

Abstract
1-L-myo-inositol-phosphate synthase (mIPS) catalyzes the first step of the unique, de novo pathway of inositol biosynthesis. However, details about the complex mIPS catalytic mechanism, which requires oxidation, enolization, intramolecular aldol cyclization, and reduction, are not fully known. To gain further insight into this mechanism, we determined the crystal structure of the wild-type mIPS from Archaeoglobus fulgidus at 1.7 Å, as well as the crystal structures of three active-site mutants. Additionally, we obtained the structure of mIPS with a trapped 5-keto-glucose-6-phosphate intermediate at 2 Å resolution by a novel (to our knowledge) process of activating the crystal at high temperature. A comparison of all of the crystal structures of mIPS described in this work suggests a novel type of catalytic mechanism that relies on the forced atomic proximity of functional groups. The lysine cluster is contained in a small volume in the active site, where random motions of these side chains are responsible for the progress of the complex multistep reaction as well as for the low rate of catalysis. The mechanism requires that functional groups of Lys-274, Lys-278, Lys-306, and Lys-367 assume differential roles in the protonation/deprotonation steps that must occur during the mIPS reaction. This mechanism is supported by the complete loss of activity of the enzyme caused by the Leu-257 mutation to Ala that releases the lysine containment.
Introduction
In recent years, inositol has rapidly gained importance as a ubiquitous, versatile molecule that is used in a variety of cellular mechanisms. In eukaryotes, inositol compounds are implicated in all aspects of cellular life, including cell division and proliferation, signal transduction, gene expression, and cellular death (1–6). A wide variety of inositol-containing compounds, such as phospholipids, also have important roles in prokaryotic and archaeal organisms. Inositol is used for cell wall biosynthesis in prokaryotes (7,8), and is an important component of phospholipids in archaea (9,10). A subclass of archaea and some thermophilic bacteria incorporate myo-inositol into small molecules as a response to osmotic or thermal stress (7,11–13).
A single de novo pathway for myo-inositol biosynthesis is known to be present in all three major kingdoms (14). The synthesis of myo-inositol proceeds through two steps. In the first step, myo-inositol-1-phosphate synthase (mIPS1) catalyzes the conversion of a D-glucose-6-phosphate (G-6-P) to L-myo-inositol-1-phosphate (I-1-P). In the second step, inositol-1-phosphate phosphatase (inositol monophosphatase) catalyzes the hydrolysis of the phosphate group to produce myo-inositol.
Early studies of mIPS (15,16) showed that the enzyme binds the pyranose form of G-6-P and converts it to I-1-P without releasing intermediates (5-keto-glucose-6-phosphate (5-keto-G-6-P), NADH, or 2-inosose-1-phosphate). Crystal structures determined for the yeast enzyme showed a tetramer with the active-site region ordered when a linear substrate analog was bound (17–19). The active site was replete with cationic residues that could aid in catalysis. However, it is difficult to fine-tune the catalytic mechanism using mutagenesis studies based on the crystal structure of the yeast mIPS with the inhibitor bound, because such a structure cannot be on the catalytic pathway.
The extremely heat-stable mIPS protein (20) from Archaeoglobus fulgidus is much more amenable to such studies. It has also been cloned, purified, biochemically characterized, and crystallized (21). The recombinant protein is also a tetramer with 44 kDa subunits, and thus is considerably smaller than the eukaryotic homolog, a tetramer with ∼60 kDa subunits. Its catalytic activity was shown to be critically dependent on divalent metal ions such as Zn2+, Mn2+, and Mg2+, but unaffected by Ca2+ (20). Despite significant differences in the proteins' length and sequence, the general architecture of the archaeal enzyme is similar to that of the eukaryotic mIPS from Saccharomyces cerevisiae (17–19) and bacterial mIPS from Mycobacterium tuberculosis (22).
The requirement of the archaeal enzyme for divalent cations allows investigators to test mutated enzymes to determine whether specific residues are required for the initial substrate oxidation step that converts G-6-P and NAD+ to 5-keto-G-6-P and NADH (23). On the basis of kinetic characterization and ligand-binding studies of eight A. fulgidus mIPS mutant proteins combined with the structures of both the yeast and archaeal proteins, Neelon et al. (23) proposed a detailed catalytic mechanism for the archaeal mIPS. Their analyses suggested that Lys-367 or Lys-274 initiates the reaction by a proton transfer, resulting in opening of the G-6-P ring. Subsequently, hydride transfer to NAD+ leads to the formation of NADH and a keto group at C5. Enolization of the C5-C6 bond and the developing negative charge is stabilized by Lys-274 or Lys-367. The electron enolate C6 subsequently attacks the C1 aldehyde to form the cyclohexane ring of inositol. The reaction is completed by oxidation of NADH coupled to the reduction of the ketone at what was the C5 of glucose, a reduction that was suggested to be facilitated by proton transfer from Lys-274. A schematic of the different chemical steps without attributing any specific side chains to a given step is shown in Fig. S1 of the Supporting Material. However, two anomalous results from that study were noted, i.e., both K367A and N255A exhibited weak G-6-P binding.
In this work, we present the structures of three of those mutant proteins: K278A, K367A, and N255A (the last two did not behave as would have been predicted from the initial crystal structure of this mIPS). We compare them with the newly obtained crystal of the wild-type (WT) enzyme refined at 1.7 Å resolution to elucidate the defects introduced by these mutations. Our comparison of these mutant protein structures with a structure of mIPS containing the 5-keto-G-6-P intermediate trapped in the active site leads us to propose a modified atomic crowding hypothesis for the catalysis in this enzyme. Random movements of amino groups of four lysine residues present in the active-site pocket facilitate all of the proton transfer steps needed for oxidation, carbon-carbon bond formation, and reduction of G-6-P to generate the final product of the reaction, I-1-P (18,23). Furthermore, the modeling of C-1 of linearized 5-keto-6-P as a gem diol suggests that a major role of the catalytic metal ion is to stabilize the aldehyde carbonyl at this position for subsequent cyclization.
