

Abstract
Aldo-keto reductase 1C3 (AKR1C3; type 5 17β-hydroxysteroid dehydrogenase) is overexpressed in castrate resistant prostate cancer (CRPC) and is implicated in the intratumoral biosynthesis of testosterone and 5α-dihydrotestosterone. Selective AKR1C3 inhibitors are required since compounds should not inhibit the highly related AKR1C1 and AKR1C2 isoforms which are involved in the inactivation of 5α-dihydrotestosterone. NSAIDs, N-phenylanthranilates in particular are potent but non-selective AKR1C3 inhibitors. Using flufenamic acid, 2-{[3-(trifluoromethyl)phenyl]amino}benzoic acid as lead compound, five classes of structural analogs were synthesized and evaluated for AKR1C3 inhibitory potency and selectivity. Structure activity relationship (SAR) studies revealed that a meta-carboxylic acid group relative to the amine conferred pronounced AKR1C3 selectivity without loss of potency, while electron withdrawing groups on the phenylamino B-ring were optimal for AKR1C3 inhibition. Lead compounds did not inhibit COX-1 or COX-2 but blocked the AKR1C3 mediated production of testosterone in LNCaP-AKR1C3 cells. These compounds offer promising leads towards new therapeutics for CRPC.
INTRODUCTION
Prostate cancer (PC) is the second most common malignancy in men and is responsible for about one in ten of all cancer related deaths.1, 2 Treatment of locally invasive PC or metastatic PC is aimed at ablation of the testicular production of androgens. This is achieved by either orchiectomy or by chemical castration. This suppression of androgen production produces a significant symptomatic improvement but is often followed by a period of therapeutic failure marked by the recurrence of a fatal, metastatic phenotype of prostate cancer, known as castration resistant prostate cancer (CRPC). CRPC is accompanied by an increase in prostatic serum antigen (an androgen induced protein), indicating that the androgen axis has been reactivated in the tumor.3-5 Two mechanisms are thought to contribute to reactivation of the androgen axis in CRPC. One mechanism involves adaptive intratumoral androgen biosynthesis in which enzymes involved in androgen biosynthesis are up-regulated within the tumor and increase the concentration of potent ligands than can activate the androgen receptor (AR). The other mechanism involves increased AR signaling which can occur through AR gene amplification. 3,4 Both mechanisms are targeted therapeutically.
The recent success of abiraterone acetate, a CYP17 hydroxylase /17,20 lyase inhibitor in Phase II and III clinical trials for CRPC and its approval by the FDA supports the involvement of androgen biosynthesis in the pathogenesis of CRPC.6-8 However, because this drug acts high up in the steroidogenic pathway by blocking the conversion of pregnenolone to dehydroepiandrosterone (DHEA) (refer to Figure 1.), it has the unintended consequence of preventing cortisol production in the adrenal and causing the accumulation of the potent mineralocorticoid, desoxycorticosterone. The formation of desoxycorticosterone is exacerbated by the over stimulation of the adrenal by adrenocorticotropic hormone (ACTH) which occurs in the absence of cortisol to block ACTH production. To prevent these side effects, the drug must be co-administered with prednisone to suppress the hypothalamo-pituitary-adrenal axis. There is therefore a need for better therapeutic agents.
Figure 1.
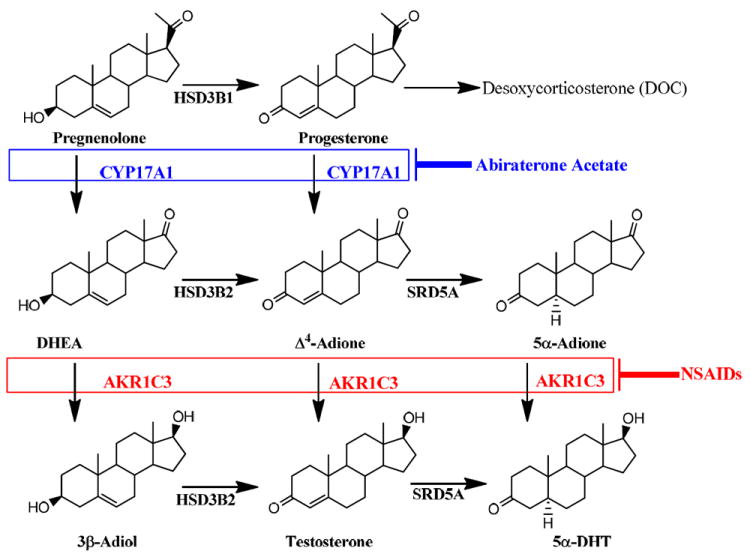
Roles of CYP17A1 and AKR1C3 in androgen biosynthesis and their inhibition by abiraterone acetate and NSAIDs. (HSD3B2 is Type 2, 3β hydroxysteroid dehydrogenase and SRD5A is 5α-reductase)
The AR can also be amplified in CRPC so that it can bind and be activated by trace ligand. The new AR antagonist MDV3100 was designed to be effective in prostate cancer cell lines in which the AR is amplified.9, 10 In the clinic, cases of abiraterone and MDV3100 resistance have emerged so that other methods of blocking the androgen axis in CRPC are required.
