

Abstract
The activation of STAT transcription factors is a critical determinant of their subcellular distribution and their ability to regulate gene expression. Yet, it is not known how activation affects the behavior of individual STAT molecules in the cytoplasm and nucleus. To investigate this issue, we injected fluorescently labeled STAT1 in living HeLa cells and traced them by single-molecule microscopy. We determined that STAT1 moved stochastically in the cytoplasm and nucleus with very short residence times (<0.03 s) before activation. Upon activation, STAT1 mobility in the cytoplasm decreased ∼2.5-fold, indicating reduced movement of STAT1/importinα/β complexes to the nucleus. In the nucleus, activated STAT1 displayed a distinct saltatory mobility, with residence times of up to 5 s and intermittent diffusive motion. In this manner, activated STAT1 factors can occupy their putative chromatin target sites within ∼2 s. These results provide a better understanding of the timescales on which cellular signaling and regulated gene transcription operate at the single-molecule level.
Introduction
Extracellular signaling to the nucleus frequently entails the activation of transcription factors at the cell membrane. These transcription factors then translocate to the nucleus, where they bind to specific sequences in the promoters of their target genes. An example is the STAT1 signaling pathway, which transmits interferon-γ (IFNγ) signals that have numerous immune-modulatory functions (1,2). STAT1 is a dimer that moves rapidly in and out of the nucleus before the cells are stimulated by interferon. This occurs independently of metabolic energy via direct contacts with proteins of the nuclear pores. After treatment of the cells with interferon, approximately one-third of the STAT1 molecules become activated, i.e., they are phosphorylated on a single tyrosine residue. This happens at interferon receptors at the cell membrane. Activation is associated with the emergence of another, parallel dimer conformation (as opposed to the antiparallel conformation of the unphosphorylated dimers). Only parallel dimers are capable of DNA binding and hence transcription activation. Another important consequence of STAT1 activation is its switch from carrier-independent to carrier-dependent nuclear import. Activated dimers expose a nuclear localization signal (NLS) that is required for their nuclear import in complex with importin transport factors (3,4). Inside the nuclei, the activated STAT1 dimers bind to promoter regions of genes containing γ-activated site (GAS) motifs and activate transcription (1,5–7). Approximately 32,000 binding sites can be occupied by STAT1 molecules upon IFNγ-stimulation, as determined by chromatin immunoprecipitation sequencing (ChIP-Seq) (8). On DNA, STAT1 dimers can interact with one another, resulting in strongly reduced dissociation from DNA, as determined in vitro. The interaction of STAT1 with chromatin in vivo presumably leads to prolonged residence times at gene promoters; however, the exact values are not known (9). A hallmark associated with STAT1 activation is the visible accumulation of activated dimers in the nucleus of cytokine-stimulated cells (7). This phenomenon is explained by the inability of phosphorylated STAT1 to exit from the nucleus, which is retained in the nucleus until dephosphorylation occurs, allowing nuclear export (10,11).
The movement of transcription factors within cells, and their encounter with and binding to their target genes are decisive for both rapid signal transmission and efficient gene regulation (12,13). Accordingly, our molecular understanding of the activity of transcription factors is only as good as our knowledge of their dynamic properties. To explore these processes, investigators have used fluorescence recovery after photobleaching (FRAP) and fluorescence correlation spectroscopy (FCS) extensively, and we owe essentially all of our current knowledge about protein dynamics to the use of these methods (14,15). A potential drawback of these techniques is that their results can vary with the theoretical model employed and the specific approach used for data analysis. The observation of transcription factors at the single-molecule level can provide a solution to this problem because molecular dynamics can be viewed directly and the results are less ambiguous (13,16). Single-molecule imaging allows one to quantify different modes of mobility and visualize the binding processes. However, in experiments with eukaryotic cells, it is difficult to obtain sufficiently long single-molecule trajectories with a high time resolution, and hence such information is rarely obtained.
In previous studies (for review, see (17)), we used sensitive high-speed microscopy in living cells, which allows the visualization and tracing of single protein molecules at physiological concentrations. Using this method, we were able to analyze biomolecular interactions with nanometer precision and millisecond time resolution. In the work presented here, we analyzed the cytoplasmic and intranuclear mobility of single fluorescently labeled STAT1 molecules before and after activation. To that end, we covalently fluorescence-labeled STAT1 molecules with the photostable dye ATTO647N (ATTO-TEC, Siegen, Germany). STAT1 was activated in vitro or in vivo by treatment of cells with interferon γ. We found that before activation occurred, STAT1 was immobilized for only tens of milliseconds irrespective of the cellular compartment. The activation step dramatically modified the transcription factor dynamics. Reduced mobility of activated STAT1 was observed in the cytoplasm, but it was most pronounced in the nucleus, where the activated transcription factors showed distinct retardation and immobilization lasting up to several seconds. Our detailed statistical trajectory analysis revealed numerous binding events of activated STAT1, and for the first time (to our knowledge) allowed the direct observation of transcription factor binding events in eukaryotic cells.
Materials and Methods
Fluorescent proteins
Wild-type STAT1α, phosphorylated STAT1-P, STAT1-tc, GST-NLS, and GST-NTF2 were prepared as previously described (7,18,19). All of the STAT1 variants and GST-NLS were labeled with amino-reactive ATTO647N. Bovine serum albumin (BSA; Sigma-Aldrich, St. Louis, MO) and NTF2 (after removing the GST tag) were labeled with amino-reactive Alexa Fluor-488 (AF488; Invitrogen, Carlsbad, CA). Electrophoretic mobility shift assays quantifying the DNA-binding activity of fluorescently labeled STAT1-P were performed with purified Tyr701-phosphorylated STAT1 protein as described previously (19). The STAT1 protein was unlabeled or AF488-labeled, and the dye/protein molar ratio was between 1 and 2. As the probe, we used the duplex oligonucleotides 5′-CGACATTTCCGTAAATCTG (the complementary strand and 5′-ACGT overhangs on both strands for radioactive labeling by the Klenow reaction are not listed; the STAT1 binding site M67 is in boldface and underlined). The reaction contained 2.5 nM STAT1 protein and 1 nM DNA. Binding activity was detected and quantified by phosphorimaging.
