

Abstract
Sudden cardiac death exhibits diurnal variation in both acquired and hereditary forms of heart disease 1, 2, but the molecular basis is unknown. A common mechanism that underlies susceptibility to ventricular arrhythmias is abnormalities in the duration (e.g. short or long QT syndromes, heart failure) 3-5 or pattern (e.g. Brugada syndrome) 6 of myocardial repolarization. Here we provide the first molecular evidence that links circadian rhythms to vulnerability in ventricular arrhythmias in mice. Specifically, we show that cardiac ion channel expression and QT interval duration (an index of myocardial repolarization) exhibit endogenous circadian rhythmicity under the control of a novel clock-dependent oscillator, Krüppel-like factor 15 (Klf15). Klf15 transcriptionally controls rhythmic expression of KChIP2, a critical subunit required for generating the transient outward potassium current (Ito). 7 Deficiency or excess of Klf15 causes loss of rhythmic QT variation, abnormal repolarization and enhanced susceptibility to ventricular arrhythmias. In sum, these findings identify circadian transcription of ion channels as a novel mechanism for cardiac arrhythmogenesis.
Sudden cardiac death from ventricular arrhythmias is the principal cause of mortality from heart disease worldwide, and remains a major unresolved public health problem. The incidence of sudden cardiac death exhibits diurnal variation in both acquired and hereditary forms of heart disease 1, 2. In the general population, the occurrence of sudden cardiac death increases sharply within a few hours of rising in the morning, and a second peak is evident in the evening hours 1. In specific hereditary disorders, e.g. Brugada syndrome, fatal ventricular arrhythmias often occur during sleep2. A common mechanism in both acquired and hereditary forms of heart disease that enhances susceptibility to ventricular arrhythmias is abnormal myocardial repolarization6. Clinically, three common types of alterations in myocardial repolarization are evident on the surface electrocardiogram (ECG). First, prolongation of repolarization is seen in acquired (e.g., heart failure) 5 and congenital disorders (e.g., long QT syndrome) 3. Second, shortening of repolarization is observed in the short QT syndrome 4. Third, early repolarization is the hallmark ECG finding in Brugada syndrome 8. Interestingly, all three modifications of repolarization increase vulnerability to ventricular arrhythmias 6. Despite rigorous investigation of the biophysical and structural characteristics of ion channels that control myocardial repolarization, the molecular basis for the diurnal variation in occurrence of ventricular arrhythmias remains unknown.
Biological processes in living organisms that oscillate with a periodicity of 24-hours are ascribed to be circadian. This cell-autonomous rhythm is coordinated by an endless negative transcriptional-translational feedback loop, commonly referred to as the biological clock 9. Several physiological parameters in the cardiovascular system such as heart rate, blood pressure, vascular tone, QT interval and ventricular effective refractory period exhibit diurnal variation 10-13. Recent studies have also identified a direct role for the biological clock in regulating cardiac metabolism, growth and response to injury 14. Previous studies have also reported that expression of repolarizing ion channels and ionic currents (Ito) exhibit diurnal changes 15. However, a potential link between circadian rhythms and arrhythmogenesis remains unknown. We made the serendipitous observation that the Krüppel-like factor 15 (Klf15) exhibits endogenous circadian rhythmicity in the heart (Fig. 1a). Gene expression microarrays in hearts of mice deficient in Klf15 led us to identify KChIP2, the regulatory β-subunit for the repolarizing transient outward potassium current (Ito) as a putative target for this factor in the heart. These observations led us to posit whether the circadian clock through Klf15 may regulate rhythmic variation in repolarization and alter susceptibility to arrhythmias.
Figure 1. Klf15 expression, ECG QTc interval and expression of repolarizing ion channels exhibit endogenous circadian rhythm.

(a) Klf15 expression exhibits endogenous circadian variation in WT hearts from mice in constant darkness (DD) (n=4 per time point) (CT – Circadian Time). (b) BMAL1 ChIP on Klf15 promoter illustrating rhythmic variation in binding of BMAL1 to the Klf15 promoter in WT hearts (n=3 per group). (c) ECG QTc interval in conscious mice exhibits endogenous circadian variation in constant darkness (n=4). (d) Representative ECG’s from conscious mice after 36 hours in constant darkness at CT 0 and CT 12 (CT - Circadian Time). (e, f) Endogenous circadian variation in transcripts for Kv4.2 and KChIP2 in WT hearts measured every four hours after 36 hours in constant darkness (n=4 per time point). Data presented as mean ± SEM.