Materials and Methods
Construction of mutants
A. fulgidus mIPS and mutant enzymes were constructed and purified as described previously (20,23). SDS-PAGE on 12% polyacrylamide gel was used to assess the purity of the mutants, and absorbance at 280 nm was used to determine the concentration of the mutants (the extinction coefficient at 280 nm is calculated as 50,210 M−1 cm−1 from the sequence). All mutants were stored at 4°C in 50 mM Tris-acetate buffer, pH 7.5.
Crystallization and structure determination of the WT and mutant enzymes
After obtaining the crystals characterized by the P1 space group as described in our previous publication (23), we initiated the search for new crystallization conditions that would allow us to obtain structures of the ternary complexes. We obtained a new crystal form from the 0.1 M HEPES, pH 7.5, 0.2 M CaCl2, 14% PEG 400, and 15% PEG 1500 mixture. Crystals appeared as long, chunky needles and were characterized by the space group I222 with a single subunit in the asymmetric part of the unit cell. These crystals diffracted to better than 1.7 Å resolution. The mutant enzymes were crystallized in a similar condition and showed an internal symmetry similar to that of the WT enzyme. The only exception was the mutant N255A, which under the same conditions formed a crystal characterized by the space group P21.
Monitoring the progress of the catalytic reaction and trapping of the intermediate
We used a spectroscopic method to monitor the progress of the reaction after the formation of NADH at ∼340 nm. In our previous work (20), we reported that the presence of divalent metal ions, such as Mg2+, is necessary for completion of the reaction. This suggested the method of trapping of the intermediate. We introduced 400 μM EDTA to prevent the reaction from going to completion. After obtaining the crystals, we soaked them in 5 mM G-6-P and subjected them to heating under oil for 1 h at 85°C.
Preparation of crystals and data collection
The crystals were stabilized in glycerol cryoprotectant and flash-frozen in a cold stream of N2. The data were collected at 100 K on a Microfocus 007 system on a RaxisIV++ imaging plate detector (Rigaku). We reduced the data using the CrystalClear software (Rigaku). We then solved the structures using the molecular replacement method implemented in the program Molrep (CCP4) with the template structure 1U1I extracted from the Protein Data Bank (PDB). The models of the WT and the mutant enzymes were refined by the program Refmac5. We used the difference electron densities (DEDs) to locate novel or missing elements of the structure. The data collection and refinement statistics are presented in Table 1.
Table 1.
Data collection and refinement statistics for the WT and mutant mIPS
| Data set | WT | K367A | K278A | N255A | WT+5-keto-G-6-P |
|---|---|---|---|---|---|
| Space group | I222 | I222 | I222 | P21 | I222 |
| Unit cell parameters | |||||
| a (Å) | 82.47 | 82.07 | 82.26 | 91.51 | 82.73 |
| b (Å) | 89.50 | 89.28 | 89.94 | 88.04 | 89.54 |
| c (Å) | 104.62 | 104.97 | 104.87 | 103.73 | 104.94 |
| β ° | 94.9 | ||||
| Diffraction limit (Å) | 1.7 | 1.9 | 2.0 | 2.6 | 2.0 |
| Number of reflections | 43,214 | 29,996 | 26,096 | 46,723 | 26,667 |
| I/s(I) (last shell) | 13.8 (2.1) | 14.4 (3.6) | 7.77 (2.4) | 5.5 (1.5) | 8.6 (1.8) |
| Complete % (last shell) | 98.1 (94.9) | 96.8 (91.8) | 97.6 (95.8) | 99.0 (90.0) | 99.6 (99.7) |
| Redundancy | 4.2 (2.3) | 3.4 (2.2) | 2.5 (1.2) | 3.1 (1.8) | 3.1 (1.9) |
| Rmerge (last shell) | 0.061 (0.34) | 0.040 (0.21) | 0.088 (0.32) | 0.174 (0.82) | 0.101 (0.81) |
| Resolution (Å) | 40.0–1.7 | 40.0–1.9 | 40.0–2.0 | 40.0–2.6 | 40.0–2.0 |
| R-factor | 0.201 | 0.177 | 0.218 | 0.188 | 0.197 |
| Rfree | 0.243 | 0.229 | 0.283 | 0.262 | 0.248 |
| Protein B-factor (Å2) | 26.6 | 21.1 | 27.7 | 15.1 | 24.8 |
| Disordered residues | 14 | 16 | 15 | 0 | 16 |
| Water molecules | 235 | 234 | 235 | 486 | 219 |
| Water B-factor (Å2) | 39.3 | 31.5 | 36.7 | 16.3 | 36.4 |
| Ligand B-factor (Å2) | |||||
| NAD | 28.3 | 14.7 | 23.7 | 17.2 | 31.9 |
| PEG | 39.3 | 38.7 | 47.7 | - | 33.6 |
| PO4/Glyc/G-6-P | 21.7 | 14.9/26.2 | 23.9/42.4 | 11.2/48.2 | 20.1/17.5 |
| RMSD | |||||
| Bond lengths (Å) | 0.016 | 0.015 | 0.018 | 0.017 | 0.016 |
| Bond angles (deg) | 1.662 | 1.533 | 1.927 | 1.773 | 1.796 |
| Chiral angles (deg) | 0.104 | 0.099 | 0.115 | 0.112 | 0.111 |
| Ramachandran | |||||
| Preferred | 97.1 | 97.2 | 97.1 | 92.1 | 97.1 |
| Allowed | 2.3 | 2.2 | 2.3 | 6.3 | 2.3 |
| Generous/disallowed | 0.6 | 0.6 | 0.6 | 1.6 | 0.6 |
Results and Discussion
Novel crystal form of the mIPS WT enzyme
The WT protein was crystallized in a novel structural arrangement characterized by the I222 space group in which the monomer is located at the intersection of two twofold symmetry axes. The organization of both the enzyme and its eukaryotic counterpart is tetrameric. Thus, the tetramer is formed from the monomer by the application of symmetry operators of the I222 space group. The monomer is subdivided into two distinct domains: the Rossmann fold domain binds the NAD+ cofactor, and the β-sheet-rich domain constitutes the tetramerization domain. The architecture is described in detail elsewhere (17,19,22). The higher resolution and quality of our data (completeness as well as Rmerge) provide interesting insights into the proposed mechanism.
