupta Rajnish A.; Tejada Lovella V.; Tong Beverly J.; Das Sanjoy K.; Morrow Jason D.; Dey Sudhansu K.; DuBois Raymond N. Cyclooxygenase-1 is overexpressed and promotes angiogenic growth factor production in ovarian cancer. Cancer Res. 2003, 63, 906–911. [PubMed] [Google Scholar]
- Kakuta H.; Fukai R.; Zheng X.; Ohsawa F.; Bamba T.; Hirata K.; Tai A. Identification of urine metabolites of TFAP, a cyclooxygenase-1 inhibitor. Bioorg. Med. Chem. Lett. 2010, 20, 1840–1843. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya D. K.; Lecomte M.; Dunn J.; Morgans D. J.; Smith W. L. Selective inhibition of prostaglandin endoperoxide synthase-1 (cyclooxygenase-1) by valerylsalicylic acid. Arch. Biochem. Biophys. 1995, 317, 19–24. [DOI] [PubMed] [Google Scholar]
- Siqueira-Junior J. M.; Peters R. R.; de Brum-Fernandes A. J.; Ribeiro-do-Valle R. M. Effects of valeryl salicylate, a COX-1 inhibitor, on models of acute inflammation in mice. Pharmacol. Res. 2003, 48, 437–443. [DOI] [PubMed] [Google Scholar]
- Labayle D.; Fischer D.; Vielh P.; Drouhin F.; Pariente A.; Bories C.; Duhamel O.; Trousset M.; Attali P. Sulindac causes regression of rectal polyps in familial adenomatous polyposis. Gastroenterology 1991, 101, 635–639. [DOI] [PubMed] [Google Scholar]
- Narisawa T. An overview on chemoprevention of colorectal cancer. Nihon Geka Gakkai zasshi 1998, 99, 362–367. [PubMed] [Google Scholar]
- Kashfi K.; Rigas B. Non-COX-2 targets and cancer: Expanding the molecular target repertoire of chemoprevention. Biochem. Pharmacol. 2005, 70, 969–986. [DOI] [PubMed] [Google Scholar]
- Piazza G. A.; Keeton A. B.; Tinsley H. N.; Gary B. D.; Whitt J. D.; Mathew B.; Thaiparambil J.; Coward L.; Gorman G.; Li Y.; Sani B.; Hobrath J. V.; Maxuitenko Y. Y.; Reynolds R. C. A novel sulindac derivative that does not inhibit cyclooxygenases but potently inhibits colon tumor cell growth and induces apoptosis with antitumor activity. Cancer Prev. Res. (Philadelphia, PA, U. S.) 2009, 2, 572–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piazza G. A.; Keeton A. B.; Tinsley H. N.; Whitt J. D.; Gary B. D.; Mathew B.; Singh R.; Grizzle W. E.; Reynolds R. C. NSAIDs: Old drugs reveal new anticancer targets. Pharmaceuticals 2010, 3, 1652–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Kingsley P. J.; Marnett L. J.; Eling T. E. The role of NAG-1/GDF15 in the inhibition of intestinal polyps in APC/Min mice by sulindac. Cancer Prev. Res. 2011, 4, 150–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbrink S. D.; Pergola C.; Buehring U.; George S.; Metzner J.; Fischer A. S.; Haefner A.-K.; Wisniewska J. M.; Geisslinger G.; Werz O.; Steinhilber D.; Maier T. J. Sulindac sulfide suppresses 5-lipoxygenase at clinically relevant concentrations. Cell. Mol. Life Sci. 2010, 67, 797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne F.; Resnick L.; Sagher D.; Brot N.; Weissbach H. Reduction of Sulindac to its active metabolite, sulindac sulfide: Assay and role of the methionine sulfoxide reductase system. Biochem. Biophys. Res. Commun. 2003, 312, 1005–1010. [DOI] [PubMed] [Google Scholar]
- Walters M. J.; Blobaum A. L.; Kingsley P. J.; Felts A. S.; Sulikowski G. A.; Marnett L. J. The influence of double bond geometry in the inhibition of cyclooxygenases by sulindac derivatives. Bioorg. Med. Chem. Lett. 2009, 19, 3271–3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusakiewicz J. J.; Felts A. S.; Mackenzie B. S.; Marnett L. J. Molecular Basis of the Time-Dependent Inhibition of Cyclooxygenases by Indomethacin. Biochemistry 2004, 43, 15439–15445. [DOI] [PubMed] [Google Scholar]
- Alcalde E.; Mesquida N.; Frigola J.; Lopez-Perez S.; Merce R. Indene-based scaffolds. Design and synthesis of novel serotonin 5-HT6 receptor ligands. Org. Biomol. Chem. 2008, 6, 3795–3810. [DOI] [PubMed] [Google Scholar]
- Felts A. S.; Siegel B. S.; Young S. M.; Moth C. W.; Lybrand T. P.; Dannenberg A. J.; Marnett L. J.; Subbaramaiah K. Sulindac Derivatives That Activate the Peroxisome Proliferator-activated Receptor gamma but Lack Cyclooxygenase Inhibition. J. Med. Chem. 2008, 51, 4911–4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musser J. H.; Kreft A. F. III; Failli A. A.; Demerson C. A.; Shah U. S.; Nelson J. A.. Substituted indole-, indene-, pyranoindole- and tetrahydrocarbazolealkanoic acid derivatives as inhibitors of PLA2 and lipoxygenase. 93-29199 5420289, 19930310, 1995.