Cells and microinjection
HeLa S3 cells were seeded on coverslips 1 day before measurements were obtained. For microinjection at the confocal laser scanning microscope (LSM510 Meta; Carl Zeiss, Jena, Germany), proteins were diluted in transport buffer (20 mM HEPES/KOH, pH 7.3, 110 mM potassium acetate, 5 mM sodium acetate, 2 mM magnesium acetate, 1 mM EGTA, and 2 mM DTT) to 7 μM. For single-molecule microscopy, proteins were diluted to 1–10 nM. Proteins were microinjected into the cytoplasm via an Eppendorf microinjector (Femtojet; Eppendorf, Hamburg, Germany) with ∼50 hPa injection pressure for 1 s. Stimulation with human IFNγ (Calbiochem, Darmstadt, Germany) was done for 30–60 min using 5 ng/ml. We performed all of the measurements at 37°C using type 37 DF immersion oil from Cargille Laboratories (Cedar Grove, NJ).
Single-molecule microscopy
We performed single-molecule imaging using an inverted microscope with a 63X NA 1.4 oil immersion objective lens and a fourfold magnifier in front of the EMCCD camera (iXon BI DV-860; Andor Technologies, Belfast, Ireland) as previously described (20). To image single fluorescent proteins, we recorded movies with 1000–2000 frames with an integration time of 5 ms at 191.6 Hz. For each protein, 20–50 movies from 10–20 cells were acquired and analyzed. We identified and tracked the single-molecule signals using Diatrack 3.02 (Semasopht, Chavannes, Switzerland). Further data processing was performed as described previously (21). After the nuclear envelope was localized in reference images, the trajectories identified by the tracking software were sorted into nucleoplasmic and cytoplasmic ones. Signals that occurred in a 10-pixel border region along both sides of the nuclear envelope were discarded to avoid evaluation of molecules during nucleocytoplasmic transport.
Heterogeneous mobility populations were analyzed by means of a jump-distance analysis as described previously (22). The probability that a diffusing molecule will be encountered within a distance r and width dr after time t from that position is given by
| (1) |
We quantified several mobility fractions by curve-fitting, considering several terms according to Eq. 1, yielding for n different species:
| (2) |
where fj designates the fractions with diffusion constants Dj.
Determination of binding duration
For the analysis of single STAT1 binding times, we used trajectories that we determined after smoothing the original data by a Gaussian kernel with a standard deviation of one pixel in x, y, and time to reduce signal fluctuations. However, long binding events were not fully recovered by Diatrack due to image noise or dye blinking. Therefore, we considered trajectories as erroneously interrupted if a subsequent position was <1 μm away from a previous segment. Such interrupted trajectories were connected manually and controlled individually by visual inspection.
Apparently immobile particles showed a certain positional variance σb2 that was due to a finite localization precision caused by amplification, background, and shot noise. This variance σb2 was independent of time and could be estimated from distinct binding events observed in the data (see Fig. S1 in the Supporting Material). From 15 such events, we deduced a value of <σb2> = 0.0005 μm2. We assumed immobility due to binding in a trajectory when the positional variance of a trajectory segment approached this <σb2>. To identify such events in the trajectory data, we related the local variance σn2 of subsequent particle positions within the temporal window of n frames to <σb2>. (A similar approach was suggested in previous studies (23–26).) We calculated a binding parameter, LE, as a moving average in windows of length n along the trajectories as follows:
| (3) |
According to this definition, LE was ≈1 for bound molecules and <1 for mobile molecules, because for the latter the variance was greater than <σb2> due to diffusional jumps between the subsequent observations. The advantage of comparing <σb2> and σn2 is that no Brownian diffusion has to be assumed for the unbound particle population.
The theoretical minimum value of LE was related to the average positional variance <σn2> of the mobile molecules, which could be related to their diffusion coefficient D by <σn2> = 4DΔt, where Δt is the time duration of the window of length n. We defined the threshold for switching between bound and mobile states to be in the middle between one and the minimum LE. When LE was plotted as a function of time, binding times could be determined directly, when <σn2> was known.
To test this approach, we performed extensive Monte Carlo simulations with particles switching between free and bound states for different ratios of immobility versus mobility, i.e., <σb2>/<σn2>, and for binding durations ranging from 10 ms to 2 s (see Fig. S2). We recovered >80% of simulated binding events correctly for <σb2>/<σn2> < 0.15, if the binding events lasted >100 ms. Also, for <σb2>/<σn2> = 0.1, we recovered approximately correct binding durations in the range of 50–800 ms, with a certain underestimation for binding times >300 ms (Fig. S3). An analysis of the trajectories derived from smoothed STAT1 data verified that <σb2>/<σn2> ≤ 0.1. Subsequently, we calculated LE for all STAT1 trajectories with at least five jumps and analyzed it as a function of time. Trajectory segments with LE greater than the threshold value, Lc = 0.5, indicated binding of STAT1 molecules. The final results were robust to its exact choice (see Table S1 and Table S2).
Results
We examined the intracellular distribution and mobility of several recombinant STAT variants. Activation of unphosphorylated wild-type STAT1 (wt-STAT1) was achieved in vitro, such that essentially all STAT1 molecules became activated (STAT1-P). Alternatively, wt-STAT1 was activated within cells by stimulation with IFNγ, resulting in the activation of ∼30% of both the endogenous native STAT1 and the microinjected recombinant STAT1. In addition, we tested truncated STAT1 (STAT1-tc) lacking both the N-domain of ∼130 residues and the C-terminal transactivation domain of ∼40 residues. Importantly, these truncations render the molecule monomeric and preclude transactivation (Fig. 1 A).
Figure 1.
