

Abstract
Alcoholic liver disease (ALD) is a major cause of morbidity and mortality worldwide. The spectrum of ALD ranges from fatty liver to alcoholic hepatitis and cirrhosis, which may eventually lead to the development of hepatocellular carcinoma. In developed countries as well as many developing nations, ALD is a major cause of end-stage liver disease that requires liver transplantation. The most effective therapy for ALD is alcohol abstinence; however, for individuals with severe forms of ALD and those in whom alcohol abstinence is not achievable, targeted therapies are absolutely necessary. In this context, advances of our understanding of the pathophysiology of ALD over the past two decades have contributed to the development of therapeutic modalities (e.g., pentoxifylline and corticosteroids) for the disease though the efficacy of the available treatments remains limited. This article is intended to succinctly review the recent experimental and clinical findings regarding the involvement of oxidative stress and redox signaling in the pathophysiology of ALD and the development of mechanistically-based antioxidant modalities to target the oxidative stress and redox signaling mechanisms of ALD. The biochemical and cellular sources of reactive oxygen and nitrogen species (ROS/RNS) and the dysregulated redox signaling pathways associated with alcohol consumption are particularly discussed to provide insight into the molecular basis of hepatic cell dysfunction and destruction as well as tissue remodeling underlying ALD.
Keywords: alcoholic liver disease, oxidative stress, redox signaling, inflammation, antioxidants
INTRODUCTION
In chemistry, an alcohol is any organic compound in which a hydroxyl functional group (-OH) is bound to a carbon atom, usually connected to other carbon or hydrogen atoms. Although alcohol is a general term for any alcoholic compound, here in this article alcohol refers specifically to ethyl alcohol or ethanol (CH3CH2OH). Alcohol has been a part of human culture since the beginning of recorded history. Excessive use of alcohol contributes substantially to the global burden of disease (4% of the total mortality) and is thus one of the largest avoidable risk factors 1. In the United States, alcoholism is also a major public health problem, and it is estimated that over 100 million Americans are alcoholic. Alcohol abuse accounts for 100,000 to 200,000 deaths annually in the United States, of which over 20,000 are attributable directly to end-stage hepatic cirrhosis and many more are the result of automobile accidents (www.cdc.gov/nchs/fastats/alcohol.htm). Because of the above, it is recommended that alcohol consumption be moderate: ≤ 2 drinks daily in men and ≤ 1 drink in women. In the United States, a “standard” drink is any drink that contains about 0.6 fluid ounces or 14 grams of “pure” ethanol. It is of note that ethanol contains 7 calories per gram.
Although excessive alcohol consumption is associated with a variety of disorders, alcoholic liver disease (ALD) is of the great health impact. As depicted in Fig. 1, ALD encompasses a spectrum of hepatic disorders including: (1) fatty liver (also known as steatosis, due to abnormal accumulation of lipids in hepatic tissue), (2) alcoholic hepatitis (due to inflammation and necrosis), and (3) cirrhosis (due to excessive fibrosis) 2. These are not necessarily distinct stages of evolution of the disease, but rather multiple stages that may be present simultaneously in a given individual. Over 90% of heavy drinkers develop fatty liver, and of those, 10-35% develop alcoholic hepatitis. About 8-20% of chronic alcoholics develop liver cirrhosis, which may eventually lead to hepatocellular carcinoma 3.
Fig. 1.
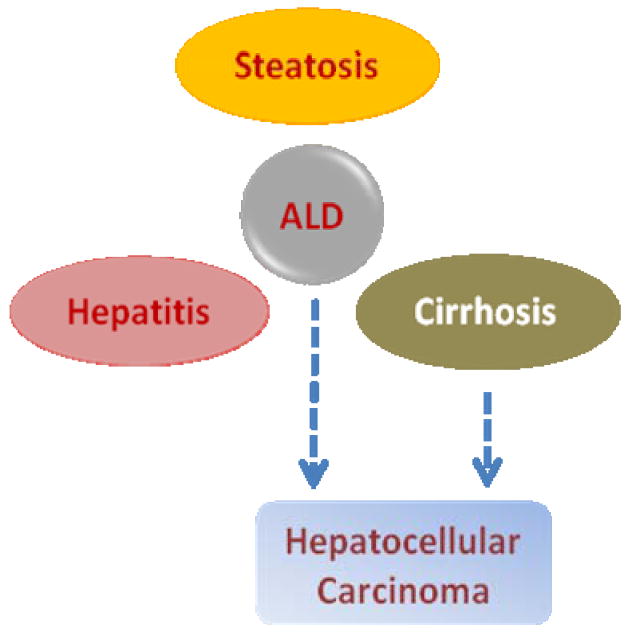
Spectrum of alcoholic liver disease (ALD). It is of note that although cirrhosis is usually the primary pathophysiological process that leads to the development of hepatocellular carcinoma, ALD may progress into liver cancer without necessarily going through cirrhosis.
PATHOPHYSIOLOGY OF ALD
Multiple pathways are involved in the genesis and progression of alcoholic fatty liver, alcoholic hepatitis, and cirrhosis. The spectrum of ALD is illustrated in Fig. 1 above and the pathophysiology summarized individually below. Alcoholic fatty liver results from excessive accumulation of triglycerides in hepatocytes.
Alcohol is metabolized in hepatocytes through oxidation to acetaldehyde and subsequently from acetaldehyde to acetate catalyzed by various enzymes or enzymatic systems, including the alcohol dehydrogenase pathway, cytochrome P4502E1 system, and catalase (Fig. 2) 4. The oxidative metabolism of alcohol generates an excess of NADH, resulting in an increased ratio of NADH to NAD+ in hepatocytes. This altered NADH/NAD+ ratio in hepatocytes leads to inhibition of fatty acid oxidation and promotion of lipogenesis. In addition, alcohol promotes lipid metabolism through inhibition of peroxisome proliferator-activated receptor α (PPARα) and AMP kinase, and via stimulation of sterol regulatory element-binding protein 1, a membrane-bound transcription factor 5, 6. In combination, these effects result in a fat-storing metabolic remodeling of the liver, which is manifested as excessive accumulation of lipids in hepatocytes. Although fatty liver resolves with abstinence, it predisposes the individuals who continue to drink excessively to more serious forms of ALD, i.e., alcoholic hepatitis and cirrhosis.
Fig. 2.