First, we explored mechanisms through which the circadian clock regulated rhythmic expression of Klf15 in the heart. Examination of the −5kb promoter region of Klf15 revealed four canonical “E-box,” regions, i.e., consensus binding sites for CLOCK and its heterodimer BMAL1, which are essential transcription factors involved in the circadian clock (Supplemental Fig. 1a inset). Consistent with this finding, Klf15-luciferase (−5kb) was activated in a dose-dependent manner by the CLOCK/BMAL1 heterodimer (Supplemental Fig. 1a). To confirm this interaction, we performed chromatin immunoprecipitation (ChIP) and identified rhythmic variation in BMAL1 binding to the Klf15 promoter in WT hearts, but not in BMAL1-null hearts (Fig. 1b). In accordance with above observations, the expression of Klf15 was disrupted in BMAL1-null and Per2/Cry1-null hearts (Supplemental Fig. 1b). Thus, our data strongly suggests that the circadian clock directly regulates the oscillation of Klf15 in the heart.
Next, to determine whether myocardial repolarization and ion channel expression exhibit “true” (endogenous) circadian rhythms i.e., oscillate in the absence of external cues such as light, wildtype (WT) mice were placed in constant darkness for 36 hours (DD) and telemetry-based ECG intervals were measured every 2 hours for 24 hours thereafter. Under these conditions, the heart rate and QT interval corrected to heart rate (QTc) were both rhythmic and exhibited true endogenous circadian rhythmicity (Fig. 1c, d). Next, to examine whether expression of repolarizing ion channels had endogenous circadian rhythms, mice were placed in constant darkness for 36 hours, and hearts harvested every four hours over a 24-hour period. The expression of the alpha-subunit for the transient outward potassium current (Ito), i.e., Kv4.2 (Fig. 1e) and the regulatory beta-subunit, i.e., KChIP2 (Fig. 1f) exhibit endogenous circadian rhythmicity, as did components of the circadian clock in the heart (Supplemental Fig. 2). In contrast, the expression of two other major repolarizing currents in the murine ventricle, Kv1.5 (the alpha subunit for the ultra-rapid delayed rectifier potassium current) and Kir2.1 (the alpha subunit for the inward rectifier potassium current) did not reveal significant rhythmic variation (Supplemental Fig. 3). In addition, we observed a 24-hour rhythm in oscillation of Bmal1, Klf15 and KChIP2 following serum shock in cultured neonatal rat ventricular myocytes (Supplemental Fig. 4). In sum, these data indicate that myocardial repolarization and the expression of some repolarizing ion channels exhibit an endogenous circadian rhythm.
Next, to elucidate the role of Klf15 in regulating rhythmic changes in repolarization, we used complementary in vivo loss and gain-of-function approaches in mice. For loss-of-function, a previously described systemic Klf15-null mouse was utilized 16; for gain-of-function, a cardiac-specific Klf15 transgenic (Klf15-Tg) mouse driven by an attenuated α-myosin heavy chain (α-MHC) promoter was developed (Supplemental Fig. 5). First, we examined if rhythmic expression of Kv4.2 or KChIP2 was altered in the Klf15-deficient state. Kv4.2 expression exhibited altered rhythmic variation in Klf15-null mice with reduced expression at ZT6, and increased expression at ZT22 when compared to WT controls (Fig. 2a). KChIP2 expression was devoid of any discernable rhythm in the Klf15-null mice along with sustained reduction at all time points (Fig. 2b,c & Supplemental Fig. 6a). Next, we examined if either Kv4.2 or KChIP2 serve as transcriptional targets for Klf15 in the heart. Adenoviral overexpression of Klf15 in neonatal rat ventricular myocytes robustly induced KChIP2, but had no effect on Kv4.2 expression (Supplemental Fig. 6b). Importantly, in Klf15-Tg hearts, a two-fold greater expression of KChIP2 with no effect on Kv4.2 expression was observed (Fig. 2d, e). Examination of the KChIP2 promoter