AKR1C3 also known as type 5, 17β-hydroxysteroid dehydrogenase (17β-HSD), is among the most highly up-regulated enzymes in CRPC and represents an alternative drug target.11, 12 AKR1C3 acts further downstream of CYP17-hydroxylase/17, 20 lyase in the androgen biosynthetic pathway in the prostate. AKR1C3 catalyzes the NADPH dependent reduction of Δ4-androstene-3, 17-dione (Δ4-AD) to yield testosterone and the NADPH dependent reduction of 5α-androstane-3,17-dione (5α-Adione) to yield 5α-dihydrotestosterone (5α-DHT).13,14 Thus all paths downstream from DHEA to the potent androgens, testosterone and 5α-DHT proceed via AKR1C3. Interestingly, following castration there is an adaptive reprogramming of androgen biosynthesis in prostate cancer metastases, which results in a 30-fold increase in the testosterone: 5α-DHT ratio.12 This occurs in concert with a substantial increase in transcript levels for AKR1C3 which is accompanied by a decrease in transcript levels of 5α-reductase type 2 and only a modest increase in transcript levels of 5α-reductase type 1.11, 12 These data suggest that the tumor is now more dependent on testosterone than 5α-DHT and that the key enzyme involved is AKR1C3. Other studies in prostate cancer cell lines and xenografts suggest that the pathway to 5α-DHT involves preferential reduction of 5α-Adione to 5α-DHT and bypasses testosterone altogether.15 However, regardless of the pathway involved, the intraprostatic production of the potent androgens requires AKR1C3. Taken together, AKR1C3 inhibition is predicted to prevent cancer cell proliferation and offers an attractive target for the treatment of CRPC.
AKR1C3 inhibitors should not exhibit inhibitory activity on the closely related isoforms, AKR1C1 (20α-HSD) and AKR1C2 (Type 3 3α-HSD). The two enzymes share > 86% sequence identity to AKR1C3, and are also involved in intraprostatic androgen metabolism. 14, 16-18 AKR1C1 and AKR1C2 catalyze the 3-ketoreduction of 5α-DHT to form 5α-androstane-3β,17β-diol (3β-Adiol) and 5α-androstane-3α,17β-diol (3α-Adiol), respectively.17,18 3β-Adiol is a pro-apoptotic ligand for estrogen receptor β, while 3α-Adiol is an inactive androgen.18, 19 Inhibition of AKR1C1 and AKR1C2 mediated 5α-DHT metabolism in the prostate will be counterproductive as it can increase the androgenic signal and deprive ERβ of its ligand. Thus the challenge is to develop AKR1C3 selective inhibitors.
AKR1C9 (rat 3α-hydroxysteroid dehydrogenase) is inhibited by non-steroidal anti-inflammatory drugs (NSAIDs) at concentrations that inhibit their accepted targets, cyclooxygenase enzymes (COX-1 and COX-2), and these observations were extended to AKR1C3.20-24 AKR1C3 inhibition might thus contribute to the non-COX dependent anti-neoplastic effects seen with several of these compounds. Indeed, Desmond et al, showed that the inhibition of AKR1C3 by indomethacin promoted the differentiation of HL-60 cell lines.25 Among the NSAIDs, N-phenylanthranilates (N-PA) are among the most potent AKR1C3 inhibitors.23, 24 While chronic COX inhibition is undesirable, these compounds represent good leads to develop potent and selective inhibitors of AKR1C3 since much is known about their pharmacokinetics and SAR required for COX inhibition.26-28 We have previously shown that substitution on the N-benzoic acid ring (A-ring) of N-PA led to potent AKR1C3 inhibitors that are devoid of inhibitory activity on both COX isoforms.23 However, these compounds retain significant inhibitory effects on other AKR1C enzymes. In the current report, we describe the detailed structure activity relationship (SAR) of a library of N-PA based compounds (Figure 2) leading to the discovery of potent inhibitors of AKR1C3 with little or no inhibitory effects on other AKR1C isoforms, COX enzymes, and other human AKR enzymes. We also show that these compounds block testosterone production in LNCaP cells genetically engineered to overexpress AKR1C3.
Figure 2.
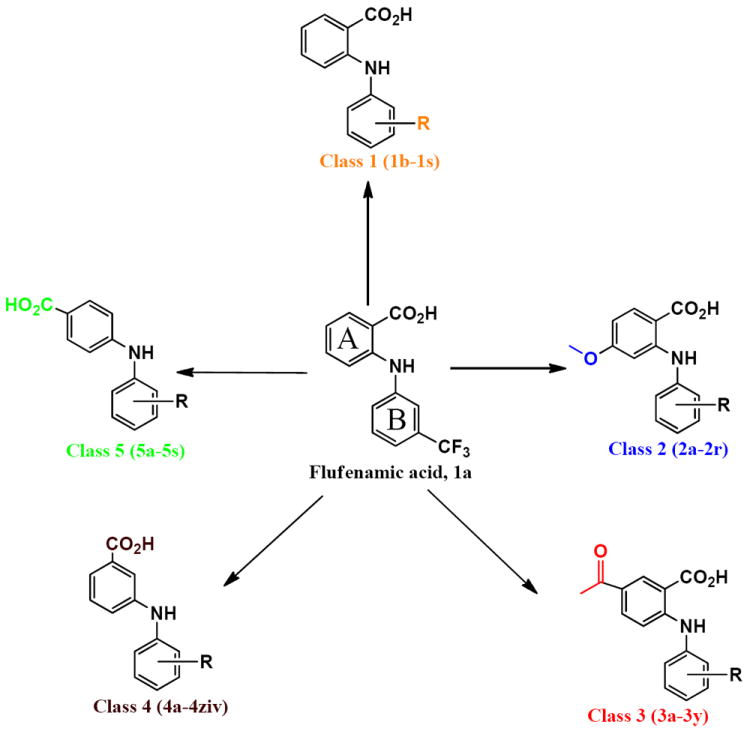
Overview of flufenamic acids analogs. Aromatic rings are designated A and B for ease of discussion in text.