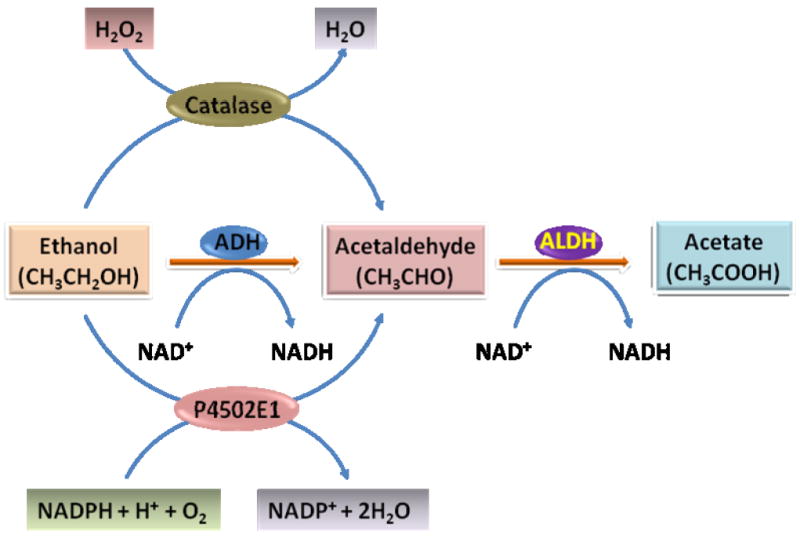
Sequential metabolism of ethanol to form acetaldehyde and acetate. As illustrated, the metabolism of ethanol occurs in the liver (also in many other types of tissues) via three enzymatic systems, ADH, P4502E1, and catalase, which are primarily located in cytosol, endoplasmic reticulum membrane (microsomes), and peroxisomes, respectively. ALDH is located in mitochondria and cytosol. ADH, alcohol dehydrogenase; ALDH, aldehyde dehydrogenase.
Alcoholic hepatitis is characterized by inflammation and necrosis 7. It is believed that alcohol compromises the intestinal barrier, which leads to increased permeability and the subsequent presence of gut bacteria-derived lipopolysaccharide (LPS) in the portal blood. LPS stimulates the Kupffer cells and possibly other types of cells in the liver, resulting in excessive release of inflammatory cytokines and reactive oxygen and nitrogen species (ROS/RNS). The dysregulated inflammatory responses are thought to primarily contribute to the hepatocyte injury and tissue necrosis. In addition, accumulation of lipids also promotes inflammation. Progression of these detrimental processes may eventually cause excessive accumulation of collagen in extracellular matrix, resulting in liver fibrosis and cirrhosis. The crucial role of inflammation in ALD is supported by the demonstrated therapeutic efficacy of anti-inflammatory drugs (e.g., glucocorticosteroids) in animal models and certain patient subgroups 7, 8. In addition to dysregulated inflammation, recent work demonstrated that oxidative stress also plays an important role in the development of alcoholic hepatitis and cirrhosis as well as fatty liver. Since the general aspects of oxidative stress mechanism of ALD have been reviewed previously 5, 9, this article is intended to focus on discussing the new findings from both animal experiments and human studies with an emphasis on novel oxidative stress and redox signaling pathways and the mechanistically-based modalities targeting these molecular pathways for the intervention of ALD. To set a stage for the discussion of the new findings, previously well-established data on oxidative stress in ALD are briefly outlined wherever pertinent.
ROLE OF OXIDATIVE STRESS IN ALD IN EXPERIMENTAL ANIMALS
Studies using animal models have contributed greatly to our understanding of how ALD develops and how the severity of liver injury is influenced by factors other than alcohol, such as nutrition, oxygen deprivation (as occurs with sleep apnea and smoking), and gene regulation. In this regard, the intragastric feeding model results in liver lesions that mimic human ALD and has been widely used 10, 11. In addition, chronic feeding of liquid diet containing ethanol is also a commonly used model of ALD 12. Animal experiments have provided important information on the pathophysiology of ALD including the causative involvement of oxidative stress and also contributed to the development of new therapeutic approaches.
Evidence for a causative role of oxidative stress in experimental ALD
As discussed below, there are four lines of experimental evidence that support a causative involvement of oxidative stress in the development of ALD in animal models.
Increased formation of ROS/RNS
The ability of acute and chronic alcohol treatment to increase the production of ROS/RNS as well as other free radical species (e.g., 1-hydroxyethyl radical) has been demonstrated in a variety of systems, including cell cultures and experimental animals, as well as in human subjects (see refs. 5 and 9; also see later sections for the discussion of human evidence). The detailed cellular sources and mechanisms of ROS/RNS formation are also described later in the manuscript.
Depletion of antioxidants and increased biomarkers of oxidative damage
Although acute exposure to alcohol may cause transient induction of certain antioxidant genes, excessive chronic exposure usually results in decreased levels of antioxidant defenses both in hepatic tissue and in blood. Of note, an early change after alcohol treatment is the depletion of mitochondrial glutathione in hepatocytes, which appears to play an important part in alcohol-induced liver injury 13. In line with the augmented formation of ROS/RNS and decreased levels of antioxidant defenses, increased levels of oxidative stress biomarkers are observed in experimental animals exposed to alcohol. These biomarkers include lipid peroxidation products (e.g., malondialdehyde, 4-hydroxy-2-nonenal, F2-isoprostanes), oxidative DNA base modifications (e.g., 8-hydroxy-2’-deoxyguanosine) and the protein adducts derived from reactive aldehydes, as well as 1-hydroxyethyl radical. Notably, the levels of the oxidative stress biomarkers are correlated with the severity of alcohol-induced liver injury. As such, oxidative stress biomarkers are frequently used to assess the liver injury as well as the protection by potential therapeutic agents, including antioxidant compounds 5, 9.
Protection by exogenous antioxidant compounds
The ability of alcohol to cause oxidative stress makes it rational to use antioxidant compounds to protect against the liver injury. Indeed, various compounds with antioxidant properties have been shown to be protective against alcohol-induced liver injury in animal models. These include antioxidant vitamins, glutathione-augmenting agents (e.g., glutathione esters, N-acetylcysteine, S-adenosyl-L-methionine), antioxidant mimetics, probucol, and dietary polyphenols 14-18. In addition, induction of endogenous antioxidants by chemical inducers also ameliorated alcohol-induced liver injury 19, 20. These findings are consistent with the concept that oxidative stress plays a causal role in experimental ALD.
Protection by genetic overexpression of endogenous antioxidant genes and sensitization by antioxidant gene knockout
In line with the protection by exogenous antioxidant compounds, overexpression of endogenous antioxidant proteins (e.g., manganese superoxide dismutase, Cu, Zn superoxide dismutase, metallothionein) via transgenic approaches or gene delivery blunted alcohol-induced liver injury in experimental animals 21-23. Of note, the protective effects were also observed with systemic administration of antioxidant proteins, such as thioredoxin 24. Conversely, gene knockout of endogenous antioxidant proteins (e.g., Cu, Zn superoxide dismutase, catalase, glutathione peroxidase-1, metallothionein) sensitized the experimental animals to alcohol-induced liver injury 25-28. More recently, it was reported that ablation of either sulfiredoxin or peroxiredoxin-1 in mice markedly aggravated alcohol-induced oxidative hepatic injury 29. Notably, peroxiredoxin-1 was found to be closely associated with cytochrome P4502E1 on the cytosolic side of the endoplasmic reticulum membrane of the hepatocytes. This close proximity of peroxiredoxin-1 and P4502E1 was suggested to be responsible for peroxiredoxin-1-mediated removal of ROS generated by P4502E1 29.