- Musso D. L.; Cochran F. R.; Kelley J. L.; McLean E. W.; Selph J. L.; Rigdon G. C.; Orr G. F.; Davis R. G.; Cooper B. R.; Styles V. L.; Thompson J. B.; Hall W. R. Indanylidenes. 1. Design and Synthesis of (E)-2-(4,6-Difluoro-1-indanylidene)acetamide, a Potent, Centrally Acting Muscle Relaxant with Antiinflammatory and Analgesic Activity. J. Med. Chem. 2003, 46, 399–408. [DOI] [PubMed] [Google Scholar]
- Romeiro Nelilma C.; Leite Ramon D. F.; Lima Lidia M.; Cardozo Suzana V. S.; de Miranda Ana L. P.; Fraga Carlos A. M.; Barreiro Eliezer J. Synthesis, pharmacological evaluation and docking studies of new sulindac analogues. Eur. J. Med. Chem. 2009, 44, 1959–1971. [DOI] [PubMed] [Google Scholar]
- Jung M.; Wahl A. F.; Neupert W.; Geisslinger G.; Senter P. D. Synthesis and activity of fluorinated derivatives of sulindac sulphide and sulindac Sulphone. Pharm. Pharmacol. Commun. 2000, 6, 217–221. [Google Scholar]
- Felts A. S.; Ji C.; Stafford J. B.; Crews B. C.; Kingsley P. J.; Rouzer C. A.; Washington M. K.; Subbaramaiah K.; Siegel B. S.; Young S. M.; Dannenberg A. J.; Marnett L. J. Desmethyl Derivatives of Indomethacin and Sulindac as Probes for Cyclooxygenase-Dependent Biology. ACS Chem. Biol. 2007, 2, 479–483. [DOI] [PubMed] [Google Scholar]
- Broyles D. A.; Carpenter B. K. Factors affecting the selection of products from a photochemically generated singlet biradical. Org. Biomol. Chem. 2005, 3, 1757–1767. [DOI] [PubMed] [Google Scholar]
- Brewster K.; Chittenden R. A.; Pinder R. M.; Skeels M. Structure of the indene-3-acetic acids. II. Reformatskii reactions of 6-(benzyloxy)-, 5,6-dimethoxy-, and 6-methoxy-1-indanones. J. Chem. Soc., Perkin Trans. 1 1972, 941–943. [Google Scholar]
- Peerboom R. A. L.; de Koning L. J.; Nibbering N. M. M. On the stabilization of carbanions by adjacent phenyl, cyano, methoxycarbonyl, and nitro groups in the gas phase. J. Am. Soc. Mass Spectrom. 1994, 5, 159–168. [DOI] [PubMed] [Google Scholar]
- Peretto I.; Radaelli S.; Parini C.; Zandi M.; Raveglia L. F.; Dondio G.; Fontanella L.; Misiano P.; Bigogno C.; Rizzi A.; Riccardi B.; Biscaioli M.; Marchetti S.; Puccini P.; Catinella S.; Rondelli I.; Cenacchi V.; Bolzoni P. T.; Caruso P.; Villetti G.; Facchinetti F.; Del Giudice E.; Moretto N.; Imbimbo B. P. Synthesis and biological activity of flurbiprofen analogues as selective inhibitors of beta -amyloid1–42 secretion. J. Med. Chem. 2005, 48, 5705–5720. [DOI] [PubMed] [Google Scholar]
- Barreca G.; Cannata V.. Stereoselective isomerization process for the preparation of cis-5-fluoro-2-methyl-1-[4-(methylthio)benzyliden]inden-3-acetic acid. 2000-677842 6355836, 20001003, 2002.