Chemistry
All compounds were synthesized via a two-step reaction involving a Buchwald-Hartwig C-N coupling followed by saponification of the formed methyl ester (Scheme 1).29-31 Step A: Briefly, the aniline (1.2 equiv), Cs2CO3 (1.4 equiv), BINAP (0.08 equiv), and Pd(OAc)2 (0.05 equiv) were added to a solution of the aryl bromide (or triflate) (1 equiv) in toluene (0.1 M) at room temperature. The reaction mixture was allowed to stir at 120 °C for 4-48 h. Once the reaction appeared to be complete by consumption of the bromide (or triflate) by TLC analysis, the mixture was allowed to cool to room temperature, diluted with EtOAc, washed with 2 M aq HCl (2×), brine, and dried over sodium sulfate. The solution was concentrated, loaded on silica gel, and purified by silica gel chromatography. Step B: KOH (2 equiv per ester) in water (0.2 M) was added to the purified methyl ester (1 equiv) in EtOH (0.2 M) at room temperature. The reaction mixture was stirred at 100 °C for 1-6 h and monitored by TLC analysis. EtOH was evaporated from the reaction mixture upon completion the resultant solution was cooled to 0 °C and acidified to pH 2 with 2 M aq HCl. In most cases, the product crystallized out of the aqueous medium upon acidification allowing the product to be collected by a simple vacuum filtration. Compounds were characterized by NMR analysis and high resolution mass spectrometry.
Scheme 1. Synthesis of N-Phenylanthranilic acid analogs.
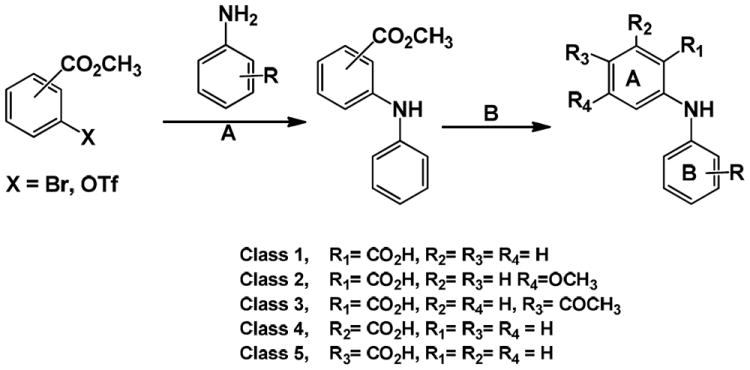
(A.) Pd(OAc)2, 2,2’-bis(diphenylphosphino)-1,1’-binaphthyl (BINAP), Cs2CO3, toluene, 120 °C (B.) KOH, EtOH, H2O, 100 °C
RESULTS
Five different classes of compounds (Figure 2) were synthesized and tested for AKR1C3 inhibitory potency and selectivity based on the following rationale. Class 1 compounds were analogs of flufenamic acid (FLU) 1a, with varied B-ring substituents, (1b-1s), so that the effect of B-ring substitution could be explored. Class 2 compounds contained a 4-OCH3 substituent on the N-benzoic acid ring (A-ring) to give compounds (2a-2r), and Class 3 compounds contained a 5-COCH3 substituent on the N-benzoic acid ring to give compounds (3a-3y), respectively. Class 2 and 3 compounds were synthesized since substitution on the N-benzoic acid ring is known to eliminate COX inhibition,23, 26-28 Class 4 and 5 compounds changed the position of the carboxyl group on the N-benzoic acid from either ortho- to meta- (Class 4), or ortho to para- (Class 5) to give compounds (4a-4ziv) and (5a-5s), respectively. This change was made since the ortho arrangement of the carboxyl group with respect to the secondary amine is essential for COX inhibition by N-PA26-28. In addition, the carboxyl group is essential for the anchoring of N-PAs to the oxyanion hole of AKR1C3. In all five compound classes, multiple substituents were introduced into the N-Phenyl (B-ring).
Screening Compounds against AKR1C Isoforms
The primary screen for the compounds was based on their ability to inhibit the NADP+dependent oxidation of S-tetralol catalyzed by AKR1C3 and AKR1C2. The selectivity for AKR1C3 was measured by comparing the ratio of inhibitory potency (IC50 value) for AKR1C2: AKR1C3, where a high value demonstrates high selectivity. We adopted this strategy based on the high (98%) sequence identity between AKR1C1 and AKR1C2. The two enzymes differ in only a single amino acid at their respective active sites,15, 32 making it likely that an inhibitor of AKR1C2 will likewise display inhibitory effects against AKR1C1. Compounds with high AKR1C3 potency and selectivity from this screen were selected for further evaluation.