The above observations from studies using antioxidant gene overexpression and/or knockout models provide the most compelling evidence for a cause and effect relationship between oxidative stress and the development of ALD. The crucial beneficial function of endogenous antioxidant defenses in ALD is further strengthened by the finding that targeted disruption of Nrf2, a central regulator of antioxidant genes, markedly aggravated alcohol-induced liver injury in mice, as indicated by increased liver necrosis, inflammation, oxidative stress, and mortality 30. As such, activation of Nrf2 signaling by pharmacologic agents or phytochemicals has been proposed as an important strategy for protecting against liver injury induced by alcohol as well as other hepatotoxicants 31 (see a later section for the further discussion of Nrf2 signaling in the molecular pathophysiology of ALD).
Sources of ROS/RNS in experimental ALD
Hepatocytes
As oxidative damage is an event predominantly in hepatocytes following alcohol administration, hepatocytes may thus be a major source of the ROS/RNS as well as other free radical species. Indeed, several intracellular pathways have been shown to contribute to the increased production of the reactive species in hepatocytes. These include mitochondria, cytochrome P4502E1, NAD(P)H oxidase (NOX), and inducible nitric oxide synthase (iNOS).
-
Mitochondria: As discussed earlier, metabolism of alcohol results in increased levels of NADH, which provide electrons for mitochondrial electron transport chain, leading to increased one-electron reduction of oxygen to superoxide. In addition, formation of acetaldehyde by alcohol was shown to cause mitochondrial damage, which may also lead to increased formation of superoxide from the electron transport chain 32, 33.
Recently, the redox-active protein p66SHC was found to be associated with mitochondria and generate ROS via electron transfer from cytochrome c 34. This redox-active protein potentiated ethanol-induced oxidative stress and mitochondrial damage in hepatocytes 35. Ethanol treatment of mice also caused induction of p66SHC in hepatic tissue, and mice deficient in p66SHC showed increased resistance to ethanol-induced liver oxidative stress, steatosis, and necrosis as compared with the wild-type counterparts 35. Interestingly, deletion of p66SHC led to marked augmentation of ethanol-mediated induction of manganese superoxide dismutase in hepatic tissue and cultured hepatocytes, suggesting p66SHC may also act as a transcriptional suppressor of liver manganese superoxide dismutase 35. Hence, the mitochondria-associated, ethanol-inducible, redox-active protein p66SHC may function as a novel source of ROS generation during ethanol consumption, contributing to the oxidative liver injury underlying ALD.
P4502E1: Cytochrome P450 enzymes, especially P4502E1 are known to produce ROS. The ability of P4502E1 to produce ROS is augmented during alcohol administration as this enzyme is highly inducible by alcohol. Both cell experiments and in vivo animal studies demonstrated P4502E1 as an important source of ROS that participate in alcohol-induced liver injury. Knockout of P4502E1 blunted alcohol-induced oxidative stress and liver injury in mice, whereas transgenic overexpression of the enzyme aggravated alcohol-induced hepatic damage, pointing to a causative involvement of P4502E1 in the pathophysiology of ALD 36.
NOX and iNOS: Although NOX and iNOS of inflammatory cells are primarily responsible for the augmented formation of ROS/RNS during inflammatory responses alcohol treatment also resulted in activation of both NOX and iNOS in hepatocytes, leading to increased production of both superoxide and nitric oxide 37, 38. Reaction of these two radical species generates the potent oxidant peroxynitrite, which has been suggested to cause mitochondrial damage underlying experimental ALD 39, 40.
Inflammatory cells
As stated above, activation of NOX and iNOS in inflammatory cells results in the formation of large amounts of ROS/RNS. Both hepatic Kupffer cells and infiltrated inflammatory cells were found to be activated to produce ROS/RNS following alcohol administration 41. Activation of Kupffer cells by LPS derived from gut bacteria is an early event leading to ALD. In this regard, depletion or inactivation of Kupffer cells ameliorated alcohol-induced inflammation, oxidative stress, and liver injury in experimental animals 41. Alcohol-induced oxidative stress and hepatic injury were also blunted in mice deficient in either p47phox or iNOS, indicating a causative role for NOX and iNOS in the pathophysiology of ALD 42-44. p47phox is an essential component of the NOX enzyme complex.
Hepatic stellate cells
In addition to hepatocytes and inflammatory cells, hepatic stellate cells may also contribute to the formation of ROS during alcohol exposure. Treatment with alcohol or its metabolite acetaldehyde was shown to elicit increased ROS formation from NOX in stellate cells. Such formed ROS as well as the ROS derived from other cellular sources, such as P4502E1 may play an important part in the transformation of stellate cells into myofibroblasts as well as the increased production of collagen, a process critically involved in liver fibrosis 5, 45. Mechanistically, it was demonstrated that induction of collagen expression by acetaldehyde in stellate cells was dependent on PPAR-γ phosphorylation induced by a hydrogen peroxide-mediated activation of the profibrogenic c-Abl signaling pathway 46. This finding also highlighted the intimate association between acetaldehyde and ROS in the molecular pathophysiology of ALD.
Mechanisms of oxidative stress-mediated injury in experimental ALD
The key molecular mechanisms underlying oxidative stress-mediated liver injury in ALD can be summarized into the following three schemes: (1) mitochondrial damage, (2) perpetuation of inflammation via dysregulated redox signaling, and (3) transformation of stellate cells and extracellular matrix remodeling associated with dysregulated redox signaling 5, 47. In addition, recent studies suggested a crucial role for dysregulated Nrf2 signaling in molecular alterations leading to experimental ALD.
Mitochondrial damage
Alcohol administration causes increased formation of ROS in mitochondria 32, 33. Such locally generated ROS as well as those derived from other cellular sources cause mitochondrial dysfunction, energy depletion, and ultimately cell death 48. The increased production of ROS/RNS also results in damage of other cellular constituents, including membrane lipids, enzymes, and nucleic acids, contributing to cell injury. Notably, alcohol treatment increases hepatic iron accumulation, which may further facilitate the production of the highly reactive hydroxyl radical, causing damage to mitochondria and other cellular constituents 49. The key role of mitochondrial oxidative stress injury in ALD is further supported by the finding that either overexpression of the mitochondrial antioxidant enzyme manganese superoxide dismutase or administration of exogenous antioxidant compounds targeted to mitochondria, such as mito-coenzyme Q (MitoQ), led to attenuation of oxidative liver injury underlying ALD 22, 50.