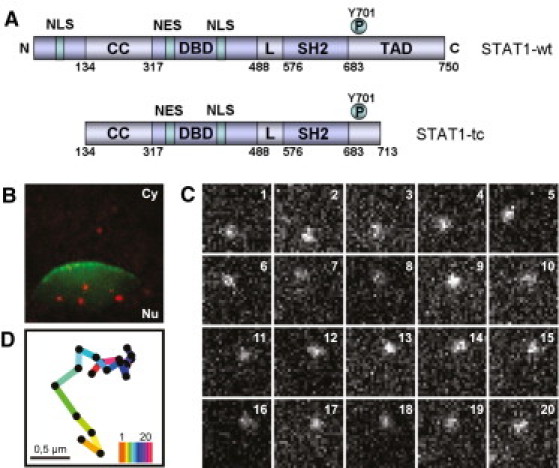
Structure and visualization of STAT1. (A) Schematic representation of the STAT1 wild-type and the truncated mutant. N, N domain; CC, coiled coil domain; NES, nuclear export signal; DBD, DNA-binding domain; L, linker domain; SH2, Src homology 2 domain; TAD, transactivation domain. (B) wt-STAT1 molecules (red) and nuclear envelope labeled by NTF2-AF488 (green) in a microinjected HeLa cell. Field size: (12.2 μm)2. (C) Sequence of 20 magnified consecutive image sections of a single wt-STAT1 molecule in the cell nucleus. Frame rate: 191.6 Hz; field size: (2.5 μm)2. (D) Complete trajectory of the wt-STAT1 molecule shown in C; the color code is according to the frame number in C.
Functionality of fluorescently labeled STAT1 variants
To assess the functionality of the ATTO647N-labeled STAT1 variants, we examined their intracellular distribution by confocal microscopy at 37°C after microinjection into the cytoplasm of HeLa cells (Fig. S4). As the positive nuclear import control, we used GST-NLS-ATTO647N, a stable dimer of ∼58 kDa that accumulates in the nucleus within minutes due to its NLS. In further agreement with expectations, STAT1-tc (a 68 kDa monomer) and wt-STAT1 (a ∼170 kDa dimer) distributed approximately evenly in both the nucleus and cytoplasm, thus confirming their ongoing nucleocytoplasmic shuttling in unstimulated cells. In contrast, incubation of cells with IFNγ for 60 min after cytoplasmic injection of wt-STAT1, and injection of in vitro activated STAT1-P, resulted in increased nuclear accumulation of STAT1. Additionally, we used gel shift experiments to assess the impact of fluorescence labeling on the DNA-binding activity of STAT1-P, and found that labeled STAT1-P retained ∼100% of the DNA-binding activity of the unlabeled STAT1-P (Fig. S5). Together, these results confirm the DNA-binding activity and undisturbed intracellular dynamics of the fluorescently labeled STAT1 proteins used in this study. Additionally, nuclear exclusion of AF488-conjugated BSA (BSA-AF488), which was coinjected in these experiments, demonstrated uncompromised nuclear envelope integrity of the cells.
Tracking of single STAT1 molecules
To investigate the intracellular mobility of STAT1, we used single-molecule fluorescence microscopy. STAT1 was microinjected at 37°C into the cytosol of HeLa cells together with fluorescently labeled NTF2, a nuclear transport factor that is concentrated at the nuclear envelope (20). To observe individual STAT1 molecules, we recorded movies using a high-speed EMCCD camera. An example frame is presented in Fig. 1 B, which shows the NTF2-AF488 fluorescence demarcating the nuclear envelope, and several wt-STAT1-ATTO647N molecules in the cytosol and nucleus. Fig. 1 C shows an image sequence revealing the dynamics of a mobile wt-STAT1 molecule in the cell nucleus. The variable signal intensities indicate that the molecule moved not only laterally but also axially, causing signal reduction and defocusing blur in some images. For a quantitative mobility analysis, single STAT1 molecules were identified, localized, and tracked in the movies. In Fig. 1 D, the trajectory of the molecule shown in Fig. 1 C is represented by dots and lines. The dots represent the various positions of the molecule along the trajectory, and the connecting lines represent the distances covered between frames. A summary of all recorded trajectories in the cells for truncated and full-length STAT1 before and after activation is shown in Fig. 2. The trajectories were plotted into the corresponding NTF2 reference images to provide a qualitative representation of the overall STAT1 dynamics, which reveal clear differences between inactive and activated STAT1. The unphosphorylated STAT1-tc and wt-STAT1 appear to be similarly mobile, and most of their tracks are seen in the cytoplasm. In contrast, upon activation, the majority of STAT1 molecules reside in the nucleus, reflecting their nuclear retention due to nuclear export block and DNA binding (7). Moreover, the activated STAT1 molecules display a striking clustering of trajectory positions, indicating an overall decrease in mobility, which is especially marked within the nucleus. Of note, this phenomenon occurred irrespective of the method by which activation of STAT1 was achieved, that is, no difference was seen between STAT1 activated in vitro and STAT1 activated in the cells by treatment with IFNγ. We therefore conclude that the mobility change was triggered solely by the phosphorylation of STAT1.
Figure 2.
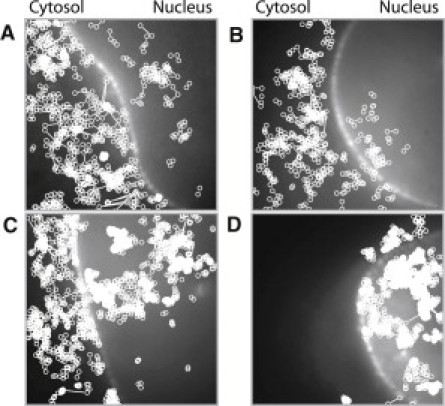
Single STAT1 molecule tracks in living cells. Compilations of all identified trajectories in example cells observed for (A) STAT1-tc, (B) wt-STAT1, (C) STAT1-P, and (D) wt-STAT1 after IFNγ activation, plotted into the NTF2 reference images (left, cytosol; right, nucleus). Field size: (12.2 μm)2.