Class 1: N-PA analogs with B-ring substitution
FLU, 1a, potently inhibits AKR1C3 with IC50 value of 51 nM but was only slightly selective for AKR1C3 yielding a ratio of IC50 values of AKR1C2: AKR1C3 equal to 7, Table 1. Removal of the trifluoromethyl group of FLU, 1a to give the unsubstituted analog compound 1b, led to a 30-fold loss of AKR1C3 potency with no real change in the AKR1C2 potency, and a reversal of selectivity. AKR1C3 potency was restored with the re-introduction of various substituents (1c-1s) on the B-ring. Notably, introduction of t-butyl group at the para position on the B-ring gave compound 1o, the most potent and selective AKR1C3 inhibitor among the monosubstituted analogs. Compound 1o displayed slightly greater AKR1C3 inhibitory potency than FLU with an IC50 value of 38 nM, which resulted in a 28-fold selectivity for AKR1C3 over AKR1C2. Compared to the unsubstituted analog 1b, this represents a 40-fold increase in inhibitory potency for AKR1C3 and a 90-fold gain in AKR1C3 selectivity. Substitution with a carboxyl group at the meta and para position on the B-ring to give the di-carboxylic acids 1i and 1p, respectively led to modest changes in AKR1C3 potency and a 8-10 fold loss in potency for AKR1C2.
Table 1.
Inhibitory properties of class 1 compounds on AKR1C3 and AKR1C2
 | ||||
|---|---|---|---|---|
| Cmpd | R | 1C3 IC50 (μM) | 1C2 IC50 (μM) | Ratio IC50 1C2:1C3 |
| FLU 1a | m-CF3 | 0.05 | 0.37 | 7 |
| 1b | -H | 1.52 | 0.44 | 0.3 |
| 1c | o-OMe | 2.22 | 1.28 | 1 |
| 1d | m-NO2 | 0.04 | 0.51 | 14 |
| 1e | m-Ac | 0.14 | 1.55 | 11 |
| 1f | m-Cl | 0.15 | 0.15 | 1 |
| 1g | m-OMe | 0.57 | 1.11 | 2 |
| 1h | m-Me | 0.24 | 0.38 | 2 |
| 1i | m-CO2H | 0.98 | 4.16 | 4 |
| 1j | p-NO2 | 0.16 | 0.28 | 1.5 |
| 1k | p-Ac | 0.13 | 0.40 | 3 |
| 1l | p-Cl | 0.07 | 0.41 | 6 |
| 1m | p-OMe | 0.09 | 0.70 | 8 |
| 1n | p-Me | 0.36 | 1.46 | 4 |
| 1o | p-t-Bu | 0.04 | 1.07 | 28 |
| 1p | p-CO2H | 2.74 | 3.26 | 1 |
| 1q | o-NO2, p-OMe | 0.03 | 0.92 | 29 |
| 1r | m,m-diOMe | 0.89 | 3.40 | 4 |
| 1s | m,m-diMe | 1.09 | 2.36 | 2 |
Introduction of an o-NO2 into 1m to give 1q led to a 3 fold increase in AKR1C3 potency but no change in AKR1C2 potency which resulted in 1q being 29-fold selective for AKR1C3. 1o and 1q were the most potent and selective AKR1C3 inhibitors in this class. Overall, B-ring substitution led to lower or similar AKR1C2 potency, but an increase in AKR1C3 potency.
Class 2 and 3: B-ring substituted 4-methoxy and 5-acetyl- 2-phenylaminobenzoates
The inhibitory profiles of class 2 and 3 compounds are shown in Tables 2 and 3, respectively. Class 2 compounds have a 4-OCH3 on the A-ring while class 3 compounds have a 5-COCH3 group on the A-ring with various B-ring substituents. The presence of a -OCH3 group para to the carboxylic acid of FLU (AKR1C3 IC50 = 51 nM) to give 2a did not alter AKR1C3 (IC50 = 60 nM) and AKR1C2 potency (IC50 = 220 nM), (Table 2). However, the introduction of an - COCH3 group meta to the carboxylic acid of FLU to give 3a led to a 14 fold loss in AKR1C3 potency and a 7 fold loss in AKR1C2 potency (Table 3).
Table 2.
Inhibitory properties of class 2 (4-Methoxy-2-(phenylamino)benzoates) on AKR1C3 and AKR1C2
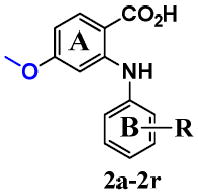 | ||||
|---|---|---|---|---|
| Cmpd | R | 1C3 IC50 (μM) | 1C2 IC50 (μM) | Ratio IC50 1C2:1C3 |
| 2a | m-CF3 | 0.06 | 0.22 | 4 |
| 2b | -H | 1.66 | 1.49 | 0.9 |
| 2c | o-OMe | 1.73 | 2.40 | 1 |
| 2d | m-NO2 | 0.08 | 0.19 | 3 |
| 2e | m-Cl | 0.16 | 0.28 | 2 |
| 2f | m-OMe | 0.57 | 1.47 | 3 |
| 2g | m-Me | 0.64 | 0.87 | 1 |
| 2h | m-CO2H | 1.45 | 8.38 | 6 |
| 2i | p-NO2 | 0.50 | 0.19 | 0.4 |
| 2j | p-Ac | 0.50 | 0.27 | 0.5 |
| 2k | p-Cl | 0.22 | 0.29 | 1 |
| 2l | p-Br | 0.15 | 0.54 | 4 |
| 2m | p-OMe | 0.25 | 0.91 | 4 |
| 2n | p-Me | 0.80 | 1.11 | 1 |
| 2o | p-t-Bu | 0.08 | 0.70 | 8 |
| 2p | p-CO2H | 0.69 | 0.98 | 1 |
| 2q | m,m-diOMe | 2.22 | 6.78 | 3 |
| 2r | m,m-diMe | 1.73 | 3.24 | 2 |
Table 3.