Perpetuation of inflammation via dysregulated redox signaling
As noted earlier, activation of inflammatory cells results in the formation of large quantities of ROS/RNS. Such formed ROS/RNS also cause dysregulated redox homeostasis, leading to the activation of NF-κB and the subsequent overexpression of proinflammatory cytokines and adhesion molecules 51, 52. Indeed, the NF-κB-regulated chemokine MCP-1 was recently shown to be essential for the continuous inflammatory responses in liver tissue and the development of hepatic steatosis in alcohol-treated mice 53. Hence, the perpetuation of inflammation and inflammatory injury may be an important molecular mechanism underlying the development and progression of ALD. Since metabolism of alcohol also leads to the increased formation of ROS independent of the activation of inflammatory cells (for example, the P4502E1 and mitochondria-derived ROS) ROS may also contribute to the initiation of the inflammatory responses by activating NF-κB and other redox-sensitive signaling molecules. In this regard, oxidative stress may contribute to the genesis of ALD. This notion is supported by observations that administration of antioxidant compounds or overexpression of endogenous antioxidant proteins is frequently able to prevent the development of ALD in experimental animals (see earlier sections on protection by exogenous antioxidant compounds and genetic overexpression of endogenous antioxidant genes).
Transformation of stellate cells and extracellular matrix remodeling associated with dysregulated redox signaling
ROS/RNS are able to induce the transformation of hepatic stellate cells into myofibroblasts, leading to excessive production and deposition of collagen in extracellular matrix. Reactive aldehydes derived from lipid peroxidation, such as 4-hydroxy-2-nonenal have been shown to also stimulate collagen production from stellate cells 54. In addition, ROS/RNS are capable of inducing proliferation and activation of other cells, such as periportal fibroblasts and fibrocytes. Furthermore, ROS/RNS may cause activation of matrix metalloproteinases, resulting in remodeling of the extracellular matrix 55. Progression of these molecular and cellular events eventually leads to excessive hepatic fibrosis and cirrhosis 56.
Hepatic Nrf2 signaling
Studies in the early 1990’s demonstrated a crucial role for a cis-acting element, named antioxidant response element (ARE) in the regulation of antioxidant gene expression 57. In 1997, M. Yamomoto and coworkers identified Nrf2 (nuclear factor-erythroid 2 p45-related factor 2) as a critical transcription factor that interacts with the ARE, leading to increased expression of various antioxidant and cytoprotective genes 58. Subsequent studies from multiple research groups have established that Nrf2 plays a central role in regulating both constitutive and inducible expression of a wide variety of antioxidant and anti-inflammatory genes in mammalian tissues/cells 59-61. As noted earlier, targeted disruption of Nrf2 in mice led to aggravation of ethanol-induced liver injury as evidenced by augmented hepatic oxidative stress, inflammatory responses, steatosis, and necrosis. The ethanol-mediated hepatic mitochondrial damage was also exacerbated in the Nrf2-knockout mice. These exacerbated deleterious responses led to accelerated liver failure and death caused by ethanol in the Nrf2-knockout mice 30. Notably, short-term ethanol treatment resulted in induction of several antioxidant and anti-inflammatory enzymes, including heme oxygenase-1, NAD(P)H:quinine oxidoreductase-1, and glutathione S-transferase in the liver tissue in an Nrf2-dependent manner 30. Hence, the induction of antioxidant and anti-inflammatory enzymes via Nrf2 signaling might act an early compensatory or adaptive mechanism to suppress ethanol-induced oxidative injury and inflammation in liver tissue, and ablation of this protective compensatory mechanism in Nrf2-knockout mice led to dramatic acceleration of ethanol-induced liver injury, hepatic failure, and death in mice 30. In this context, a recent study reported that ethanol-induced upregulation of heme oxygenase-1 expression was mediated by both Nrf2 signaling and hypoxia-inducible factor-1α (HIF-1α), and the upregulation of heme oxygenase-1 attenuated inflammatory cytokine expression in ALD 62. Nrf2 was also found to be induced by overexpression of P4502E1 in liver tissue and cultured hepatocytes, and the augmented Nrf2-dependent antioxidants attenuated P4502E1-mediated oxidative hepatic injury 63.
Taken together, the above observations demonstrated a crucial role for Nrf2 signaling (and possibly other signaling pathways) in suppressing the oxidative and inflammatory pathophysiology of ALD (Fig. 3). Hence, disruption of the normal functionality of Nrf2 signaling may be a potential mechanism leading to increased susceptibility to ALD. Likewise, augmentation of the hepatic Nrf2 signaling by pharmacological approaches or gene therapy, as mentioned earlier, may represent an effective strategy for the intervention of the oxidative stress pathophysiology of ALD.
Fig. 3.

Involvement of Nrf2 signaling in the pathophysiology of alcoholic liver disease (ALD). Activation of Nrf2 signaling and Nrf2-dependent antioxidants and anti-inflammatory enzymes may act as a compensatory protective mechanism in ALD. Consequently, comprised Nrf2 functionality may aggravate the pathophysiology of ALD. See text for more detailed description. ROS, reactive oxygen species; LPS, lipopolysaccharide.
Summary of the role of oxidative stress in experimental ALD
Alcohol causes augmented formation of ROS/RNS from multiple sources, including hepatocyes, inflammatory cells, and other cell types in liver tissue. The augmented production of ROS/RNS and the consequent depletion of antioxidants together lead to marked oxidative stress that contributes to both the genesis and progression of ALD in animal models (Fig. 4). The causative involvement of oxidative stress in ALD is further supported by the demonstrated efficacy of both exogenous and endogenous antioxidants in protecting against alcohol-induced liver injury. These findings in experimental animals have prompted the investigation of the causative role of oxidative stress in human ALD and the antioxidant-based modalities for the disease intervention.
Fig. 4.
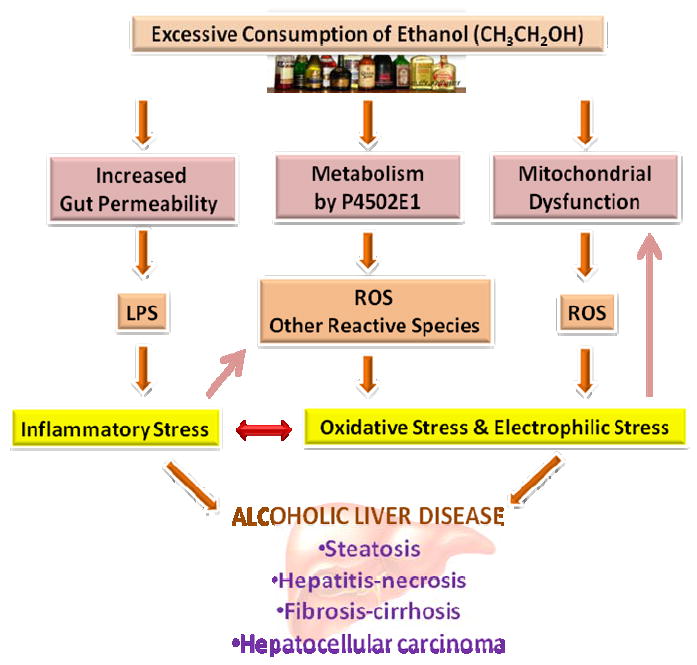
Schematic illustration of the overall involvement of oxidative/electrophilic stress and dysregulated inflammation in the pathophysiology of alcoholic fatty liver disease. See text for more detailed description. LPS, lipopolysaccharide; ROS, reactive oxygen species.