- Liedtke A. J.; Keck P. R. W. E. F.; Lehmann F.; Koeberle A.; Werz O.; Laufer S. A. Arylpyrrolizines as Inhibitors of Microsomal Prostaglandin E2 Synthase-1 (mPGES-1) or as Dual Inhibitors of mPGES-1 and 5-Lipoxygenase (5-LOX). J. Med. Chem. 2009, 52, 4968–4972. [DOI] [PubMed] [Google Scholar]
- Hutchinson J. H.; Li Y.; Arruda J. M.; Baccei C.; Bain G.; Chapman C.; Correa L.; Darlington J.; King C. D.; Lee C.; Lorrain D.; Prodanovich P.; Rong H.; Santini A.; Stock N.; Prasit P.; Evans J. F. 5-Lipoxygenase-Activating Protein Inhibitors: Development of 3-[3-tert-Butylsulfanyl-1-[4-(6-methoxy-pyridin-3-yl)-benzyl]-5-(pyridin-2-ylmethoxy)-1H-indol-2-yl]-2,2-dimethylpropionic Acid (AM103). J. Med. Chem. 2009, 52, 5803–5815. [DOI] [PubMed] [Google Scholar]
- Bundgaard H.; Nielsen N. M.. Ester prodrug derivatives of carboxylic acid drugs. 1987-DK104 8801615, 19870825, 1988.
- Stock N.; Munoz B.; Wrigley J. D. J.; Shearman M. S.; Beher D.; Peachey J.; Williamson T. L.; Bain G.; Chen W.; Jiang X.; St-Jacques R.; Prasit P. The geminal dimethyl analogue of Flurbiprofen as a novel Abeta 42 inhibitor and potential Alzheimer's disease modifying agent. Bioorg. Med. Chem. Lett. 2006, 16, 2219–2223. [DOI] [PubMed] [Google Scholar]
- Hutchinson D. K.; Bellettini J. R.; Betebenner D. A.; Bishop R. D.; Borchardt T. B.; Bosse T. D.; Cink R. D.; Flentge C. A.; Gates B. D.; Green B. E.; Hinman M. M.; Huang P. P.; Klein L. L.; Krueger A. C.; Larson D. P.; Leanna M. R.; Liu D.; Madigan D. L.; McDaniel K. F.; Randolph J. T.; Rockway T. W.; Rosenberg T. A.; Stewart K. D.; Stoll V. S.; Wagner R.; Yeung M. C.. Preparation of fused thiadiazinediones, particularly dioxothiadiazinylnaphthalenones, as antiviral agents for the treatment of infections involving RNA-containing viral species such as hepatitis B and C and HIV. 2004-925072 20050107364, 20040824, 2005.
- Allegretti M.; Bertini R.; Cesta M. C.; Bizzarri C.; Di Bitondo R.; Di Cioccio V.; Galliera E.; Berdini V.; Topai A.; Zampella G.; Russo V.; Di Bello N.; Nano G.; Nicolini L.; Locati M.; Fantucci P.; Florio S.; Colotta F. 2-Arylpropionic CXC Chemokine Receptor 1 (CXCR1) Ligands as Novel Noncompetitive CXCL8 Inhibitors. J. Med. Chem. 2005, 48, 4312–4331. [DOI] [PubMed] [Google Scholar]
- Halder S.; Satyam A. Accidental discovery of a 'longer-range' vinylogous Pummerer-type lactonization: Formation of sulindac sulfide lactone from sulindac. Tetrahedron Lett. 2011, 52, 1179–1182. [Google Scholar]
- Barreiro E. J.; Lima M. E. F. The synthesis and anti-inflammatory properties of a new sulindac analog synthesized from natural safrole. J. Pharm. Sci. 1992, 81, 1219–1222. [DOI] [PubMed] [Google Scholar]
- Piazza G. A.; Rahm A. L. K.; Krutzsch M.; Sperl G.; Paranka N. S.; Gross P. H.; Brendel K.; Burt R. W.; Alberts D. S.; Pamukcu R.; Ahnen D. J. Antineoplastic drugs sulindac sulfide and sulfone inhibit cell growth by inducing apoptosis. Cancer Res. 1995, 55, 3110–3116. [PubMed] [Google Scholar]
- Williams C. S.; Goldman A. P.; Sheng H.; Morrow J. D.; DuBois R. N. Sulindac sulfide, but not sulindac sulfone, inhibits colorectal cancer growth. Neoplasia (New York) 1999, 1, 170–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter M.; Weiss M.; Weinberger I.; Furstenberger G. Marian, B. Growth inhibition and induction of apoptosis in colorectal tumor cells by cyclooxygenase inhibitors. Carcinogenesis 2001, 22, 17–25. [DOI] [PubMed] [Google Scholar]
- Tegeder I.; Pfeilschifter J.; Geisslinger G. Cyclooxygenase-independent actions of cyclooxygenase inhibitors. FASEB J. 2001, 15, 2057–2072. [DOI] [PubMed] [Google Scholar]