Quantitative mobility analysis
We next sought to quantify STAT1 mobility. To that end, in all of the recorded movies we measured the jump distances covered by the transcription factors between successive frames, and plotted the results in histograms (Fig. 3 A). For molecules performing purely Brownian diffusion, such jump-distance histograms can be described by Eq. 1. Within cells, however, even noninteracting molecules do not show such one-component diffusion, due to the complex intracellular environment (27,28). Therefore, we expected that an interacting molecule such as STAT1 would require several diffusion constants to adequately describe its mobility. We found that numerous trajectories contained mobile and immobile phases. Accordingly, for all experiments, a set of four mobility fractions was required corresponding to a total of eight fitting parameters. To minimize parameter correlations between the fitting parameters, we reduced the number of free parameters by determining the prevailing diffusion coefficients Di in a first series of fits. In a previous work (29), we introduced this data analysis approach and demonstrated its adequacy. The results for the four Di-values were averaged over the experiments, which yielded the following values: D1 = 0.12 μm2/s, D2 = 0.475 μm2/s, D3 = 3 μm2/s, and D4 = 12 μm2/s. D1, which corresponded to a localization precision of σ = SQRT(4 × D1 × 0.005 s) ≈ 50 nm, characterized immobile molecules, and D4 corresponded to mobile molecules. The intermediate diffusion coefficients D2 and D3 reflected the different extents to which binding interactions took place in the cells.
Figure 3.
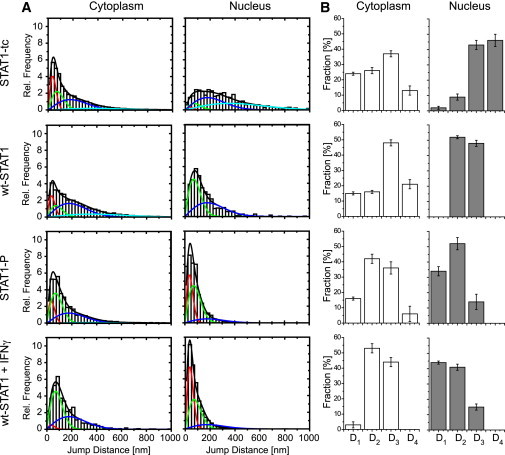
Mobility of the STAT1 variants in living cells. (A) Jump-distance distributions for the indicated STAT1 variants in cytoplasm and nucleus. The distributions in A were fitted using a four-component model with D1 = 0.12 μm2/s (red), D2 = 0.475 μm2/s (green), D3 = 3 μm2/s (blue), and D4 = 12 μm2/s (turquoise). Colored lines show mobility fractions, and the black lines show their sum. (B) Corresponding sizes of the fractions for D1–D4.
Dynamics of STAT1 before and after activation
To simplify comparisons between the STAT1 variants, we present the relative contributions of the four components (D1–D4) to STAT1 mobility in Fig. 3 B. In addition, Table 1 shows average values for the mobile components (D2–D4) to facilitate comparisons of our data with previous measurements of protein mobility obtained with classical methods such as FRAP and FCS. As shown in Fig. 3 B, in the cytoplasm the mobility patterns of STAT1-tc and wt-STAT1 are very similar, with all four mobility fractions each contributing ≥10%. Within nuclei, STAT1-tc was highly mobile, as 80% of the observed jumps corresponded to D3 and D4. Wt-STAT1 showed a trend to lower nuclear mobility, because there were almost no molecules with the D4 component. However, molecules with a D1 diffusion coefficient indicative of immobile STAT1 were essentially absent for both unphosphorylated STAT1 proteins. These data indicate that the monomer-to-dimer transition of STAT1 apparently has little effect on mobility. In contrast, phosphorylation, whether achieved in vitro or after IFNγ stimulation of cells, significantly reduced STAT1 mobility. Of note, this occurred in both the cytoplasm and the nucleus. However, the reduction was more pronounced in the nucleus, where ∼40% of the jumps indicated virtual immobility (D1) and another 40% showed quite hindered movements (D2). Accordingly, the fractions of more-mobile molecules characterized by D3 and D4 were strongly diminished or absent, respectively, after activation.
Table 1.
Single-molecule jump-distance analysis for STAT1 variants in cytosol and nucleus
| Cytosol | A1 (%) of D1 = 0.12 μm2/s | A2 (%) of D2 = 0.475 μm2/s | A3 (%) of D3 = 3μm2/s | A4 (%) of D4 = 12 μm2/s | Dmob (μm2/s) |
|---|---|---|---|---|---|
| STAT1-tc | 24 ± 1 | 26 ± 2 | 37 ± 2 | 13 ± 3 | 3.7 |
| wt-STAT1 | 15 ± 1 | 16 ± 1 | 48 ± 2 | 21 ± 3 | 4.7 |
| wt-STAT1-P | 16 ± 1 | 42 ± 3 | 36 ± 4 | 6 ± 5 | 2.4 |
| wt-STAT1+IFNγ | 3 ± 2 | 53 ± 3 | 44 ± 3 | 0 | 1.6 |
| Nucleus | A1 (%) of D1 = 0.12 μm2/s | A2 (%) of D2 = 0.475 μm2/s | A3 (%) of D3 = 3 μm2/s | A4 (%) of D4 = 12 μm2/s | Dmob (μm2/s) |
| STAT1-tc | 2 ± 1 | 9 ± 2 | 43 ± 3 | 46 ± 4 | 7 |
| wt-STAT1 | 0 | 52 ± 1 | 48 ± 2 | 0 | 1.7 |
| wt-STAT1-P | 34 ± 3 | 52 ± 4 | 14 ± 5 | 0 | 1 |
| wt-STAT1+IFNγ | 44 ± 1 | 41 ± 2 | 15 ± 2 | 0 | 1.1 |
A, relative fraction; D, diffusion coefficient; Dmob, average of the mobile components D2–D4.