ROLE OF OXIDATIVE STRESS IN ALD IN HUMANS
Evidence for a role of oxidative stress in human ALD
Increased ROS/RNS formation and biomarkers of oxidative stress
Excessive consumption of alcohol led to increases in biomarkers of oxidative stress in plasma and urine, including the lipid peroxidation products F2-isoprostanes and 4-hydroxy-2-nonenal 64,65. The oxidative stress biomarkers were closely associated with the severity of liver injury. Notably, acute alcohol consumption in healthy individuals also resulted in increases in the oxidative stress biomarkers, suggesting that oxidative stress may precede the development of ALD 66. In addition, levels of antioxidants, such as glutathione, vitamin C, and vitamin E were decreased in patients with ALD as compared with normal subjects. As discussed above, metabolism of alcohol forms 1-hydroxyethyl radical in experimental systems. The protein adduct of 1-hydroxyethyl radical was also detected in patients with alcoholic cirrhosis 67.
Antioxidant gene polymorphisms
A number of studies have demonstrated that polymorphisms of glutathione S-transferase (GST), especially the null-genotypes of GSTM1 and GSTT1 were associated with increased risk of developing ALD as well as hepatocellular carcinoma 68, 69. These findings are consistent with the notion that GST enzymes play an important role in the detoxification of reactive aldehydes, including those that participate in alcohol-induced liver injury. In addition to GST, a Val-Ala polymorphism in manganese superoxide dismutase (MnSOD) was associated with elevated risk of developing cirrhosis in French alcoholics and increased rates of hepatocellular carcinoma development and death in cirrhotic patients 70. Notably, the polymorphisms in antioxidant genes may interact with those in other xenobiotic biotransforming enzymes, resulting in augmented risk of developing ALD. For example, combination of the GSTM1-null genotype and a polymorphism of P4502E1 (causing increased P4502E1 activity) resulted in increased risk of developing alcoholic liver cirrhosis 71. Combination of GG-myeloperoxidase (MPO) genotype (leading to high MPO expression) and at least one Ala-MnSOD allele markedly elevated the risk of hepatocellular carcinoma occurrence and death in patients with alcoholic cirrhosis 72.
Antioxidant intervention
Although the experimental evidence supporting a protective role of antioxidants in alcohol-induced liver injury is extensive, the effectiveness of antioxidant therapy remains unclear in human patients with ALD. Randomized controlled clinical trials on using antioxidant vitamin E or an antioxidant cocktail (containing antioxidant vitamins, N-acetylcysteine, and coenzyme Q among others) have failed to show an efficacy in improving the disease conditions in ALD patients 73-75. The causes of the apparent discrepancy between the experimental studies and clinical trials are not clear. Potential reasons may include the doses of antioxidants used in the trials, the duration and time-window of the intervention, the patients’ status of oxidative stress and antioxidants, as well as the lack of a profound understanding of the pharmacokinetic and pharmacodynamic activities of the antioxidant compounds in patients with ALD. In this context, S-adenosyl-L-methionine is a well-characterized antioxidant compound that functions as a methylating agent, a precursor of glutathione, and a modulator of proinflammatory cytokine expression. A previous randomized controlled trial demonstrated that patients with ALD treated with S-adenosyl-L-methionine showed improved mortality and decreased need for liver transplantation 76. Although S-adenosyl-L-methionine showed promising effects, the exact contribution of the antioxidant activity of this compound to the therapeutic activity in ALD remains to be determined. Moreover, the clinical efficacy of S-adenosyl-L-methionine as a therapy for ALD needs to be further established in large-scale well-designed trials.
Summary and future perspectives on studies of ALD in humans
Compared with the substantial experimental evidence supporting a causative role of oxidative stress in ALD and the benefits of antioxidant-based modalities in disease intervention in animal models, evidence from studies in human subjects is less compelling. Nevertheless, there is accumulating evidence indicating a possible causative involvement of oxidative stress in the pathophysiology of human ALD. Future studies should focus on developing novel antioxidant compounds with well-characterized pharmacokinetic and pharmacodynamic profiles, and testing them in selected patient populations with overt oxidative stress and/or antioxidant deficiency. Such a subpopulation targeting strategy has been shown to be effective in the intervention of other oxidative stress-associated diseases, such as cardiovascular disorders and diabetes 77-79. In addition to the therapeutic intervention, future efforts should also be devoted to the development of antioxidant-based strategies for primary prevention of ALD in susceptible or high risk individuals.
Acknowledgments
ZH is supported by NIH grant DK81905; YL is supported by a grant from AICR and NIH HL93557.