Inhibitory properties of class 3 compounds (5-acetyl-2-(phenylamino)benzoates) on AKR1C3 and AKR1C2
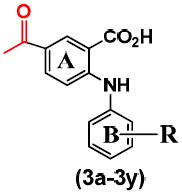 | ||||
|---|---|---|---|---|
| Cmpd | R | 1C3 IC50 (μM) | 1C2 IC50 (μM) | Ratio IC50 1C2:1C3 |
| 3a | m-CF3 | 0.73 | 2.67 | 4 |
| 3b | -H | 2.53 | 3.81 | 2 |
| 3c | o-NO2 | 1.11 | 3.97 | 4 |
| 3d | o-Ac | 4.44 | 12.7 | 3 |
| 3e | o-CF3 | 1.35 | 2.07 | 2 |
| 3f | o-OMe | 3.06 | 8.97 | 3 |
| 3g | m-NO2 | 0.98 | 3.17 | 3 |
| 3h | m-Ac | 0.65 | 1.88 | 3 |
| 3i | m-Cl | 1.16 | 2.55 | 2 |
| 3j | m-OMe | 2.26 | 3.58 | 2 |
| 3k | m-Me | 1.94 | 2.84 | 1 |
| 3l | p-NO2 | 1.17 | 1.33 | 1 |
| 3m | p-Ac | 0.47 | 18.7 | 40 |
| 3n | p-CF3 | 0.14 | 2.73 | 19 |
| 3o | p-Cl | 0.60 | 2.23 | 4 |
| 3p | p-Br | 0.25 | 0.49 | 2 |
| 3q | p-OMe | 0.61 | 0.91 | 1 |
| 3r | p-Me | 1.65 | 8.23 | 5 |
| 3s | p-t-Bu | 0.28 | 7.05 | 25 |
| 3t | p-CO2H | 1.55 | 18.9 | 12 |
| 3u | o-NO2, p-OMe | 0.26 | 3.93 | 15 |
| 3v | m,m-diCF3 | 1.76 | 16.0 | 9 |
| 3w | m,m-diOMe | 0.90 | 26.8 | 30 |
| 3x | m,m-diMe | 1.82 | 10.6 | 6 |
| 3y | m,m-diCO2H | 0.61 | 31.6 | 56 |
Removal of the -CF3 group from 2a and 3a to give 2b and 3b, respectively led to pronounced reduction in AKR1C3 potency and a slight loss of AKR1C2 potency. B-ring substitution in class 2 and 3 compounds improved AKR1C3 inhibition over the corresponding unsubstituted analogs (2b and 3b) following a trend that was noted with FLU and the class 1 compounds. Compound 2a (m-CF3) was the most potent inhibitor while 2o (p-t-Bu) was the most selective AKR1C3 inhibitor in the class 2 compounds. Within the class 3 compounds, compound 3n (p-CF3) was the most potent inhibitor while 3y (m,m-diCO2H) was the most selective for AKR1C3. The AKR1C3 inhibition and selectivity of compounds in the two classes do not appear to display a clear preference for substitution at any particular B–ring position.
With the exception of 3y which displayed 56 fold selectivity for AKR1C3, class 2 and 3 compounds exhibited only slightly better inhibitory activity on AKR1C3 than AKR1C2 and were not significantly selective for AKR1C3.
Class 4: B-ring substituted 3-phenylaminobenzoates
The inhibitory potency of the A-ring m-CO2H analogs is shown in Table 4. Movement of the A-ring carboxyl group of FLU from the ortho to the meta position relative to the amine to give 4a resulted in a 6-fold and 43-fold loss of AKR1C3 and AKR1C2 potency, respectively. This translates to 50-fold selectivity for AKR1C3, a remarkable increase over FLU. The AKR1C3 inhibitory potency of 4a and the substituted B-ring analogs, 4c-4ziv were mostly higher than the unsubstituted analog 4b while AKR1C2 potency was mostly unaltered or lowered.
Table 4.