Mobility of individual STAT1 molecules in the nucleus
To evaluate the binding events, we developed a statistical analysis in which immobilization in a trajectory was reported by a parameter LE, which approached one for bound phases and zero for mobile trajectory segments. The calculation and analysis of LE along the trajectory allowed us to reliably identify the immobilization and binding phases, respectively. To confirm this, we performed extensive numerical simulations of particles with diffusion and binding periods (Fig. S2 and Fig. S3). In our data analysis, trajectory segments with LE > 0.5 were defined to indicate a binding event (see Materials and Methods, and further details in the Supporting Material). Fig. 4 A shows example trajectories representing intranuclear mobility of STAT1-tc, wt-STAT1, STAT1-P, and IFNγ-activated wt-STAT1. For STAT1-tc and wt-STAT1, trajectory segments indicative of immobilized or slow-moving molecules are few and short. Contrary, activated STAT1 showed repeated and long-lasting immobilizations, indicating strong binding at sites in the nucleus. Examples of the data analysis are shown in Fig. 4, B–E, where the LE-values are plotted along the time line of the trajectories shown in Fig. 4 A. The trajectory segments, where LE > 0.5, are color-coded in these time lines and correspondingly marked in Fig. 4 A.
Figure 4.
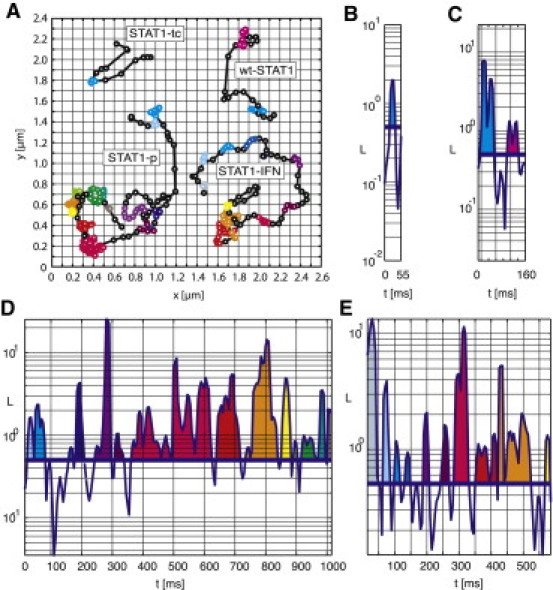
STAT1 trajectories in cell nuclei and the corresponding LE plots. (A) Nuclear trajectories of STAT1-tc, wt-STAT1, STAT1-P, and wt-STAT1 upon IFNγ activation. Colored trajectory sections indicate immobile STAT1 (LE > 0.5). (B–E) Corresponding LE plots for the trajectories shown in A with (B) STAT1-tc, (C) wt-STAT1, (D) STAT1-P, and (E) wt-STAT1 upon IFNγ activation. The same color code was used in A and B–E.
Estimation of binding duration
Finally, as described in the Materials and Methods section, we deduced the actual durations of binding events by analyzing LE along all trajectories obtained in the various experiments. The respective distributions of binding times for the four STAT1 variants in cytoplasm and nucleus are shown in Fig. 5. For all STAT1 variants, activated or not, we observed only a few and relatively short binding events in the cytoplasm (Fig. 5, A–D). These distributions were fitted by monoexponential decay functions. We obtained a mean binding duration of τcyt = 0.03 ± 0.015 s. However, due to the small number of observed binding events, the fits were not satisfactory and the results were not very reliable. For the same reason, it was not possible to determine the duration of binding events in the nucleus for STAT1-tc and wt-STAT1 before activation (Fig. 5, E and F). In stark contrast, repeated nuclear binding events lasting up to 5 s were observed for STAT1 after its activation. A two-component exponential decay function was required for a satisfactory description of the corresponding binding-time histograms (Fig. 5, G and H), yielding time constants that did not differ significantly for the two experiments, namely, a short τ1 = 0.02 ± 0.01 s and a long τ2 = 0.5 ± 0.4 s. All of the fitting results for the binding-time histograms, together with statistical data on the trajectories, are reported in Table S1. Finally, we tested the dependence of the binding time τ2 on the LE threshold chosen for detecting bound states. Reassuringly, no significant dependence of τ2 on the value of LE threshold within the error range of the fit was found for values ranging from 0.3 to 0.7 (see Table S2).
Figure 5.
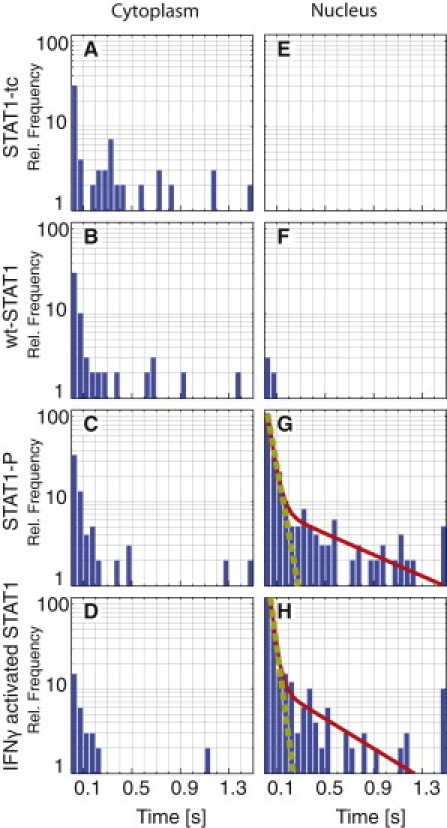
STAT1 binding in cytoplasm and nucleus. (A–H) The histograms display the distribution of binding durations as detected with the LE plots. The referenced data are indicated in the figure (top and left). The dashed line shows the result of a fit by a single exponential decay, and the solid line represents the sum of two fitted exponential decay functions. The fitting results are summarized in Table S1. Data for IFNγ-activated Stat1 in the cytoplasm (D), nuclear STAT1-tc (E), and nuclear wt-STAT1 (F) were too sparse for meaningful fitting.