References
- 1.Rehm J, Mathers C, Popova S, Thavorncharoensap M, Teerawattananon Y, Patra J. Global burden of disease and injury and economic cost attributable to alcohol use and alcohol-use disorders. Lancet. 2009;373:2223–33. doi: 10.1016/S0140-6736(09)60746-7. [DOI] [PubMed] [Google Scholar]
- 2.O’Shea RS, Dasarathy S, McCullough AJ. Alcoholic liver disease. Am J Gastroenterol. 2010;105:14–32. doi: 10.1038/ajg.2009.593. quiz 3. [DOI] [PubMed] [Google Scholar]
- 3.Altamirano J, Bataller R. Alcoholic liver disease: pathogenesis and new targets for therapy. Nat Rev Gastroenterol Hepatol. 2011;8:491–501. doi: 10.1038/nrgastro.2011.134. [DOI] [PubMed] [Google Scholar]
- 4.Lieber CS. Ethanol metabolism, cirrhosis and alcoholism. Clin Chim Acta. 1997;257:59–84. doi: 10.1016/s0009-8981(96)06434-0. [DOI] [PubMed] [Google Scholar]
- 5.Beier JI, McClain CJ. Mechanisms and cell signaling in alcoholic liver disease. Biol Chem. 2010;391:1249–64. doi: 10.1515/BC.2010.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gyamfi MA, Wan YJ. Pathogenesis of alcoholic liver disease: the role of nuclear receptors. Exp Biol Med (Maywood) 2010;235:547–60. doi: 10.1258/ebm.2009.009249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lucey MR, Mathurin P, Morgan TR. Alcoholic hepatitis. N Engl J Med. 2009;360:2758–69. doi: 10.1056/NEJMra0805786. [DOI] [PubMed] [Google Scholar]
- 8.Frazier TH, Stocker AM, Kershner NA, Marsano LS, McClain CJ. Treatment of alcoholic liver disease. Therap Adv Gastroenterol. 2011;4:63–81. doi: 10.1177/1756283X10378925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cederbaum AI, Lu Y, Wu D. Role of oxidative stress in alcohol-induced liver injury. Arch Toxicol. 2009;83:519–48. doi: 10.1007/s00204-009-0432-0. [DOI] [PubMed] [Google Scholar]
- 10.Nanji AA, French SW. Animal models of alcoholic liver disease--focus on the intragastric feeding model. Alcohol Res Health. 2003;27:325–30. [PMC free article] [PubMed] [Google Scholar]
- 11.Siegmund SV, Haas S, Singer MV. Animal models and their results in gastrointestinal alcohol research. Dig Dis. 2005;23:181–94. doi: 10.1159/000090165. [DOI] [PubMed] [Google Scholar]
- 12.Lieber CS, DeCarli LM, Sorrell MF. Experimental methods of ethanol administration. Hepatology. 1989;10:501–10. doi: 10.1002/hep.1840100417. [DOI] [PubMed] [Google Scholar]
- 13.Mantena SK, King AL, Andringa KK, Eccleston HB, Bailey SM. Mitochondrial dysfunction and oxidative stress in the pathogenesis of alcohol- and obesity-induced fatty liver diseases. Free Radic Biol Med. 2008;44:1259–72. doi: 10.1016/j.freeradbiomed.2007.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ronis MJ, Butura A, Sampey BP, et al. Effects of N-acetylcysteine on ethanol-induced hepatotoxicity in rats fed via total enteral nutrition. Free Radic Biol Med. 2005;39:619–30. doi: 10.1016/j.freeradbiomed.2005.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yanardag R, Ozsoy-Sacan O, Ozdil S, Bolkent S. Combined effects of vitamin C, vitamin E, and sodium selenate supplementation on absolute ethanol-induced injury in various organs of rats. Int J Toxicol. 2007;26:513–23. doi: 10.1080/10915810701707296. [DOI] [PubMed] [Google Scholar]
- 16.Kono H, Arteel GE, Rusyn I, Sies H, Thurman RG. Ebselen prevents early alcohol-induced liver injury in rats. Free Radic Biol Med. 2001;30:403–11. doi: 10.1016/s0891-5849(00)00490-1. [DOI] [PubMed] [Google Scholar]
- 17.Song Z, Zhou Z, Chen T, et al. S-adenosylmethionine (SAMe) protects against acute alcohol induced hepatotoxicity in mice small star, filled. J Nutr Biochem. 2003;14:591–7. doi: 10.1016/s0955-2863(03)00116-5. [DOI] [PubMed] [Google Scholar]
- 18.Ajmo JM, Liang X, Rogers CQ, Pennock B, You M. Resveratrol alleviates alcoholic fatty liver in mice. Am J Physiol Gastrointest Liver Physiol. 2008;295:G833–42. doi: 10.1152/ajpgi.90358.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mandal P, Roychowdhury S, Park PH, Pratt BT, Roger T, Nagy LE. Adiponectin and heme oxygenase-1 suppress TLR4/MyD88-independent signaling in rat Kupffer cells and in mice after chronic ethanol exposure. J Immunol. 2010;185:4928–37. doi: 10.4049/jimmunol.1002060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bao W, Li K, Rong S, et al. Curcumin alleviates ethanol-induced hepatocytes oxidative damage involving heme oxygenase-1 induction. J Ethnopharmacol. 2010;128:549–53. doi: 10.1016/j.jep.2010.01.029. [DOI] [PubMed] [Google Scholar]
- 21.Wheeler MD, Kono H, Yin M, et al. Delivery of the Cu/Zn-superoxide dismutase gene with adenovirus reduces early alcohol-induced liver injury in rats. Gastroenterology. 2001;120:1241–50. doi: 10.1053/gast.2001.23253. [DOI] [PubMed] [Google Scholar]
- 22.Wheeler MD, Nakagami M, Bradford BU, et al. Overexpression of manganese superoxide dismutase prevents alcohol-induced liver injury in the rat. J Biol Chem. 2001;276:36664–72. doi: 10.1074/jbc.M105352200. [DOI] [PubMed] [Google Scholar]
- 23.Zhou Z, Sun X, James Kang Y. Metallothionein protection against alcoholic liver injury through inhibition of oxidative stress. Exp Biol Med (Maywood) 2002;227:214–22. doi: 10.1177/153537020222700310. [DOI] [PubMed] [Google Scholar]
- 24.Cohen JI, Roychowdhury S, DiBello PM, Jacobsen DW, Nagy LE. Exogenous thioredoxin prevents ethanol-induced oxidative damage and apoptosis in mouse liver. Hepatology. 2009;49:1709–17. doi: 10.1002/hep.22837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kessova IG, Ho YS, Thung S, Cederbaum AI. Alcohol-induced liver injury in mice lacking Cu, Zn-superoxide dismutase. Hepatology. 2003;38:1136–45. doi: 10.1053/jhep.2003.50450. [DOI] [PubMed] [Google Scholar]
- 26.Curry-McCoy TV, Osna NA, Nanji AA, Donohue TM., Jr Chronic ethanol consumption results in atypical liver injury in copper/zinc superoxide dismutase deficient mice. Alcohol Clin Exp Res. 2010;34:251–61. doi: 10.1111/j.1530-0277.2009.01088.x. [DOI] [PubMed] [Google Scholar]
- 27.Kim SJ, Lee JW, Jung YS, et al. Ethanol-induced liver injury and changes in sulfur amino acid metabolomics in glutathione peroxidase and catalase double knockout mice. J Hepatol. 2009;50:1184–91. doi: 10.1016/j.jhep.2009.01.030. [DOI] [PubMed] [Google Scholar]