Inhibitory properties of class 4 (3-(phenylamino)benzoates) on AKR1C3 and AKR1C2
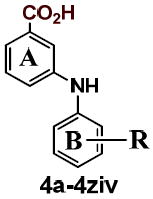 | ||||
|---|---|---|---|---|
| Cmpd | R | 1C3 IC50 (μM) | 1C2 IC50 (μM) | Ratio IC50 1C2:1C3 |
| 4a | m-CF3 | 0.32 | 16 | 50 |
| 4b | -H | 0.94 | 12.9 | 14 |
| 4c | o-NO2 | 0.15 | 4.77 | 32 |
| 4d | o-Ac | 0.96 | 32.6 | 34 |
| 4e | o-CF3 | 0.56 | 15.1 | 27 |
| 4f | o-OMe | 1.65 | 26.6 | 16 |
| 4g | m-NO2 | 0.29 | 19.3 | 66 |
| 4h | m-Ac | 0.56 | 36.6 | 65 |
| 4i | m-Cl | 0.46 | 7.17 | 16 |
| 4j | m-OMe | 1.19 | 15.2 | 13 |
| 4k | m-Me | 1.88 | 22.9 | 12 |
| 4l | m-CO2H | 3.57 | >100 | >28.0 |
| 4m | p-NO2 | 0.03 | 3.38 | 101 |
| 4n | p-Ac | 0.05 | 19.5 | 364 |
| 4o | p-CF3 | 0.06 | 15.4 | 249 |
| 4p | p-Cl | 0.14 | 19.1 | 140 |
| 4q | p-Br | 0.13 | 18.1 | 146 |
| 4r | p-OMe | 0.49 | 11.5 | 23 |
| 4s | p-Me | 0.70 | 56.2 | 80 |
| 4t | p-t-Bu | 0.28 | 31.1 | 111 |
| 4u | p-CO2H | 1.64 | 19.9 | 12 |
| 4v | o,o-diMe | 2.91 | 10.3 | 4 |
| 4w | o-CO2H, p-Ac | 0.04 | 6.41 | 164 |
| 4x | o-NO2, p-CF3 | 0.03 | 4.35 | 132 |
| 4y | o-NO2, p-OMe | 0.04 | 5.56 | 139 |
| 4z | o-CF3, p-NO2 | 0.04 | 1.78 | 51 |
| 4zi | m,m-diCF3 | 0.25 | 19.2 | 78 |
| 4zii | m,m-diOMe | 0.36 | 56.2 | 156 |
| 4ziii | m,m, diMe | 1.08 | 44.6 | 41 |
| 4ziv | m,m-diCO2H | >30 | >100 | ND |
Compared to the class 1 analogs, movement of the -CO2H to the meta position (class 4) generally resulted in similar or slightly weaker AKR1C3 inhibitory activity. However, when EWG are placed on the B-ring, remarkable selectivity and potency was observed for the inhibition of AKR1C3 with some compounds yielding IC50 values in the low nanomolar range and greater than 200 fold selectivity for AKR1C3.
The AKR1C3 inhibitory potency of the class 4 compounds were strongly influenced by B-ring substitution and displayed strong positional effects, with the para substituted analogs having the highest inhibitory potency and selectivity for AKR1C3. By contrast, B-ring substitution did not display any positional preference on AKR1C2 inhibitory potency. The rank order of AKR1C3 inhibitory potency and selectivity seen with all B-ring substituents was ortho ≤ meta < para such that -CF3 group at the ortho, meta, para positions gave compounds 4e, 4a and 4o with IC50 values for AKR1C3 of 560 nM, 319 nM and 62 nM, and selectivity ratios of 27, 50 and 249, respectively.
At each of the B-ring positions tested, introduction of electron withdrawing groups (EWG) other than the carboxyl group gave better AKR1C3 inhibitors than electron donating groups (EDG). In particular, the electron withdrawing -NO2 group gave the most potent AKR1C3 inhibitors at each B-ring position tested e.g. compounds 4c, 4g and 4m with NO2-substitution at ortho, meta, para positions gave IC50 values of 150 nM, 290 nM and 33 nM, respectively. Compound 4n with a p-COCH3 on the B-ring was the most selective compound in this class. It displayed IC50 value for AKR1C3 of 54 nM and IC50 value for AKR1C2 of 19.47 μM translating to 360 fold selectivity for AKR1C3.
Introduction of a second substituent on the B-ring had varied results depending on the position of the substitution. With the exception of 4z (o-CF3, p-NO2), analogs with bis-substituents on the ortho and para positions (4w-4y) gave potent AKR1C3 inhibitors with IC50 values of 30- 40 nM with over 100 fold selectivity for AKR1C3. The addition of a p-CF3 and p-OCH3 to 4c (o-NO2) to give compounds 4x and 4y, respectively resulted in about a 5-fold increase in AKR1C3 potency with little or no change in the AKR1C2 potency. Likewise, relative to their corresponding un-nitrated analogs (4o and 4r, respectively) 4x and 4y displayed greater AKR1C3 inhibitory potency. Interestingly, 4x and 4y were also better AKR1C2 inhibitors than their un-nitrated analogs. Reversing the position of the -CF3 and -NO2 groups of 4x to give 4z did not have any effect on AKR1C3 inhibition, however, there was an increase in the AKR1C2 inhibitory potency. Our studies on B-ring substitution in this class shows that the addition of an ortho substituent to a para- substituted analog consistently increases the inhibitory potency of para substituted analog for AKR1C2 while increasing or having no effect on AKR1C3 inhibition.
Class 5: B-ring substituted 4-phenylaminobenzoates
The inhibitory properties of the class 5 analogs on AKR1C3 and AKR1C2 are shown in Table 5. The movement of the -CO2H group of FLU to the para position on the A-ring to give 5a led to a 10 fold loss of inhibitory activity on AKR1C3 and 30 fold loss of inhibitory activity on AKR1C2, respectively (Table 5). This translates to 20 fold selectivity for AKR1C3. Introduction of B-ring substituents (5c-5s) produced only modest changes in AKR1C3 potency.
Table 5.