Discussion
To elucidate the dynamics of extracellular signaling to the nucleus, we observed the mobility of STAT1 transcription factors in the cytoplasm and nucleus. Measurements were made in the resting state and after activation by IFNγ in living cells. STAT-P molecules, which were activated in vitro with epidermal growth factor-receptor kinase, served as a positive control. A truncation mutant, STAT-tc, which is monomeric and hence does not bind to cognate STAT1 recognition sites, was used as a negative control. All measurements were performed at physiological temperature to ensure proper functioning of the signal transduction pathway. Upon injection in the cytoplasm, the ATTO647N-labeled STAT1 variants acquired the nucleocytoplasmic distribution expected for unlabeled STAT1; moreover, responsiveness to IFNγ treatment became readily apparent as increased accumulation in the nucleus. Together with their unchanged DNA binding, these results indicate that the labeled molecules used in our experiments were fully functional.
We analyzed the molecular dynamics of STAT1 transcription factors using high-speed single-molecule microscopy. None of the STAT1 variants examined showed completely free diffusion under any condition tested, indicating that these proteins are subject to numerous nonspecific binding processes. STAT1 mobility was described satisfactorily by four diffusion coefficients: D1 = 0.12 μm2/s, D2 = 0.475 μm2/s, D3 = 3 μm2/s and D4 = 12 μm2/s. D1 meant that the molecules moved a distance of ∼50 nm in the time of 5 ms that was required to obtain a single image. It should be kept in mind that even completely fixed molecules display a virtual movement, which is due to the nonzero localization precision of the fitting procedure used to define the positions of single particles. According to our previous experiments, a mean localization precision of 50 nm is realistic for single-molecule imaging experiments performed at 37°C and an image integration time of 5 ms (17). Therefore, molecules moving with D1 corresponded to the immobile fraction (as detectable, e.g., by FRAP experiments). The other end of the spectrum is defined by D4, which corresponded to mobile molecules exhibiting the diffusion speed of noninteracting protein molecules (for review, see van Royen et al. (30)). The mobility components D2 and D3 indicated that STAT1 molecules interacted in numerous ways with the complex and heterogeneous intracellular environment. As was previously observed for other cases, a single-molecule mobility analysis thus cannot be performed with only a single diffusion term (21,29,31).
Dimerization of STAT1 had little influence on mobility. STAT1-tc and wt-STAT1 have molecular masses of ∼70 and ∼90 kDa, respectively (with the latter being dimeric). Yet, despite their difference in mass, their cytoplasmic mobilities were essentially identical, as both moved with an effective mobility of 3.7 and 4.7 μm2/s, respectively, in the cytosol (Table 1). In the nucleus, STAT1-tc and the dimers of wt-STAT1 had effective diffusion coefficients of 7 and 1.7 μm2/s, respectively. To put these values in perspective, we compared the STAT1 variants with proteins of comparable masses, namely, dimers and pentamers of green fluorescent protein (GFP). For these inert GFP polymers, we determined diffusion coefficients of 17 μm2/s and 7.7 μm2/s, respectively, in cell nuclei using FCS (27). The comparison indicates that the intracellular mobility of the transcription factor variants is determined by molecular interactions rather than by molecular mass.
In stark contrast to this, STAT1 activation, and hence dimer conformation, strongly affected intracellular mobility. Regardless of whether activation was achieved in vitro or after treatment of the cells with IFNγ, we found a strongly reduced mobility of STAT1. Although the effects were most striking in the nucleus, the activated STAT1 also showed a reduced mobility in the cytoplasm. Consequently, the time required to travel the ∼10 μm from the cell membrane, where phosphorylation takes place, to the nuclear envelope increased ∼2.5-fold from 12 to 32 s. The most likely explanation for this observation is that activated STAT1 assembles an import complex involving importin α and β, which approximately doubles the mass of the STAT1 dimer (32). In support of this reasoning, the activation-induced retardation is not observed in FRAP experiments with STAT1 mutants that harbor a defective nuclear import signal (F. Antunes and U. Vinkemeier, unpublished). These results suggest that nuclear import is likewise retarded after STAT1 activation.
The activation-induced retardation and immobilization of STAT1 was most striking in the nucleus, suggesting a role for DNA binding. A detailed analysis of the immobile trajectory segments of activated STAT1 showed that these molecules performed numerous binding events that were interrupted by diffusion phases in rapid succession. We like to describe this behavior of activated STAT1 in the nucleus as a saltatory motion. We extracted two separate binding times, τ1 = 0.02 ± 0.01 s and τ2 = 0.5 ± 0.4 s, for activated STAT1. τ1 was seen in both cytoplasm and nuclear compartments, and also for nonactivated STAT1 molecules. It probably corresponds to apparent immobilizations or short stops that occur as a result of the stochastic nature of the movement in a complex environment. In contrast, the long binding time, τ2, is specific for activated STAT1 in the nucleus. This could reflect either unspecific DNA binding during a one-dimensional sliding process in the search for specific target sites, or the actual trapping at those targets.
The strong reduction of STAT1 nuclear mobility reported here appears to contrast with previous FRAP measurements, which indicated barely detectable effects of cytokine-treatment on STAT1 mobility (9,33). Of note, we used extremely low (picomolar) concentrations of labeled STAT1 that did not saturate the available nuclear binding sites, but may have been saturated by overexpressed STAT1 in the previous experiments. The binding time of 0.5 s is on the order of magnitude of values that were previously determined for different transcription factors with the use of FRAP or FCS (e.g., 2.5 s for p53 or the glucocorticoid receptor) (14,34,35). Obviously, transcription factors remain highly mobile after their activation, and perform diffusion interrupted by binding events.