region revealed numerous consensus Krüppel binding sites, i.e., C(A/T)CCC (Supplemental Fig. 7a). The activity of KChIP2-luciferase was induced by full-length KLF15, but not by a mutant that lacked the zinc-finger DNA binding domain (Supplemental Fig. 7b). To identify the specific Klf15 binding site, deletion constructs of the KChIP2 promoter were generated, and transcriptional activity mapped to the proximal 555 bases (Supplemental Fig. 7a). Mutation of one Krüppel-binding site within this region (Δ1) was sufficient to cause complete loss of activity in the full-length KChIP2 promoter (Supplemental Fig. 7c). Chromatin immunoprecipitation of Flag-KLF15 from Klf15-Tg hearts confirmed enrichment of KLF15 on the endogenous KChIP2 promoter (Fig. 2f). Importantly, the oscillation of several components of the core clock machinery was minimally affected in the Klf15-deficient state (Supplemental Fig. 8). In addition, the expression levels of clock genes in Klf15-Tg hearts were similar to their controls at ZT6 (Supplemental Fig. 8). This suggested that the endogenous clock is dependent on Klf15 in orchestrating rhythmic changes in KChIP2 expression. Consistent with this observation, the expression of Klf15 (Supplemental Fig. 1b) and KChIP2 (supplemental Fig. 9) were altered in a similar fashion in Bmal1-null and Per2/Cry1-null mice. These data support the notion that KChIP2 is a direct transcriptional target for Klf15 in the heart.
Figure 2. Klf15 regulates KChIP2 expression in the heart.

(a) Kv4.2 mRNA expression exhibits diurnal rhythm in WT mice, but in Klf15-null hearts the rhythm is abnormal with reduced expression at ZT6 (ZT- Zeitgeber Time), and increased expression at ZT22 (n=4 per time point per group). (b) KChIP2 mRNA expression exhibits exhibit no rhythmic variation in Klf15-deficient mice, with significant reduction at all time points (n=4 per time point per group). (c) KChIP2 protein expression exhibits no variation across 12 hours in Klf15-null hearts. (d, e) Klf15-Tg mice hearts express higher levels of KChIP2 mRNA and protein. (f) Chromatin immunoprecipitation with Flag antibody illustrating enrichment of Flag-KLF15 on the KChIP2 promoter (n=3 per group). Data presented as mean ± SEM, *p<0.05.
We next examined if Klf15-dependent regulation of KChIP2 could be responsible for rhythmic day/night variation in myocardial repolarization. Interestingly, analysis of telemetry based ECG’s revealed that rhythmic QTc interval variation was indeed abrogated in both Klf15-null and Klf15-Tg mice (Fig. 3 a-d). In the Klf15 deficient state, the ECG QTc interval was prolonged in the dark phase and failed to oscillate (Fig. 3a, c). This occurred despite similar heart rates to their WT counterparts (Supplemental Fig. 11). In contrast, the Klf15-Tg mice had persistently short QT interval with no rhythmic day/night variation (Fig. 3b, d). Again, this occurred despite minimal difference in heart rates when compared to WT (Non-Tg) controls (Supplemental Fig. 11). Next, we examined in isolated myocytes if transient outward current (Ito fast) dependent changes in repolarization were responsible for the aforementioned ECG changes in Klf15-null and Klf15-Tg mice. In Klf15-null mice, there was a marked reduction in Ito fast density (Fig. 3e) and prolongation of action potential duration (APD) (Fig. 3g). In contrast, Klf15-Tg mice exhibited significant increase in Ito fast density (Fig. 3f) with dramatic shortening of APD (Fig. 3h). In the Klf15-Tg mice, in addition to short QT intervals, we observed ST-segment changes suggestive of early-repolarization that are similar to ECG findings in Brugada syndrome 8 (Fig. 3b, arrows). In sum, our data suggests that Klf15-dependent transcriptional regulation of rhythmic KChIP2 expression in murine hearts plays a central role in rhythmic variation in ventricular repolarization.
Figure 3. Klf15 deficiency or excess modulates rhythmic variation in repolarization.