Several structural features are important for the function of this enzyme. The first one is the arrangement of the cis-conformer of the peptide bond connecting Gly-223 and Asn-224. This backbone conformation is combined with the left-hand turn conformation of the next residue, Asp-225, which is catalytically important (23). The entire conformational arrangement allows for continuation of uninterrupted β-sheet hydrogen bonds at the crucial turn bracketing the active site. This arrangement exposes Asp-225 to the active site and allows for the direct bonding of Asp-225 with Lys-367, and the nicotinamide ring of NAD+ (Fig. S2 A). This unique local conformation suggests evolutionary conservation, and has been observed in other mIPS structures refined at higher resolution (18,22).
The root mean-square deviation (RMSD) between Cα positions of the P1 and I222 crystal forms was <0.4 Å and indicated a very similar conformation. However, the I222 form allowed for unequivocal tracing of density for elements of the backbone that were weakly defined in the previous structure (21). In this new structure, the fragment 100–120 between helices C and D is very well ordered and forms an extended loop, closing the active-site pocket on the reverse side from the substrate access helix I.
Helix D, which directly follows the loop, contains a lysine residue (Lys-135) that in this crystal structure is coordinated to a PEG molecule that forms a donut shape around the amino-terminal group of lysine (Fig. S2 B). Several neighboring residues are disordered. Altogether, 16 residues (residues 13, 25, 81, 84, 89, 96, 132, 136, 140, 237, 247, 283, 292, 293, 387, and 392) were found to be in multiple conformers, constituting four patches on the surface of the molecule. The presence of the PEG molecule and the modeling of the disordered residues testify to the high quality of our model and the reliability of all of the structural components refined in the mutant and catalytic intermediate models.
This crystal form of A. fulgidus mIPS clearly shows inorganic phosphate (Pi) in the active site, bound at the turn of the helix that directly follows the Rossman fold domain. The WT enzyme active site is filled with water molecules and four crucial Lys residues (residues 274, 278, 306, and 367) are clearly visible. The structure contains ∼200 water molecules. The superposition of the Cα trace for the WT and the mutant enzymes is presented in Fig. 1. This figure also shows the conformational changes at the active site in individual mutant structures. The extensive network of hydrogen bonds that connect these and other critical residues implicated in the proton shuttling seen in the WT enzyme is presented in Fig. 2.
Figure 1.
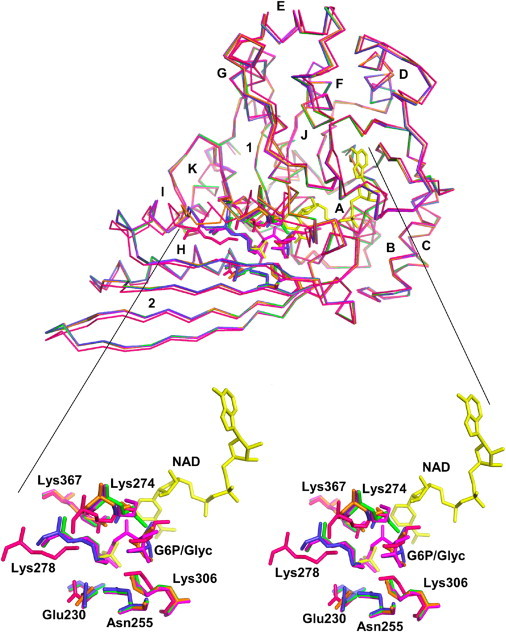
(Top) Superposition of Cα representations of the mutant structures onto the WT structure. (Bottom) A stereo view of a close-up of the active site. The WT model is shown in green, K367A is blue, K278A is orange, and the N255A mutant enzyme is red. The N255A structure shows an incompletely engaged helix I with Lys-274 and Lys-278 shifted. The model of the intermediate is shown in pink.
Figure 2.
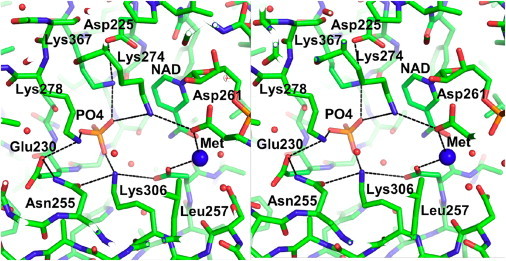
Stereo image of the active site of the WT enzyme, showing key catalytic residues involved in the charge relay system that is important in the catalytic cycle. The large blue sphere represents a water molecule that was postulated to be substituted by the metal ion required for completion of the catalytic cycle.
Mutant mIPS crystal structures
To study the roles of individual residues in the activity of the enzyme, we generated a series of mutations as previously described (23). In the series of A. fulgidus mIPS proteins with active-site mutations that were originally generated, only one, N255A, had significant residual activity (23). For the inactive archaeal enzymes, we used intrinsic fluorescence and 31P NMR studies with EDTA present to determine whether substrates could bind to the enzyme, and, if so, whether the initial oxidation of G-6-P occurred (monitored by generation of bound NADH). Structures for three of those mutant proteins, N255A, K278A, and K367A, have now been obtained.