It is tempting to speculate about the biological meaning of the observed nuclear binding time, τ2. The following considerations are based on an analysis by Schmid and Bucher (8) of public ChIP-Seq data defining the genome-wide distribution of STAT1 protein in IFNγ-stimulated HeLa cells. According to this analysis, activated STAT1 reproducibly occupies ∼32,000 sites (defined as the binding sites occupied by more than five ChIP-Seq tags (8)) in the diploid human genome outside repeats, such that there are ∼30 such sites per μm3 in the cell nucleus with a total volume of Vn ≈ 1000 μm3. Thus, several hundred specific STAT1 binding sites theoretically can be seen in each microscopic image. Therefore, we consider τ2 = 0.5 s to represent the binding time of the endogenous STAT1 at its specific binding sites. How long does it then take for activated STAT1 to find these sites? For a diffusion-controlled reaction, the search time of a protein with diffusion coefficient D for targeting a binding site of size a is ts = V/(4π D a) (36). If we consider only mobile, activated STAT1 with Dmob (Table 1) and a nuclear volume of Vn, assume a target size of 1 nm and at least 32,000 high-affinity STAT1 binding sites within nonrepetitive genomic DNA (8), this yields a search time of ∼2.5 s for a single activated STAT1 dimer. Upon stimulation of cells with IFNγ, up to 20,000 endogenous STAT1 dimers are activated (37). If we describe the STAT1 binding kinetics to promoter sites by simple chemical kinetics using the association constant ka = 4π D a and the dissociation constant kd = 1/τ2, we can conclude that within 2 s the activated STAT1 dimers occupy >99% of the ∼3100 binding sites, which are occupied in equilibrium. It should be noted in this regard that several hundred interferon-regulated STAT1 target genes are known, which is about two orders of magnitude lower than the 32,000 nuclear binding sites that in ChIP experiments are often found to be occupied by STAT1. Thus, numerous STAT1 binding sites are located outside promoters of STAT1-regulated genes (38). Therefore, it remains to be determined whether the binding duration τ2 indeed reflects the binding to sites where STAT1-mediated transcriptional activation can occur, and whether the observed binding describes the actual duration of transcription activation. To discriminate between these possibilities, in future work we will consider the role of cooperative DNA binding, which reduces the dissociation of STAT1 from DNA in vitro and in vivo (7). Our results indicate that cooperativity is likely to affect the mobility of STAT1. This was suggested by the observation that the immobile fraction of nuclear STAT1 is significantly increased when STAT1 is activated in living cells by IFNγ, rather than in vitro, indicating that the presence of excess activated endogenous STAT1 molecules increases the immobilization of the injected probe molecules. In conclusion, our data provide the first view (to our knowledge) of the behavior of individual transcription factors before and during the transmission of extracellular signals to the nucleus of eukaryotic cells.
Acknowledgments
This study was supported by grants from Deutsche Forschungsgemeinschaft (KU 975/4-2 and UV218/2-3) and the Biotechnology and Biological Sciences Research Council (BB/GO019290/1) to U.V.
Contributor Information
Uwe Vinkemeier, Email: uwe.vinkemeier@nottingham.ac.uk.
Ulrich Kubitscheck, Email: u.kubitscheck@uni-bonn.de.
Supporting Material
References
- 1.Levy D.E., Darnell J.E., Jr. Stats: transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 2.Meyer T., Vinkemeier U. Nucleocytoplasmic shuttling of STAT transcription factors. Eur. J. Biochem. 2004;271:4606–4612. doi: 10.1111/j.1432-1033.2004.04423.x. [DOI] [PubMed] [Google Scholar]
- 3.Melen K., Kinnunen L., Julkunen I. Arginine/lysine-rich structural element is involved in interferon-induced nuclear import of STATs. J. Biol. Chem. 2001;276:16447–16455. doi: 10.1074/jbc.M008821200. [DOI] [PubMed] [Google Scholar]
- 4.Meyer T., Begitt A., Vinkemeier U. Constitutive and IFN-γ-induced nuclear import of STAT1 proceed through independent pathways. EMBO J. 2002;21:344–354. doi: 10.1093/emboj/21.3.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chatterjee-Kishore M., Wright K.L., Stark G.R. How Stat1 mediates constitutive gene expression: a complex of unphosphorylated Stat1 and IRF1 supports transcription of the LMP2 gene. EMBO J. 2000;19:4111–4122. doi: 10.1093/emboj/19.15.4111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kovarik P., Mangold M., Decker T. Specificity of signaling by STAT1 depends on SH2 and C-terminal domains that regulate Ser727 phosphorylation, differentially affecting specific target gene expression. EMBO J. 2001;20:91–100. doi: 10.1093/emboj/20.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meyer T., Marg A., Vinkemeier U. DNA binding controls inactivation and nuclear accumulation of the transcription factor Stat1. Genes Dev. 2003;17:1992–2005. doi: 10.1101/gad.268003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schmid C.D., Bucher P. MER41 repeat sequences contain inducible STAT1 binding sites. PLoS ONE. 2010;5:e11425. doi: 10.1371/journal.pone.0011425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lödige I., Marg A., Vinkemeier U. Nuclear export determines the cytokine sensitivity of STAT transcription factors. J. Biol. Chem. 2005;280:43087–43099. doi: 10.1074/jbc.M509180200. [DOI] [PubMed] [Google Scholar]
- 10.Pemberton L.F., Paschal B.M. Mechanisms of receptor-mediated nuclear import and nuclear export. Traffic. 2005;6:187–198. doi: 10.1111/j.1600-0854.2005.00270.x. [DOI] [PubMed] [Google Scholar]
- 11.ten Hoeve J., de Jesus Ibarra-Sanchez M., Shuai K. Identification of a nuclear Stat1 protein tyrosine phosphatase. Mol. Cell. Biol. 2002;22:5662–5668. doi: 10.1128/MCB.22.16.5662-5668.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hager G.L., McNally J.G., Misteli T. Transcription dynamics. Mol. Cell. 2009;35:741–753. doi: 10.1016/j.molcel.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li G.W., Elf J. Single molecule approaches to transcription factor kinetics in living cells. FEBS Lett. 2009;583:3979–3983. doi: 10.1016/j.febslet.2009.11.035. [DOI] [PubMed] [Google Scholar]