- 28.Zhou Z, Sun X, Lambert JC, Saari JT, Kang YJ. Metallothionein-independent zinc protection from alcoholic liver injury. Am J Pathol. 2002;160:2267–74. doi: 10.1016/S0002-9440(10)61174-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bae SH, Sung SH, Cho EJ, et al. Concerted action of sulfiredoxin and peroxiredoxin I protects against alcohol-induced oxidative injury in mouse liver. Hepatology. 2011;53:945–53. doi: 10.1002/hep.24104. [DOI] [PubMed] [Google Scholar]
- 30.Lamle J, Marhenke S, Borlak J, et al. Nuclear factor-eythroid 2-related factor 2 prevents alcohol-induced fulminant liver injury. Gastroenterology. 2008;134:1159–68. doi: 10.1053/j.gastro.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 31.Klaassen CD, Reisman SA. Nrf2 the rescue: effects of the antioxidative/electrophilic response on the liver. Toxicol Appl Pharmacol. 2010;244:57–65. doi: 10.1016/j.taap.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boveris A, Fraga CG, Varsavsky AI, Koch OR. Increased chemiluminescence and superoxide production in the liver of chronically ethanol-treated rats. Arch Biochem Biophys. 1983;227:534–41. doi: 10.1016/0003-9861(83)90482-4. [DOI] [PubMed] [Google Scholar]
- 33.Kukielka E, Dicker E, Cederbaum AI. Increased production of reactive oxygen species by rat liver mitochondria after chronic ethanol treatment. Arch Biochem Biophys. 1994;309:377–86. doi: 10.1006/abbi.1994.1127. [DOI] [PubMed] [Google Scholar]
- 34.Giorgio M, Migliaccio E, Orsini F, et al. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell. 2005;122:221–33. doi: 10.1016/j.cell.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 35.Koch OR, Fusco S, Ranieri SC, et al. Role of the life span determinant P66(shcA) in ethanol-induced liver damage. Lab Invest. 2008;88:750–60. doi: 10.1038/labinvest.2008.44. [DOI] [PubMed] [Google Scholar]
- 36.Lu Y, Wu D, Wang X, Ward SC, Cederbaum AI. Chronic alcohol-induced liver injury and oxidant stress are decreased in cytochrome P4502E1 knockout mice and restored in humanized cytochrome P4502E1 knock-in mice. Free Radic Biol Med. 2010;49:1406–16. doi: 10.1016/j.freeradbiomed.2010.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.De Minicis S, Brenner DA. Oxidative stress in alcoholic liver disease: role of NADPH oxidase complex. J Gastroenterol Hepatol. 2008;23(Suppl 1):S98–103. doi: 10.1111/j.1440-1746.2007.05277.x. [DOI] [PubMed] [Google Scholar]
- 38.Spitzer JA, Zheng M, Kolls JK, Vande Stouwe C, Spitzer JJ. Ethanol and LPS modulate NF-kappaB activation, inducible NO synthase and COX-2 gene expression in rat liver cells in vivo. Front Biosci. 2002;7:a99–108. doi: 10.2741/A744. [DOI] [PubMed] [Google Scholar]
- 39.Yang ES, Lee JH, Park JW. Ethanol induces peroxynitrite-mediated toxicity through inactivation of NADP+-dependent isocitrate dehydrogenase and superoxide dismutase. Biochimie. 2008;90:1316–24. doi: 10.1016/j.biochi.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 40.Larosche I, Letteron P, Berson A, et al. Hepatic mitochondrial DNA depletion after an alcohol binge in mice: probable role of peroxynitrite and modulation by manganese superoxide dismutase. J Pharmacol Exp Ther. 2010;332:886–97. doi: 10.1124/jpet.109.160879. [DOI] [PubMed] [Google Scholar]
- 41.Ajakaiye M, Jacob A, Wu R, Nicastro JM, Coppa GF, Wang P. Alcohol and hepatocyte-Kupffer cell interaction (review) Mol Med Report. 2011;4:597–602. doi: 10.3892/mmr.2011.471. [DOI] [PubMed] [Google Scholar]
- 42.Kono H, Rusyn I, Yin M, et al. NADPH oxidase-derived free radicals are key oxidants in alcohol-induced liver disease. J Clin Invest. 2000;106:867–72. doi: 10.1172/JCI9020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McKim SE, Gabele E, Isayama F, et al. Inducible nitric oxide synthase is required in alcohol-induced liver injury: studies with knockout mice. Gastroenterology. 2003;125:1834–44. doi: 10.1053/j.gastro.2003.08.030. [DOI] [PubMed] [Google Scholar]
- 44.Venkatraman A, Shiva S, Wigley A, et al. The role of iNOS in alcohol-dependent hepatotoxicity and mitochondrial dysfunction in mice. Hepatology. 2004;40:565–73. doi: 10.1002/hep.20326. [DOI] [PubMed] [Google Scholar]
- 45.Nieto N, Friedman SL, Greenwel P, Cederbaum AI. CYP2E1-mediated oxidative stress induces collagen type I expression in rat hepatic stellate cells. Hepatology. 1999;30:987–96. doi: 10.1002/hep.510300433. [DOI] [PubMed] [Google Scholar]
- 46.Ceni E, Crabb DW, Foschi M, et al. Acetaldehyde inhibits PPARgamma via H2O2-mediated c-Abl activation in human hepatic stellate cells. Gastroenterology. 2006;131:1235–52. doi: 10.1053/j.gastro.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 47.Dey A, Cederbaum AI. Alcohol and oxidative liver injury. Hepatology. 2006;43:S63–74. doi: 10.1002/hep.20957. [DOI] [PubMed] [Google Scholar]
- 48.Sato N. Central role of mitochondria in metabolic regulation of liver pathophysiology. J Gastroenterol Hepatol. 2007;22(Suppl 1):S1–6. doi: 10.1111/j.1440-1746.2007.04963.x. [DOI] [PubMed] [Google Scholar]
- 49.Kohgo Y, Ohtake T, Ikuta K, Suzuki Y, Torimoto Y, Kato J. Dysregulation of systemic iron metabolism in alcoholic liver diseases. J Gastroenterol Hepatol. 2008;23(Suppl 1):S78–81. doi: 10.1111/j.1440-1746.2007.05290.x. [DOI] [PubMed] [Google Scholar]
- 50.Chacko BK, Srivastava A, Johnson MS, et al. Mitochondria-targeted ubiquinone (MitoQ) decreases ethanol-dependent micro and macro hepatosteatosis. Hepatology. 2011;54:153–63. doi: 10.1002/hep.24377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zima T, Kalousova M. Oxidative stress and signal transduction pathways in alcoholic liver disease. Alcohol Clin Exp Res. 2005;29:110S–5S. doi: 10.1097/01.alc.0000189288.30358.4b. [DOI] [PubMed] [Google Scholar]
- 52.Jaruga B, Hong F, Kim WH, Sun R, Fan S, Gao B. Chronic alcohol consumption accelerates liver injury in T cell-mediated hepatitis: alcohol disregulation of NF-kappaB and STAT3 signaling pathways. Am J Physiol Gastrointest Liver Physiol. 2004;287:G471–9. doi: 10.1152/ajpgi.00018.2004. [DOI] [PubMed] [Google Scholar]
- 53.Mandrekar P, Ambade A, Lim A, Szabo G, Catalano D. An essential role for MCP-1 in alcoholic liver injury: Regulation of pro-inflammatory cytokines and hepatic steatosis. Hepatology. 2011 doi: 10.1002/hep.24599. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zamara E, Novo E, Marra F, et al. 4-Hydroxynonenal as a selective pro-fibrogenic stimulus for activated human hepatic stellate cells. J Hepatol. 2004;40:60–8. doi: 10.1016/s0168-8278(03)00480-x. [DOI] [PubMed] [Google Scholar]