Inhibitory properties of class 5 compounds 4-(phenylamino) benzoates on AKR1C3 and AKR1C2
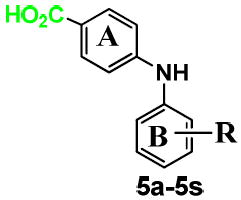 | ||||
|---|---|---|---|---|
| Cmpd | R | 1C3 IC50 (μM) | 1C2 IC50 (μM) | Ratio IC50 1C2:1C3 |
| 5a | m-CF3 | 0.50 | 11.1 | 20 |
| 5b | -H | 2.79 | 3.0 | 1 |
| 5c | o-OMe | 3.39 | 29.3 | 9 |
| 5d | m-NO2 | 4.21 | >100 | >24 |
| 5e | m-Cl | ND | ND | NA |
| 5f | m-OMe | 2.65 | 84.4 | 32 |
| 5g | m-Me | 1.67 | 21.5 | 13 |
| 5h | m-CO2H | 1.64 | 19.9 | 12 |
| 5i | p-NO2 | 1.32 | 11.1 | 8 |
| 5j | p-Ac | 2.50 | 24.5 | 9.8 |
| 5k | p-Cl | 1.05 | 20.0 | 21 |
| 5l | p-Br | 1.18 | >30 | >25 |
| 5m | p-OMe | 2.21 | 22.9 | 10 |
| 5n | p-Me | 1.89 | 48.2 | 26 |
| 5o | p-t-Bu | 0.93 | 10.3 | 11 |
| 5p | p-CO2H | 2.37 | 6.30 | 3 |
| 5q | o-NO2, p-OMe | 2.06 | 59.2 | 29 |
| 5r | m,m di-Me | 0.87 | 49.3 | 57 |
| 5s | m,m di-OMe | 2.53 | 69.9 | 28 |
However, there was a profound loss of AKR1C2 potency with the B-ring substituted analogs relative to the unsubstituted compound 5b. The p-CO2H analogs displayed significantly lower AKR1C3 inhibitory potency and an even more profound reduction in AKR1C2 potency relative to the class 1 analogs. For example, the m-NO2 analog (5d) was about 100 fold less potent as an AKR1C3 inhibitor and 200 fold weaker as an AKR1C2 inhibitor than the corresponding class 1 analog, 1d. No positional preference for B-ring substitution was observed in the class 5 compounds to achieve AKR1C3 inhibition. With an IC50 value of 500 nM, 5a, was the most potent AKR1C3 inhibitor in this class whereas 5r (m,m-diCH3), was the most selective displaying a 57 fold preference for AKR1C3.
Effect on other AKR1C, AKR1B isoforms and COX enzymes
The most promising AKR1C3 inhibitors are shown in Table 6. All the compounds displayed low nanomolar IC50 values for AKR1C3 that were at least two orders of magnitude lower than the corresponding values obtained for AKR1C1, AKR1C2 and AKR1C4. Of the other AKR1C isoforms, the IC50 value for AKR1C4 inhibition was consistently higher than those obtained for both AKR1C1 and AKR1C2. The compounds were also selective for AKR1C3 over AKR1B1 and AKR1B10, since they only inhibited the NADPH dependent reduction of DL-glyceraldehyde catalyzed by AKR1B1 and AKR1B10 at high concentrations with IC50 values generally greater than 10 μM, much higher than the IC50 values obtained for Zopolrestat, a known inhibitor of both AKR1B1 and AKR1B10 (Table 6).33
Table 6.
Inhibitory properties of compounds on other human AKR enzymes
| IC50 Values (μM) | ||||||
|---|---|---|---|---|---|---|
| a Compds | b AKR1C3 | AKR1C1 | AKR1C2 | AKR1C4 | AKR1B1 | AKR1B10 |
| 4m | 0.03 | 6.74 (204) | 3.38 (101) | 32.7 (988) | > 50 c | 46.0 |
| 4n | 0.05 | 15.6 (292) | 19.5 (364) | 25.7 (477) | 38.60 | 37.1 |
| 4o | 0.06 | 22.7 (368) | 15.4 (249) | 62.7 (1015) | > 50 c | > 50 c |
| 4p | 0.14 | 30.2 (222) | 19.1 (140) | 48.7 (357) | ND | ND |
| 4q | 0.13 | 31.3 (250) | 18.1 (145) | 91.4 (728) | > 50 c | > 50 c |
| 4t | 0.28 | 35.6 (126) | 31.1 (110) | 39.1 (139) | > 50 c | 15 |
| 4w | 0.04 | 30.5 (787) | 6.41 (165) | 28.7 (739) | 27.7 | 16.2 |
| 4x | 0.03 | 4.23 (132) | 4.35 (136) | 5.50 (172) | 50 | ND |
| 4y | 0.04 | 6.32 (173) | 5.56 (139) | 20.4 (560) | 42.7 | 39.3 |
| Zopolrestat | NA | NA | NA | NA | 0.027 | 2.36 |
Compounds have no significant effect on NADPH fluorescence at the concentrations tested. Values in brackets represent fold selectivity for AKR1C3. IC50 values for AKR1C3 and AKR1C2 are taken Table 4.