- 14.Mueller F., Mazza D., McNally J.G. FRAP and kinetic modeling in the analysis of nuclear protein dynamics: what do we really know? Curr. Opin. Cell Biol. 2010;22:403–411. doi: 10.1016/j.ceb.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Royen M.E., Farla P., Houtsmuller A.B. Fluorescence recovery after photobleaching (FRAP) to study nuclear protein dynamics in living cells. Methods Mol. Biol. 2009;464:363–385. doi: 10.1007/978-1-60327-461-6_20. [DOI] [PubMed] [Google Scholar]
- 16.Elf J., Li G.W., Xie X.S. Probing transcription factor dynamics at the single-molecule level in a living cell. Science. 2007;316:1191–1194. doi: 10.1126/science.1141967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Siebrasse J.P., Grünwald D., Kubitscheck U. Single-molecule tracking in eukaryotic cell nuclei. Anal. Bioanal. Chem. 2007;387:41–44. doi: 10.1007/s00216-006-0763-0. [DOI] [PubMed] [Google Scholar]
- 18.Marg A., Shan Y., Vinkemeier U. Nucleocytoplasmic shuttling by nucleoporins Nup153 and Nup214 and CRM1-dependent nuclear export control the subcellular distribution of latent Stat1. J. Cell Biol. 2004;165:823–833. doi: 10.1083/jcb.200403057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vinkemeier U., Cohen S.L., Darnell J.E., Jr. DNA binding of in vitro activated Stat1 α, Stat1 β and truncated Stat1: interaction between NH2-terminal domains stabilizes binding of two dimers to tandem DNA sites. EMBO J. 1996;15:5616–5626. [PMC free article] [PubMed] [Google Scholar]
- 20.Siebrasse J.P., Kubitscheck U. Single molecule tracking for studying nucleocytoplasmic transport and intranuclear dynamics. Methods Mol. Biol. 2009;464:343–361. doi: 10.1007/978-1-60327-461-6_19. [DOI] [PubMed] [Google Scholar]
- 21.Speil J., Kubitscheck U. Single ovalbumin molecules exploring nucleoplasm and nucleoli of living cell nuclei. Biochim. Biophys. Acta. 2010;1803:396–404. doi: 10.1016/j.bbamcr.2009.10.010. [DOI] [PubMed] [Google Scholar]
- 22.Siebrasse J.P., Veith R., Kubitscheck U. Discontinuous movement of mRNP particles in nucleoplasmic regions devoid of chromatin. Proc. Natl. Acad. Sci. USA. 2008;105:20291–20296. doi: 10.1073/pnas.0810692105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meilhac N., Le Guyader L., Destainville N. Detection of confinement and jumps in single-molecule membrane trajectories. Phys. Rev. E. 2006;73:011915. doi: 10.1103/PhysRevE.73.011915. [DOI] [PubMed] [Google Scholar]
- 24.Saxton M.J. Lateral diffusion in an archipelago. Single-particle diffusion. Biophys. J. 1993;64:1766–1780. doi: 10.1016/S0006-3495(93)81548-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saxton M.J. Single-particle tracking: effects of corrals. Biophys. J. 1995;69:389–398. doi: 10.1016/S0006-3495(95)79911-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Simson R., Sheets E.D., Jacobson K. Detection of temporary lateral confinement of membrane proteins using single-particle tracking analysis. Biophys. J. 1995;69:989–993. doi: 10.1016/S0006-3495(95)79972-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bancaud A., Huet S., Ellenberg J. Molecular crowding affects diffusion and binding of nuclear proteins in heterochromatin and reveals the fractal organization of chromatin. EMBO J. 2009;28:3785–3798. doi: 10.1038/emboj.2009.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Veith R., Sorkalla T., Kubitscheck U. Balbiani ring mRNPs diffuse through and bind to clusters of large intranuclear molecular structures. Biophys. J. 2010;99:2676–2685. doi: 10.1016/j.bpj.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grünwald D., Martin R.M., Cardoso M.C. Probing intranuclear environments at the single-molecule level. Biophys. J. 2008;94:2847–2858. doi: 10.1529/biophysj.107.115014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Royen M.E., Zotter A., Houtsmuller A.B. Nuclear proteins: finding and binding target sites in chromatin. Chromosome Res. 2011;19:83–98. doi: 10.1007/s10577-010-9172-5. [DOI] [PubMed] [Google Scholar]
- 31.Goulian M., Simon S.M. Tracking single proteins within cells. Biophys. J. 2000;79:2188–2198. doi: 10.1016/S0006-3495(00)76467-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nardozzi J., Wenta N., Cingolani G. Molecular basis for the recognition of phosphorylated STAT1 by importin α5. J. Mol. Biol. 2010;402:83–100. doi: 10.1016/j.jmb.2010.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lillemeier B.F., Köster M., Kerr I.M. STAT1 from the cell membrane to the DNA. EMBO J. 2001;20:2508–2517. doi: 10.1093/emboj/20.10.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hinow P., Rogers C.E., DiBenedetto E. The DNA binding activity of p53 displays reaction-diffusion kinetics. Biophys. J. 2006;91:330–342. doi: 10.1529/biophysj.105.078303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mueller F., Wach P., McNally J.G. Evidence for a common mode of transcription factor interaction with chromatin as revealed by improved quantitative fluorescence recovery after photobleaching. Biophys. J. 2008;94:3323–3339. doi: 10.1529/biophysj.107.123182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Halford S.E., Marko J.F. How do site-specific DNA-binding proteins find their targets? Nucleic Acids Res. 2004;32:3040–3052. doi: 10.1093/nar/gkh624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wenta N., Strauss H., Vinkemeier U. Tyrosine phosphorylation regulates the partitioning of STAT1 between different dimer conformations. Proc. Natl. Acad. Sci. USA. 2008;105:9238–9243. doi: 10.1073/pnas.0802130105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robertson G., Hirst M., Jones S. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat. Methods. 2007;4:651–657. doi: 10.1038/nmeth1068. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.