(a, b) Representative ECGs from WT vs. Klf15-null, and WT (Non-Tg) vs. Klf15-Tg at ZT2 and ZT14. Note ST segment abnormalities in Klf15-Tg mice (arrow) (c) QTc interval exhibits 24-hour rhythm in WT mice, this rhythm is abrogated with prolonged QTc in the dark phase in Klf15-null mice (n=4 WT and n=4 Klf15-null). (d) Klf15-Tg mice exhibit persistently short QT intervals with no day/night rhythmic variation, when compared to WT (Non-Tg) controls (n=3 WT and n=4 Klf15-Tg). (e, f) Representative outward current recordings from all study groups and summary data for the amplitude of the fast component of outward current (Ito fast) measured at 60mV with average time of decay of 45±5ms (WT, n=10; Klf15-null, n=13; WT (non-Tg), n=14; and Klf15-Tg, n=19). (g, h) Representative ventricular action potentials from all study groups with summary data in bar graphs (WT, n=10; Klf15-null, n=13; WT (non-Tg), n=14; and Klf15-Tg, n=19).). Data presented as mean ± SEM, *p<0.05.
Next, we examined if excessive prolongation or shortening of repolarization could alter arrhythmia susceptibility and survival. Klf15-null mice exhibit no spontaneous arrhythmias on ECG telemetry, hence we utilized intra-cardiac programmed electrical stimulation to examine for arrhythmia susceptibility. In contrast to WT mice, a marked increase in occurrence of ventricular arrhythmias was observed in Klf15-null mice (Fig 4a). Importantly, Klf15-Tg mice exhibit spontaneous ventricular arrhythmias on ECG telemetry (Fig. 4b), and succumb to ~ 35% mortality by 4 months of age (3/8 deaths in Klf15-Tg vs. 0/8 deaths in WT Non-Tg controls, data not shown). As the Klf15-null mice exhibit no evidence of overt ventricular dysfunction, apoptosis or fibrosis 16, 17 in the basal state, the enhanced susceptibility to arrhythmias is likely primarily driven by abnormalities in repolarization. In sum, our studies demonstrate that both deficiency and excess of Klf15 impair temporal variation in cardiac repolarization and greatly increase susceptibility to arrhythmias.
Figure 4. Klf15 deficiency or excess increases susceptibility to ventricular arrhythmias.
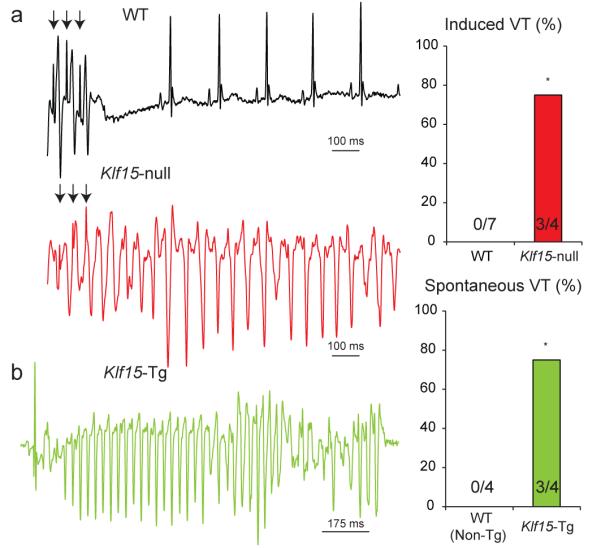
(a) Programmed electrical stimulation in WT and Klf15-null mice. Onset of ventricular tachycardia is illustrated following premature stimuli (arrows) in Klf15-null mice, and the summary data are provided (*p<0.05). (b) Occurrence of spontaneous ventricular arrhythmia in Klf15-Tg mice, and summary data are provided (*p<0.05).