Two mutant enzymes, K367A and K278A, crystallized in the same arrangement as the WT (space group I222), whereas the third (N255A) crystallized in a form that is characterized by the P21 space group containing full tetramer in the asymmetric part of the unit cell. The RMSD of the mutants and WT structures were <0.4 Å, suggesting high structural similarity. All of the mutant proteins contained a glycerol molecule bound at the active site. Additionally, two out of four active sites of the N255A mutant bound the potassium ion in a fashion similar to that observed in the P1 crystal form. The overall backbone structures of the three proteins and the active sites of the mutant enzymes are shown in Fig. 1. The glycerol molecule is bound at the subpocket where the sugar moiety of the phosphorylated inositol is expected to bind (Fig. S3). All of the mutant protein models also include a bound PEG molecule at the active site. One of the most striking observations is that the conformation of the bound glycerol is different in every mutant model (Fig. S3). Furthermore, these conformational differences are coupled to changes in side-chain orientations in the active-site pocket when one of the residues is substituted by alanine. The refinement statistics are presented in Table 1, and key distances between protein residues and the bound glycerol molecule, as well as the nicotinamide group of the NAD+, are presented in Table S1.
The conformations of the active-site Lys are clearly different in the mutant enzymes compared with the WT protein (Fig. 1). Lys-367 and Lys-306 have the same Cα positions in all of the models, but the amino-terminal groups are shifted by ∼2 Å, whereas Lys-274 and Lys-278 amino groups are shifted by up to 4 Å (Fig. 1). The structure of the N255A mutant enzyme shows the largest divergence in the region of helix I, which is shifted significantly by ∼3 Å (Fig. S2). This is an important observation because N255A is the only mutant enzyme with residual catalytic activity (0.09% of the activity of the WT enzyme (23)). The conformation of Lys-306, which is different in N255A, would be consistent with its role as a proton shuttle between Asn-255 and Asp-261 (Fig. 1 and Fig. S3).
Another notable change in the N255A structure is a small conformational transition of the entire protein. The NAD+-binding domain is rotated slightly away from the tetramerization β-sheet domain in the N255A mutant enzyme. This small angular movement, which is clearly visible in Fig. 1, likely corresponds to different packing arrangements in different space groups. It also testifies to the global conformational changes that happen during catalysis. The close-up views of well-defined active sites of mutant structures with bound phosphate and glycerol molecules are presented in Fig. S3. As mentioned above, the active sites contain glycerol molecules that provide a model of binding modes of the sugar moiety of the substrate, G-6-P.
Trapping and crystal structure of 5-keto-G-6-P bound to mIPS
Previous studies of the recombinant WT enzyme (20,23) showed that in the absence of divalent cations and the presence of excess substrate (typically 5 mM G-6-P and 0.3 mM NAD+), mIPS could oxidize G-6-P and generate NADH when the solution was incubated above 80°C (usually for 10–30 min). We detected NADH after cooling the sample to room temperature and monitoring the absorbance at 340 nm. The NADH absorbance was stable at room temperature for at least 6 h. However, a significant loss of the NADH signal (typically >60%) occurred upon addition of Mg2+ (1 mM) followed by reheating of the reaction mixture above 80°C for 30 min, and then cooling of the protein. The significant absorbance drop at 340 nm indicated that the divalent cation binding to the protein allowed the reaction to proceed to completion resulting in production of I-1-P (Fig. 3). Entry of the metal ion into the sequestered active site is expected to be slow, which makes it difficult to achieve full oxidation of NAD+ under these conditions.
Figure 3.
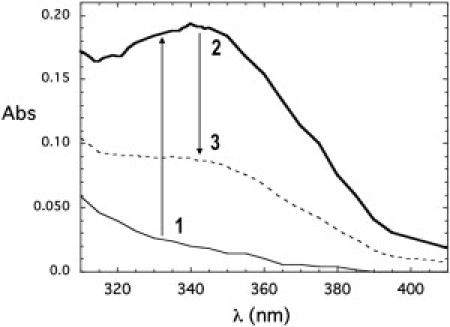
Absorbance spectra illustrating accumulation of the intermediate in the absence of a divalent metal ion. Spectra were obtained at room temperature for 30 mM IPS in 50 mM Tris, pH 7.5, with 300 μM NAD+ and 400 μM EDTA before (thin solid line) and after (bold line) the addition of 5 mM G-6-P and incubation at 85°C for 30 min. The dashed line shows the absorbance when 1 mM MgCl2 was added to the sample, which was then reheated to 85°C for 30 min and then cooled. The arrows and numbers depict the course of the experiment in which EDTA causes accumulation of the intermediate, and addition of metal ions carries the reaction through to completion, reversing the blocking effect of the EDTA.
These observations suggested a novel method of trapping the WT mIPS after binding of the substrate in one of intermediate conformational states before it reached the final aldol cyclization and reduction steps. We found that after the mIPS crystals were heated under oil for 1 h at 85°C in the presence of substrate (1 mM G-6-P) and then suddenly frozen at 100 K, electron density appeared in the active site, suggesting that an appreciable amount of the substrate/intermediate was trapped. The modeling of disordered residues and solvent molecules led to a DED that was clearly defined at the level of 2.5 σ. Such a low level of DED suggested incomplete occupancy of the intermediate. Indeed, our attempts to refine different forms of the intermediate led to the conclusion that ∼35% of these molecules were bound. The remaining 65% of active sites contained a free phosphate group and a water molecule. Nevertheless, multiple modeling attempts combined with careful refinement showed a clear conformation for the bound intermediate. The omit map calculated without the G-6-P is shown in Fig. 4 with the final refined model of the intermediate. The position of the phosphate group of the intermediate is shifted slightly from that of free phosphate (21), whereas the linear polyol moiety of the intermediate superposes well with the different conformations of glycerol found in the active sites of the mutant enzymes (Fig. 1 and Fig. S3). The cofactor in this structure is also bent, confirming that it is NADH (Fig. S4).