- 55.Svineng G, Ravuri C, Rikardsen O, Huseby NE, Winberg JO. The role of reactive oxygen species in integrin and matrix metalloproteinase expression and function. Connect Tissue Res. 2008;49:197–202. doi: 10.1080/03008200802143166. [DOI] [PubMed] [Google Scholar]
- 56.Cohen JI, Nagy LE. Pathogenesis of alcoholic liver disease: interactions between parenchymal and non-parenchymal cells. J Dig Dis. 2011;12:3–9. doi: 10.1111/j.1751-2980.2010.00468.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rushmore TH, Morton MR, Pickett CB. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J Biol Chem. 1991;266:11632–9. [PubMed] [Google Scholar]
- 58.Itoh K, Chiba T, Takahashi S, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–22. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 59.Jia Z, Zhu H, Trush MA, Misra HP, Li Y. Generation of superoxide from reaction of 3H-1,2-dithiole-3-thione with thiols: implications for dithiolethione chemoprotection. Mol Cell Biochem. 2008;307:185–91. doi: 10.1007/s11010-007-9598-z. [DOI] [PubMed] [Google Scholar]
- 60.Maher J, Yamamoto M. The rise of antioxidant signaling--the evolution and hormetic actions of Nrf2. Toxicol Appl Pharmacol. 2010;244:4–15. doi: 10.1016/j.taap.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 61.Zhu H, Jia Z, Misra BR, et al. Nuclear factor E2-related factor 2-dependent myocardiac cytoprotection against oxidative and electrophilic stress. Cardiovasc Toxicol. 2008;8:71–85. doi: 10.1007/s12012-008-9016-0. [DOI] [PubMed] [Google Scholar]
- 62.Yeligar SM, Machida K, Kalra VK. Ethanol-induced HO-1 and NQO1 are differentially regulated by HIF-1alpha and Nrf2 to attenuate inflammatory cytokine expression. J Biol Chem. 2010;285:35359–73. doi: 10.1074/jbc.M110.138636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gong P, Cederbaum AI. Nrf2 is increased by CYP2E1 in rodent liver and HepG2 cells and protects against oxidative stress caused by CYP2E1. Hepatology. 2006;43:144–53. doi: 10.1002/hep.21004. [DOI] [PubMed] [Google Scholar]
- 64.Aleynik SI, Leo MA, Aleynik MK, Lieber CS. Increased circulating products of lipid peroxidation in patients with alcoholic liver disease. Alcohol Clin Exp Res. 1998;22:192–6. [PubMed] [Google Scholar]
- 65.Meagher EA, Barry OP, Burke A, et al. Alcohol-induced generation of lipid peroxidation products in humans. J Clin Invest. 1999;104:805–13. doi: 10.1172/JCI5584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Alatalo P, Koivisto H, Kultti J, Bloigu R, Niemela O. Evaluation of reference intervals for biomarkers sensitive to alcohol consumption, excess body weight and oxidative stress. Scand J Clin Lab Invest. 2010;70:104–11. doi: 10.3109/00365510903548818. [DOI] [PubMed] [Google Scholar]
- 67.Clot P, Bellomo G, Tabone M, Arico S, Albano E. Detection of antibodies against proteins modified by hydroxyethyl free radicals in patients with alcoholic cirrhosis. Gastroenterology. 1995;108:201–7. doi: 10.1016/0016-5085(95)90025-x. [DOI] [PubMed] [Google Scholar]
- 68.Ladero JM, Martinez C, Garcia-Martin E, et al. Polymorphisms of the glutathione S-transferases mu-1 (GSTM1) and theta-1 (GSTT1) and the risk of advanced alcoholic liver disease. Scand J Gastroenterol. 2005;40:348–53. doi: 10.1080/00365520510012109. [DOI] [PubMed] [Google Scholar]
- 69.Khan AJ, Choudhuri G, Husain Q, Parmar D. Polymorphism in glutathione-S-transferases: a risk factor in alcoholic liver cirrhosis. Drug Alcohol Depend. 2009;101:183–90. doi: 10.1016/j.drugalcdep.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 70.Nahon P, Sutton A, Pessayre D, et al. Genetic dimorphism in superoxide dismutase and susceptibility to alcoholic cirrhosis, hepatocellular carcinoma, and death. Clin Gastroenterol Hepatol. 2005;3:292–8. doi: 10.1016/s1542-3565(04)00718-9. [DOI] [PubMed] [Google Scholar]
- 71.Khan AJ, Ruwali M, Choudhuri G, Mathur N, Husain Q, Parmar D. Polymorphism in cytochrome P450 2E1 and interaction with other genetic risk factors and susceptibility to alcoholic liver cirrhosis. Mutat Res. 2009;664:55–63. doi: 10.1016/j.mrfmmm.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 72.Nahon P, Sutton A, Rufat P, et al. Myeloperoxidase and superoxide dismutase 2 polymorphisms comodulate the risk of hepatocellular carcinoma and death in alcoholic cirrhosis. Hepatology. 2009;50:1484–93. doi: 10.1002/hep.23187. [DOI] [PubMed] [Google Scholar]
- 73.Mezey E, Potter JJ, Rennie-Tankersley L, Caballeria J, Pares A. A randomized placebo controlled trial of vitamin E for alcoholic hepatitis. J Hepatol. 2004;40:40–6. doi: 10.1016/s0168-8278(03)00476-8. [DOI] [PubMed] [Google Scholar]
- 74.Stewart S, Prince M, Bassendine M, et al. A randomized trial of antioxidant therapy alone or with corticosteroids in acute alcoholic hepatitis. J Hepatol. 2007;47:277–83. doi: 10.1016/j.jhep.2007.03.027. [DOI] [PubMed] [Google Scholar]
- 75.Moreno C, Langlet P, Hittelet A, et al. Enteral nutrition with or without N-acetylcysteine in the treatment of severe acute alcoholic hepatitis: a randomized multicenter controlled trial. J Hepatol. 2010;53:1117–22. doi: 10.1016/j.jhep.2010.05.030. [DOI] [PubMed] [Google Scholar]
- 76.Mato JM, Camara J, Fernandez de Paz J, et al. S-adenosylmethionine in alcoholic liver cirrhosis: a randomized, placebo-controlled, double-blind, multicenter clinical trial. J Hepatol. 1999;30:1081–9. doi: 10.1016/s0168-8278(99)80263-3. [DOI] [PubMed] [Google Scholar]
- 77.Steinberg D. The LDL modification hypothesis of atherogenesis: an update. J Lipid Res. 2009;50(Suppl):S376–81. doi: 10.1194/jlr.R800087-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sola S, Mir MQ, Cheema FA, et al. Irbesartan and lipoic acid improve endothelial function and reduce markers of inflammation in the metabolic syndrome: results of the Irbesartan and Lipoic Acid in Endothelial Dysfunction (ISLAND) study. Circulation. 2005;111:343–8. doi: 10.1161/01.CIR.0000153272.48711.B9. [DOI] [PubMed] [Google Scholar]
- 79.Tardif JC, Gregoire J, Schwartz L, et al. Effects of AGI-1067 and probucol after percutaneous coronary interventions. Circulation. 2003;107:552–8. doi: 10.1161/01.cir.0000047525.58618.3c. [DOI] [PubMed] [Google Scholar]