While our finding of circadian control of KChIP2 by Klf15 establishes the principle that circadian rhythms may contribute to arrhythmogenesis, we note that Klf15 minimally affects Kv4.2 expression that also exhibits circadian rhythm (Fig. 1f). However, Kv4.2 expression was disrupted in Bmal1-null and Per2/Cry1-null hearts suggestive of a direct regulation by the circadian clock (Supplemental Fig. 12). Consistent with this observation, cardiomyocytes from Bmal1-null mice exhibit marked action potential prolongation due to near complete elimination of the fast component of the transient outward potassium current (Supplemental Fig. 13). This raises the possibility that additional factors – perhaps components of the circadian clock or as yet unidentified transcriptional regulators – may also impact on temporal variation in electrophysiological parameters and arrhythmogenesis. Future studies in cardiac specific deletion of clock components would be necessary to confirm if the ion channel rhythms are cell autonomous, and their role in regulating cardiac electrophysiology.
In summary, our study provides the first mechanistic link between endogenous circadian rhythms and cardiac electrical instability most often associated with sudden cardiac death in humans (Supplemental Fig. 14). Specifically, we demonstrate that Klf15-dependent rhythmic transcription of KChIP2 regulates the duration and pattern of repolarization and susceptibility to arrhythmias in mice. Since the occurrence of sudden cardiac death in acquired and hereditary forms of human heart disease follows a distinct diurnal pattern 1, 2, these observations offer novel insights into heretofore unrecognized triggers of electrical instability in the heart. However, in contrast to murine repolarization which is largely dependent on Ito, human repolarization occurs through a complex interplay of multiple repolarizing ionic currents. Thus, additional studies will be needed to develop a comprehensive understanding of the link between the circadian clock and electrophysiological properties of the human heart. Nonetheless, these data may provide a mechanistic foundation for future efforts to prevent or treat cardiac arrhythmias by modulating the circadian clock through behavioral or pharmacological means.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Dr. Alfred F. Connors Jr., for support, Mike Mustar for artistic illustrations, Yingjie Cui for experimental assistance, and to members of the Jain laboratory for helpful discussions. Funding sources: Heart Rhythm Society Fellowship (D.J), NIH grants HL094660 (D.J.), HL066991 (M.D.M), HL086614 (S.M.H.), AHA post-doctoral grant (N.S), HL089598, HL091947 (X.H.W), HL76446 (S.A.S.), HL102241 (K.H), HL054807 (D.S.R) and HL075427, HL076754, HL084154, HL086548, HL097595 (M.K.J.), SNF grant 31003A/131086 (U.A.), M01-RR02635 (BWH), Leducq Foundation grants ENAFRA Network 07CVD03 (S.D.) and CNRS (S.D.).
Footnotes
METHODS SUMMARY (detailed methods in supplemental section):
Mice used in the present study, mRNA quantification using Real-Time PCR, promoter reporter analysis, Western immunoblotting, chromatin immunoprecipitation, telemetry ECG and interval analysis, isolated myocyte studies for action potential/Ito measurements, in vivo electrophysiological studies for arrhythmia susceptibility, cosinor analysis for rhythm assessment, and statistical methods are detailed in the supplemental methods section.
AUTHOR CONTRIBUTIONS:
D.J and M.K.J designed the research; D.J, S.M.H, X.W, M.D.M, J.A.R, Y.L, B.L.E and M.J.C acquired the data; J.G, A.S, J.R and R.V.K contributed critical reagents; D.J, N.S, S.D, R.V.K, S.A.S, U.A, X.H.T.W, D.S.R and M.K.J supervised the research; D.J, S.M.H, X.W, M.D.M, J.A.R, K.H, B.L.E, E.F, S.A.S, U.A, X.H.T.W, D.S.R and M.K.J analyzed and interpreted the data; D.J and M.K.J wrote the manuscript.