Figure 4.
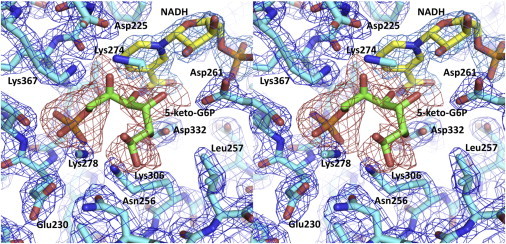
Close-up view of the active site depicting the bound 5-keto-G-6-P intermediate. The 2Fo-Fc electron density covering the model (in blue) is contoured at 1.5 σ, and the Fo-Fc omit map (in red) is contoured at 2.5 σ. The model of the intermediate was refined at the 0.35 occupancy level.
The general orientation of the bound keto-intermediate is quite similar to the linear inhibitor bound to the yeast mIPS (18). In the structure of yeast mIPS with the inhibitor, the molecule is in a more extended conformation in which the distance between C1 and C6 carbons is >5 Å. In the structure of the archaeal mIPS with the intermediate trapped, the ligand is in a much more curved conformation that clearly represents noncyclized sugar. The observation of the open form in protein without the catalytic divalent cation strongly suggests that the major role of the divalent cation is to stabilize the negative charge developing on the C1 aldehyde oxygen. The interactions of different side chains with the intermediate are slightly altered compared with structures with Pi and glycerol. For instance, amino groups of Lys-274, -367, and -278 are in different locations than in the WT model.
The most interesting feature of the intermediate is that C1 is better refined as a gem-diol than as an aldehyde form. There are clearly two electron dense atoms attached to this carbon, and a better fit is found for tetrahedral geometry. In contrast, C5 of the intermediate clearly has trigonal geometry and a single oxygen atom bound to it. Gem-diols have been proposed as reaction intermediates in several C-C cleavage enzymes (24–26). In such cases, the enzymes clearly enhance hydration of the ketone. A different case is seen with substrate bound to the reversible enzyme triosephosphate isomerase, where a significant amount of the gem-diol of DHAP was shown bound to the enzyme (identified by 13C solid-state NMR (27)). However, a mechanistically important role for this species was previously ruled out. The observation of a C1 gem-diol for the 5-keto-G-6-P intermediate bound to mIPS falls into the latter category, and strongly supports the role of the catalytic divalent metal ion in stabilizing the aldehyde form of C1 in the intermediate.
Atomic proximity and lysine side-chain motions drive the catalytic reaction
Direct comparisons of the mutant structures with the structure of the trapped intermediate suggest several modifications of the initially proposed mechanism for this enzyme (23). Lysine residues are strategically located to participate in every step of the catalysis in a concerted fashion. This explains why the enzyme is practically inactive after any one of these residues is replaced with alanine. A comparison of all of the crystal models described in this work (Fig. 1 and Fig. S3) shows the changeable positions and conformational states of the terminal amino group of Lys in the vicinity of the active site. Even a small and relatively remote mutation from the location of the particular lysine amino group influences its conformational state. Many of the key lysine residues (Lys-274, Lys-306, and Lys-367) adopt different conformations in the three mutants compared with the WT. Lys-367 changes its coordination from hydrogen bonding with Asn-225 in the WT structure to interacting with the phosphate group in several of the mutant structures. Similar observations can be made about Lys-306 and Lys-274. This emphasizes the coupled mobility of the side chains and hints that assigning a specific role to a given lysine may not truly reflect what happens during catalysis.
Upon sequestration of the substrate in the active-site cavity, the proximity of all of the positively charged atoms (Nε of Lys) is significant (<6 Å). Such close proximity of four Lys residues in the active site of an enzyme is fairly unusual. To buttress our interesting and novel (to our knowledge) observation, we carried out a review of the entire PDB for the rare occurrence of four Lys amino groups contained in a 7 Å diameter sphere, using as a template the volume occupied by the Lys in our mIPS WT structure (28). Fewer than 30 cases of four Lys residues at these distances (in >73,000 structures) occurred, and only two cases of four Lys were observed within the matrix of the enzyme and not on the surface: 1), the CMP complex of phosphopantothenoylcysteine (PPC) synthetase from Escherichia coli (29); and 2), the inositol hexakisphosphate (IP6) binding site in Clostridium difficile toxin A that induces autoprocessing of the toxin (30). The four Lys residues in PPC synthetase occur in a motif that is present in all such enzymes. The positive charges of the lysines used to anchor the nucleotide are balanced to some extent by nearby negative residues. Of more interest, only one of these four Lys residues is ordered in the crystals; the other three are disordered. In the second case, the IP6 site has numerous Lys and Arg residues that serve to bind that very anionic ligand. It is worth noting that for this protein there was no crystallization in the absence of IP6, even though the protein is well folded. The IP6 site is separated from the toxin active site by a β-flap structure, and likely through this interaction modulates the catalysis at the peptide cleavage site. A third structure, the inorganic phosphatase from Anaplasma phagocytophilum, was also highlighted, but it is unpublished. Here there are two Pi ions bound per monomer in the tetramer and no analysis of structure in terms of function. Aside from these examples, this searching procedure recovered all IPS structures (these were not counted in the final results). Our review of the structural database results in two key points: 1), the observations for mIPS are novel and rare; and 2), the mIPS enzyme is a complex enzyme that works according to very unique principles.