- Kundu N.; Fulton A. M. Selective cyclooxygenase (COX)-1 or COX-2 inhibitors control metastatic disease in a murine model of breast cancer. Cancer Res. 2002, 62, 2343–2346. [PubMed] [Google Scholar]
- Chen G.; Fei S. Non-steroidal anti-inflammatory drugs (NSAIDs) inhibiting the proliferation of human colorectal cancer cells. Nanjing Yike Daxue Xuebao 2005, 25, 923–927. [Google Scholar]
- Daikoku T.; Wang D.; Tranguch S.; Morrow J. D.; Orsulic S.; DuBois R. N.; Dey S. K. Cyclooxygenase-1 is a potential target for prevention and treatment of ovarian epithelial cancer. Cancer Res. 2005, 65, 3735–3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell J. A.; Akarasereenont P.; Thiemermann C.; Flower R. J.; Vane J. R. Selectivity of nonsteroidal antiinflammatory drugs as inhibitors of constitutive and inducible cyclooxygenase. Proc. Natl. Acad. Sci. U.S.A. 1993, 90, 11693–11697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gierse J. K.; McDonald J. J.; Hauser S. D.; Rangwala S. H.; Koboldt C. M.; Seibert K. A single amino acid difference between cyclooxygenase-1 (COX-1) and -2 (COX-2) reverses the selectivity of COX-2 specific inhibitors. J. Biol. Chem. 1996, 271, 15810–15814. [DOI] [PubMed] [Google Scholar]
- De Leval X.; Delarge J.; Devel P.; Neven P.; Michaux C.; Masereel B.; Pirotte B.; David J. L.; Henrotin Y.; Dogne J. M. Evaluation of classical NSAIDs and COX-2 selective inhibitors on purified ovine enzymes and human whole blood. Prostaglandins, Leukotrienes Essent. Fatty Acids 2001, 64, 211–216. [DOI] [PubMed] [Google Scholar]
- Kitamura T.; Kawamori T.; Uchiya N.; Itoh M.; Noda T.; Matsuura M.; Sugimura T.; Wakabayashi K. Inhibitory effects of mofezolac, a cyclooxygenase-1 selective inhibitor, on intestinal carcinogenesis. Carcinogenesis 2002, 23, 1463–1466. [DOI] [PubMed] [Google Scholar]
- Kino Y.; Kojima F.; Kiguchi K.; Igarashi R.; Ishizuka B.; Kawai S. Prostaglandin E2 production in ovarian cancer cell lines is regulated by cyclooxygenase-1, not cyclooxygenase-2. Prostaglandins, Leukotrienes Essent. Fatty Acids 2005, 73, 103–111. [DOI] [PubMed] [Google Scholar]
- Li W.; Xu R.-j.; Lin Z.-y.; Zhuo G.-c.; Zhang H.-h. Effects of a cyclooxygenase-1-selective inhibitor in a mouse model of ovarian cancer, administered alone or in combination with ibuprofen, a nonselective cyclooxygenase inhibitor. Med. Oncol. (Totowa, NJ, U. S.) 2009, 26, 170–177. [DOI] [PubMed] [Google Scholar]
- Picot D.; Loll P. J.; Garavito R. M. The x-ray crystal structure of the membrane protein prostaglandin H2 synthase-1. Nature (London, United Kingdom) 1994, 367, 243–249. [DOI] [PubMed] [Google Scholar]
- Harman C. A.; Turman M. V.; Kozak K. R.; Marnett L. J.; Smith W. L.; Garavito R. M. Structural Basis of Enantioselective Inhibition of Cyclooxygenase-1 by S-alpha -Substituted Indomethacin Ethanolamides. J. Biol. Chem. 2007, 282, 28096–28105. [DOI] [PubMed] [Google Scholar]
- Perissutti E.; Fiorino F.; Renner C.; Severino B.; Roviezzo F.; Sautebin L.; Rossi A.; Cirino G.; Santagada V.; Caliendo G. Synthesis of 2-Methyl-3-indolylacetic Derivatives as Anti-Inflammatory Agents That Inhibit Preferentially Cyclooxygenase 1 without Gastric Damage. J. Med. Chem. 2006, 49, 7774–7780. [DOI] [PubMed] [Google Scholar]
- Bikzhanova G. A.; Toulokhonova I. S.; Gately S.; West R. Novel silicon-containing drugs derived from the indomethacin scaffold: Synthesis, characterization and evaluation of biological activity. Silicon Chem. 2007, 3, 209–217. [Google Scholar]
- Boltze K. H.; Brendler O.; Jacobi H.; Opitz W.; Raddatz S.; Seidel P. R.; Vollbrecht D. Chemical structure and antiinflammatory activity in the group of substituted indole-3-acetic acids. Arzneim.-Forsch. 1980, 30, 1314–1325. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.