REFERENCES
- 1.Muller JE, Ludmer PL, Willich SN, Tofler GH, Aylmer G, Klangos I, Stone PH. Circadian variation in the frequency of sudden cardiac death. Circulation. 1987;75(1):131–138. doi: 10.1161/01.cir.75.1.131. [DOI] [PubMed] [Google Scholar]
- 2.Matsuo K, Kurita T, Inagaki M, Kakishita M, Aihara N, Shimizu W, Taguchi A, Suyama K, Kamakura S, Shimomura K. The circadian pattern of the development of ventricular fibrillation in patients with Brugada syndrome. European heart journal. 1999;20(6):465–470. doi: 10.1053/euhj.1998.1332. [DOI] [PubMed] [Google Scholar]
- 3.Goldenberg I, Moss AJ. Long QT syndrome. Journal of the American College of Cardiology. 2008;51(24):2291–2300. doi: 10.1016/j.jacc.2008.02.068. [DOI] [PubMed] [Google Scholar]
- 4.Patel U, Pavri BB. Short QT syndrome: a review. Cardiol Rev. 2009;17(6):300–303. doi: 10.1097/CRD.0b013e3181c07592. [DOI] [PubMed] [Google Scholar]
- 5.Tomaselli GF, Marban E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovascular research. 1999;42(2):270–283. doi: 10.1016/s0008-6363(99)00017-6. [DOI] [PubMed] [Google Scholar]
- 6.Antzelevitch C. Role of spatial dispersion of repolarization in inherited and acquired sudden cardiac death syndromes. Am J Physiol Heart Circ Physiol. 2007;293(4):H2024–2038. doi: 10.1152/ajpheart.00355.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuo HC, Cheng CF, Clark RB, Lin JJ, Lin JL, Hoshijima M, Nguyen-Tran VT, Gu Y, Ikeda Y, Chu PH, Ross J, Giles WR, Chien KR. A defect in the Kv channel-interacting protein 2 (KChIP2) gene leads to a complete loss of I(to) and confers susceptibility to ventricular tachycardia. Cell. 2001;107(6):801–813. doi: 10.1016/s0092-8674(01)00588-8. [DOI] [PubMed] [Google Scholar]
- 8.Antzelevitch C, Yan GX. J wave syndromes. Heart Rhythm. 2010;7(4):549–558. doi: 10.1016/j.hrthm.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reppert SM, Weaver DR. Coordination of circadian timing in mammals. Nature. 2002;418(6901):935–941. doi: 10.1038/nature00965. [DOI] [PubMed] [Google Scholar]
- 10.Bexton RS, Vallin HO, Camm AJ. Diurnal variation of the QT interval--influence of the autonomic nervous system. Br Heart J. 1986;55(3):253–258. doi: 10.1136/hrt.55.3.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kong TQ, Jr., Goldberger JJ, Parker M, Wang T, Kadish AH. Circadian variation in human ventricular refractoriness. Circulation. 1995;92(6):1507–1516. doi: 10.1161/01.cir.92.6.1507. [DOI] [PubMed] [Google Scholar]
- 12.Martino TA, Sole MJ. Molecular time: an often overlooked dimension to cardiovascular disease. Circulation research. 2009;105(11):1047–1061. doi: 10.1161/CIRCRESAHA.109.206201. [DOI] [PubMed] [Google Scholar]
- 13.Paschos GK, FitzGerald GA. Circadian clocks and vascular function. Circulation research. 2010;106(5):833–841. doi: 10.1161/CIRCRESAHA.109.211706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Durgan DJ, Young ME. The cardiomyocyte circadian clock: emerging roles in health and disease. Circulation research. 2010;106(4):647–658. doi: 10.1161/CIRCRESAHA.109.209957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamashita T, Sekiguchi A, Iwasaki YK, Sagara K, Iinuma H, Hatano S, Fu LT, Watanabe H. Circadian variation of cardiac K+ channel gene expression. Circulation. 2003;107(14):1917–1922. doi: 10.1161/01.CIR.0000058752.79734.F0. [DOI] [PubMed] [Google Scholar]
- 16.Haldar SM, Lu Y, Jeyaraj D, Kawanami D, Cui Y, Eapen SJ, Hao C, Li Y, Doughman YQ, Watanabe M, Shimizu K, Kuivaniemi H, Sadoshima J, Margulies KB, Cappola TP, Jain MK. Klf15 deficiency is a molecular link between heart failure and aortic aneurysm formation. Sci Transl Med. 2010;2(26):26ra26. doi: 10.1126/scitranslmed.3000502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang B, Haldar SM, Lu Y, Ibrahim OA, Fisch S, Gray S, Leask A, Jain MK. The Krüppel-like factor KLF15 inhibits connective tissue growth factor (CTGF) expression in cardiac fibroblasts. Journal of molecular and cellular cardiology. 2008;45(2):193–197. doi: 10.1016/j.yjmcc.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.