Therefore, the small volume occupied by the four Lys amino groups in the native mIPS is a relatively rare occurrence. How does this help us understand the different mutant binding and kinetic properties? In the two Lys mutant structures (K367A and K278A), when one Lys is removed, only three Lys occupy the same volume but with a different conformer for Lys-274, which has its amino group oriented away from the Pi- and glycerol-binding region. In contrast, in the N255A mutant enzyme (the only one with residual activity), despite the incomplete closure of the active site, all amino groups occupy the same volume. In the same way, the trapped intermediate has all four Lys amino groups interacting with substrate in a small volume. This strongly suggests that it is the relatively tight cluster of Lys amino groups that is needed for catalysis. Removal of one Lys stops catalysis. All four Lys side chains must be present to provide the necessary rearrangements of the amino groups for the reaction to proceed. Both N255A and Lys-367 have reduced affinities for G-6-P, and for N255A this is consistent with involvement in stabilizing the acyclic form of the substrate. However, N255A is active whereas K367A is not. The difference in their structures is the clustering and orientation of the Lys amino groups: the cluster of Lys in N255A resembles that of the native enzyme and the bound intermediate structures. The diverse rearrangements of side chains in different A. fulgidus mIPS mutant structures also offer a simple explanation for the somewhat puzzling behavior of the inactive L257A mutant enzyme (23), which does not bind substrate. Removal of Leu-257 releases the crowding or containment element needed for Lys-306 to perform its duties as a proton shuttle and its role in the final closure of the ring. We expect that Leu-257 should also influence the rate of closure of the active site, an event that is necessary to encapsulate the substrate and carry out the catalysis.
For this particular enzyme, what would appear to be crucial is the confinement of these four amino groups in a relatively small volume. Proximity and containment are not new concepts (31); however, in our proposal we give them a new meaning. These terms are usually discussed in reference to the formation of near attack complexes (32), preorganization of active-site residues (33,34), and the ability of an enzyme to undergo rapid changes between different conformations (35,36). Modern molecular orbital theory has confirmed the heuristic findings about directionality and distance dependence. However, the atomic crowding we invoke in explaining mIPS catalysis has a different meaning: containment that places in the specified space a collection of similar reactive groups with significant flexibility in orientation, along with substrates, and allows them to react. The key to this view is that an individual side chain may not have a single specified role but instead can assume different roles in the protonation/deprotonation steps that must occur during the mIPS reaction course. This definition of atomic crowding dispenses with the idea that a particular group must have a precisely defined function.
Indeed, in mIPS, the cluster of Lys in the mIPS active site provides the heterogeneity in conformations that is needed for all of the different steps in the chemical transformation of G-6-P. For this enzyme, the entire catalytic cycle depends more on acquiring the necessary state of containment/confinement of the four Lys amino groups (which will also require other residues, such as Leu-257) than on the conformation of a single residue (e.g., in N255A, the only mutant enzyme with residual activity). Once the substrate binds, the four cationic Lys residues are held in this small volume by Leu-257, which acts like a plug. If you remove the plug, the charged groups, driven by electrostatic repulsion, are no longer contained in that small volume, and they become an ineffective tool in driving the reaction. Side-chain crowding can also facilitate the proximity of the C1, C6, and carbon-carbon bond formation. In the intermediate structure in the absence of divalent cation, C1 and C6 are not close enough to form a carbon-carbon bond. Adding one more positive element to the reaction volume not only stabilizes the aldehyde at C1 but is also likely to alter side-chain motions and conformations that in turn are coupled to the substrate/intermediate conformer. It matters less which exact Lys side chain participates in a given protonation/deprotonation step than the fact that the substrate is contained in the right electrostatic environment for the new bond to form. By design, such a catalytic cycle is relatively slow and can be described as a reaction propelled by atomic side-chain containment. The kcat for the A. fulgidus enzyme at 90°C is ∼10 s−1 (significantly faster than for the yeast enzyme, whose kcat at 37°C is 0.6 s−1 in the absence of its activating ammonium ion, which enhances the rate fourfold). Indeed, these turnover numbers are relatively low.
Amended catalytic mechanism
The general picture described above can provide further insight into the roles of the individual Lys residues in the catalytic cycle of A. fulgidus mIPS. Several of these residues have slightly different roles from what was originally proposed, largely because there were no known structures with any intermediate on the pathway to product. The current modified mechanism emphasizes that the flexibility of the Lys side chains and the closeness of the amino groups can allow facile protonation and deprotonation of different oxygen and hydroxyl groups along the reaction pathway, converting a glucose molecule to an inositol one.
The first step of transferring the proton to O5 to open the glucose ring is mediated by a coordinated action of Lys-278 and Lys-306 (either of these two side chains can effectively transfer a proton to O1 of the substrate). Subsequently, Lys-367 and Lys-274 in concert (again both Lys residues are capable of extracting the proton from O5) transfer the proton from O5 to Asp-225. This facilitates the attack of NAD+ on carbon C5 and extraction of the proton and transfer to NAD+, which in turn facilitates the formation of the keto form of the C-O bond at position 5. Subsequent delocalization of the electron and transfer to C-C bond at positions 5 and 6 results in the creation of the enol intermediate, which is necessary for closing the inositol ring (mediated by Lys-306 and/or Lys-278). To execute this last step, the aldehyde oxygen at C1 must be stabilized. This step is presumably influenced by the metal ion bound via Asp-332 and Asp-261, which stabilizes the glucose carbonyl group at C1, and also polarizes the C1 oxygen so that the C1-C6 bond can be formed. In the absence of a divalent cation, a gem-diol forms at C1, preventing any further steps. When the cation is present, one of the motile Lys can return the proton to the inositol O6 (which was glucose C1). The bound NADH is used to reduce the inositol C2 position (substrate glucose C5), with final proton transfers executed by a combination of the amino groups of Lys-274 and Lys-306. Given the different conformations of the many Lys residues in the mutant enzymes, as well as their placement in the structure with the trapped intermediate, it is difficult to predict which exact Lys residues are protonated at a given stage; however, it explains why they work in tandem (i.e., in pairs) in every individual step of the reaction. The overall charge and flexibility of these side chains, and the large number of water molecules in the enclosed active-site cavity suggest that small motions of side chains could easily alter local pKa values as required to move protons around the substrate as the glucose is converted to an inositol ring. Thus, it is this atomic crowding forcing a close proximity of amino groups of Lys that provides a significant part of the driving force for this complex reaction. The proposal for the amended reaction scheme can be found in Fig. S5.
The results presented above for mIPS suggest that other enzymes with slow turnover rates, such as RuBisCO, might operate by the same underlying principle. Paradoxically, or perhaps by design, RuBisCO uses a similar chemical rearrangement (an enolization reaction) to carry out its complex catalytic reaction. Our redefined atomic proximity hypothesis may also partially explain why it is so much more difficult to reengineer active sites of some enzymes that rely on this type of mechanism using mutagenesis.
Coordinates
The coordinates and structure factors have been deposited in the PDB (accession codes: WT-3QVS, complex with the intermediate-3QVT, mutant K278A-3QVW, mutant K367A-3QVX, and mutant N255A-3QW2).
Acknowledgments
This work was supported by the Energy Biosciences Program, Office of Science, U.S. Department of Energy (DE-FG02-91ER20025 to M.F.R.) and the National Institutes of Health (R01-GM64481 to B.S.).
Supporting Material
References
- 1.Barker C.J., Berggren P.O. The role of inositol and the principles of labelling, extraction, and analysis of inositides in mammalian cells. Methods Mol. Biol. 2010;645:1–19. doi: 10.1007/978-1-60327-175-2_1. [DOI] [PubMed] [Google Scholar]
- 2.Berridge M.J. Inositol trisphosphate and calcium signalling mechanisms. Biochim. Biophys. Acta. 2009;1793:933–940. doi: 10.1016/j.bbamcr.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 3.Tsui M.M., York J.D. Roles of inositol phosphates and inositol pyrophosphates in development, cell signaling and nuclear processes. Adv. Enzyme Regul. 2010;50:324–337. doi: 10.1016/j.advenzreg.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Okada M., Ye K. Nuclear phosphoinositide signaling regulates messenger RNA export. RNA Biol. 2009;6:12–16. doi: 10.4161/rna.6.1.7439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shears S.B. Diphosphoinositol polyphosphates: metabolic messengers? Mol. Pharmacol. 2009;76:236–252. doi: 10.1124/mol.109.055897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Corda D., Zizza P., Mariggiò S. The glycerophosphoinositols: cellular metabolism and biological functions. Cell. Mol. Life Sci. 2009;66:3449–3467. doi: 10.1007/s00018-009-0113-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roberts M.F. Inositol in bacteria and archaea. Subcell. Biochem. 2006;39:103–133. doi: 10.1007/0-387-27600-9_5. [DOI] [PubMed] [Google Scholar]
- 8.Guerin M.E., Korduláková J., Jackson M. Molecular basis of phosphatidyl-myo-inositol mannoside biosynthesis and regulation in mycobacteria. J. Biol. Chem. 2010;285:33577–33583. doi: 10.1074/jbc.R110.168328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morii H., Kiyonari S., Koga Y. A novel biosynthetic pathway of archaetidyl-myo-inositol via archaetidyl-myo-inositol phosphate from CDP-archaeol and D-glucose 6-phosphate in methanoarchaeon Methanothermobacter thermautotrophicus cells. J. Biol. Chem. 2009;284:30766–30774. doi: 10.1074/jbc.M109.034652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koga Y., Nishihara M., Akagawa-Matsushita M. Ether polar lipids of methanogenic bacteria: structures, comparative aspects, and biosyntheses. Microbiol. Rev. 1993;57:164–182. doi: 10.1128/mr.57.1.164-182.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ciulla R.A., Burggraf S., Roberts M.F. Occurrence and role of di-myo-inositol-11′-phosphate in Methanococcus igneus. Appl. Environ. Microbiol. 1994;60:3660–3664. doi: 10.1128/aem.60.10.3660-3664.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martins L.O., Carreto L.S., Santos H. New compatible solutes related to Di-myo-inositol-phosphate in members of the order Thermotogales. J. Bacteriol. 1996;178:5644–5651. doi: 10.1128/jb.178.19.5644-5651.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lamosa P., Gonçalves L.G., Santos H. Occurrence of 1-glyceryl-1-myo-inosityl phosphate in hyperthermophiles. Appl. Environ. Microbiol. 2006;72:6169–6173. doi: 10.1128/AEM.00852-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Michell R.H. Inositol derivatives: evolution and functions. Nat. Rev. Mol. Cell Biol. 2008;9:151–161. doi: 10.1038/nrm2334. [DOI] [PubMed] [Google Scholar]
- 15.Barnett J.E., Rasheed A., Corina D.L. Partial reactions of D-glucose 6-phosphate-1L-myoinositiol 1-phosphate cyclase. Biochem. J. 1973;131:21–30. doi: 10.1042/bj1310021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wong Y.H., Sherman W.R. Anomeric and other substrate specificity studies with myo-inositol-1-P synthase. J. Biol. Chem. 1985;260:11083–11090. [PubMed] [Google Scholar]
- 17.Stein A.J., Geiger J.H. The crystal structure and mechanism of 1-L-myo-inositol-1-phosphate synthase. J. Biol. Chem. 2002;277:9484–9491. doi: 10.1074/jbc.M109371200. [DOI] [PubMed] [Google Scholar]
- 18.Jin X., Foley K.M., Geiger J.H. The structure of the 1L-myo-inositol-1-phosphate synthase-NAD+-2-deoxy-D-glucitol 6-(E)-vinylhomophosphonate complex demands a revision of the enzyme mechanism. J. Biol. Chem. 2004;279:13889–13895. doi: 10.1074/jbc.M308986200. [DOI] [PubMed] [Google Scholar]
- 19.Kniewel R., Buglino J.A., Lima C.D. Structural analysis of Saccharomyces cerevisiae myo-inositol phosphate synthase. J. Struct. Funct. Genomics. 2002;2:129–134. doi: 10.1023/a:1021293408654. [DOI] [PubMed] [Google Scholar]
- 20.Chen L., Zhou C., Roberts M.F. Inositol-1-phosphate synthase from Archaeoglobus fulgidus is a class II aldolase. Biochemistry. 2000;39:12415–12423. doi: 10.1021/bi001517q. [DOI] [PubMed] [Google Scholar]
- 21.Stieglitz K.A., Yang H., Stec B. Reaching for mechanistic consensus across life kingdoms: structure and insights into catalysis of the myo-inositol-1-phosphate synthase (mIPS) from Archaeoglobus fulgidus. Biochemistry. 2005;44:213–224. doi: 10.1021/bi048267o. [DOI] [PubMed] [Google Scholar]
- 22.Norman R.A., McAlister M.S., McDonald N.Q. Crystal structure of inositol 1-phosphate synthase from Mycobacterium tuberculosis, a key enzyme in phosphatidylinositol synthesis. Structure. 2002;10:393–402. doi: 10.1016/s0969-2126(02)00718-9. [DOI] [PubMed] [Google Scholar]
- 23.Neelon K., Wang Y., Roberts M.F. Probing the mechanism of the Archaeoglobus fulgidus inositol-1-phosphate synthase. J. Biol. Chem. 2005;280:11475–11482. doi: 10.1074/jbc.M500469200. [DOI] [PubMed] [Google Scholar]
- 24.Li J.-J., Li C., Bugg T.D. Evidence for a gem-diol reaction intermediate in bacterial C-C hydrolase enzymes BphD and MhpC from 13C NMR spectroscopy. Biochemistry. 2006;45:12461–12469. doi: 10.1021/bi0612519. [DOI] [PubMed] [Google Scholar]
- 25.Han Y., Joosten H.-J., Dunaway-Mariano D. Oxaloacetate hydrolase, the C-C bond lyase of oxalate secreting fungi. J. Biol. Chem. 2007;282:9581–9590. doi: 10.1074/jbc.M608961200. [DOI] [PubMed] [Google Scholar]
- 26.Lorimer G.H., Andrews T.J., Schloss J.V. 2′-Carboxy-3-keto-D-arabinitol 1,5-bisphosphate, the six-carbon intermediate of the ribulose bisphosphate carboxylase reaction. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1986;313:397–407. [Google Scholar]
- 27.Rozovsky S., McDermott A.E. Substrate product equilibrium on a reversible enzyme, triosephosphate isomerase. Proc. Natl. Acad. Sci. USA. 2007;104:2080–2085. doi: 10.1073/pnas.0608876104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shirvanyants D., Alexandrova A.N., Dokholyan N.V. Rigid substructure search. Bioinformatics. 2011;27:1327–1329. doi: 10.1093/bioinformatics/btr129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stanitzek S., Augustin M.A., Steinbacher S. Structural basis of CTP-dependent peptide bond formation in coenzyme A biosynthesis catalyzed by Escherichia coli PPC synthetase. Structure. 2004;12:1977–1988. doi: 10.1016/j.str.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 30.Pruitt R.N., Chagot B., Lacy D.B. Structure-function analysis of inositol hexakisphosphate-induced autoprocessing in Clostridium difficile toxin A. J. Biol. Chem. 2009;284:21934–21940. doi: 10.1074/jbc.M109.018929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koshland D.E., Jr. The active site and enzyme action. Adv. Enzymol. Relat. Subj. Biochem. 1960;22:45–97. doi: 10.1002/9780470122679.ch2. [DOI] [PubMed] [Google Scholar]
- 32.Bruice T.C. A view at the millennium: the efficiency of enzymatic caralysis. Acc. Chem. Res. 2002;3:139–148. doi: 10.1021/ar0001665. [DOI] [PubMed] [Google Scholar]
- 33.Warshel A. Computer simulations of enzyme catalysis: methods, progress, and insights. Annu. Rev. Biophys. Biomol. Struct. 2003;32:425–443. doi: 10.1146/annurev.biophys.32.110601.141807. [DOI] [PubMed] [Google Scholar]
- 34.Shurki A., Strajbl M., Warshel A. How much do enzymes really gain by restraining their reacting fragments? J. Am. Chem. Soc. 2002;124:4097–4107. doi: 10.1021/ja012230z. [DOI] [PubMed] [Google Scholar]
- 35.McGeagh J.D., Ranaghan K.E., Mulholland A.J. Protein dynamics and enzyme catalysis: insights from simulations. Biochim. Biophys. Acta. 2011;1814:1077–1092. doi: 10.1016/j.bbapap.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 36.Ma B., Nussinov R. Enzyme dynamics point to stepwise conformational selection in catalysis. Curr. Opin. Chem. Biol. 2010;14:652–659. doi: 10.1016/j.cbpa.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.