Values represent average of two or more independent experiments,
less than 40% inhibition at 50 μM
ND- Not determined, NA- Not applicable
Since N-PA analogs are used clinically for their ability to inhibit COX enzymes, lead compounds were tested against COX-1 and COX-2. FLU displayed IC50 values of 2.23 μM for COX 1 and 16 nM for COX-2, respectively, (Table 7) and these values are consistent with the finding that N-phenylanthranilates are more selective inhibitors for COX-2 over COX-1.34 Lead compounds showed little or no inhibitory effects on COX-1 or COX-2 catalyzed conversion of arachidonic acid to PGH2 at 100 μM with the exception of 4m which inhibited COX-1 and COX-2 with IC50 values of 30.7 μM and 0.74 μM, respectively.
Table 7.
Inhibitory properties of compounds on COX enzymes
| Cmpds | COX-1 IC50 (μM) | COX-2 IC50 (μM) |
|---|---|---|
| FLU | 2.23 | 0.016 |
| 4m | 30.76 | 0.74 |
| 4n | >100 a | >100 a |
| 4o | >100 a | >100 a |
| 4p | >100 b | ND |
| 4q | >100 b | >100 a |
| 4t | >100 b | >100 b |
| 4w | >100 a | >100 b |
| 4x | >100 a | >100 b |
| 4y | >100 a | >100 a |
less than 10 % inhibition at 100 μM
between 10 and 40% inhibition at 100 μM
Inhibition of Testosterone formation in LNCaP-AKR1C3 cells
Metabolism studies of 0.1 μM [14C]-Δ4-AD were conducted in LNCaP-AKR1C3 cells over 48 h in the presence and absence of 4o. In the absence of the inhibitor, Δ4-AD was rapidly consumed and was metabolized to water soluble conjugates. Treatment of the aqueous phase with β-glucuronidase generated a significant peak of testosterone which was extracted and identified upon radiochromatography by co-migration with the synthetic standard. Relative to the control, compound 4o (10 μM) produced a near complete inhibition of testosterone production in treated cells Figure 3. Almost identical results were observed with the lead compounds 4m, 4n and 4w.
Figure 3.
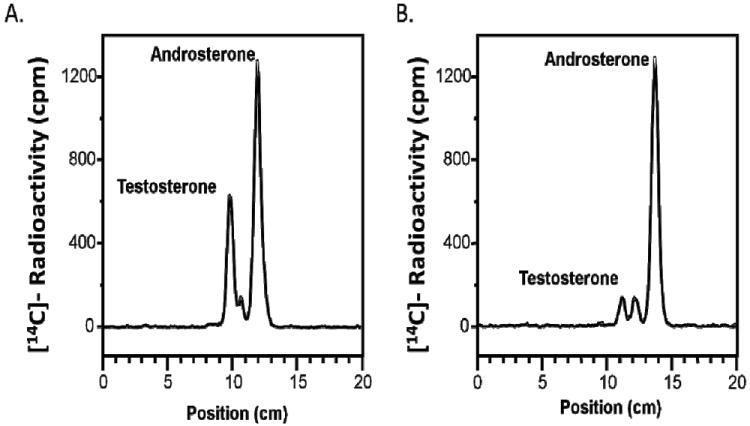
TLC radiochromatogram showing inhibition of testosterone formation in LNCaP-AKR1C3 cells by compound 4o. Metabolism of 0.1 μM [14C]-Δ4-AD in the absence (A) and presence (B) of 4o. The TLC radiochromatogram is representative of the result observed with the lead compounds 4m, 4n, and 4w.
DISCUSSION
We describe the synthesis and screening of five classes of phenyl-aminobenzoates based on flufenamic acid. Compounds in class 4 which contain a meta-carboxylic acid on the N-benzoic acid A-ring and an EWG on the para-position of the phenylamino B-ring were among the most potent and selective inhibitors of AKR1C3 reported to date. Nine lead compounds with nanomolar potency for AKR1C3 and greater than 100 fold selectivity for AKR1C3 over other AKR1C isoforms were identified. These compounds did not inhibit other human AKR isoforms or the anti-inflammatory drug targets COX-1 or COX-2. These compounds also blocked the formation of testosterone in LNCaP-AKR1C3 cells.
Structural Considerations
Properties of phenylaminobenzoate inhibitors of AKR1C3 can be explained in part by the X-ray structure of the AKR1C3·NADP+·flufenamic acid ternary complex (PDB# 1S2C).35 FLU interacts with the AKR1C3 via hydrogen bonding and hydrophobic interactions. Notably, the carboxylic acid on the A-ring of FLU is hydrogen bonded at the oxyanion hole which is comprised of the highly conserved catalytic tetrad of Asp50, Tyr 55, Lys84 and His117 and the cofactor found in AKRs. In addition, the secondary amine group is involved in intramolecular hydrogen bonding with the A-ring carboxylic acid and inter-molecular hydrogen bonding with the carboxamide group of the cofactor (Figure 4). The B-ring projects into a distinct subpocket (SP1) so that the m-CF3 group forms a hydrogen bond with the phenolic group of Tyr 216. 35, 36 The SP1 subpocket is larger and less discriminating in AKR1C3 when compared to the other AKR1C enzymes. The SP1 pocket is also lined by amino acids that can form polar interactions (S118, S308 and Y319) in AKR1C3 relative to AKR1C1 and AKR1C2 where the relevant amino acids are F118, L308 and F319.23, 36 Inhibitors with bulky and/ or polar functional groups on the B-ring that are capable of interacting with SP1 residues are expected to display greater selectivity for AKR1C3.
Figure 4.
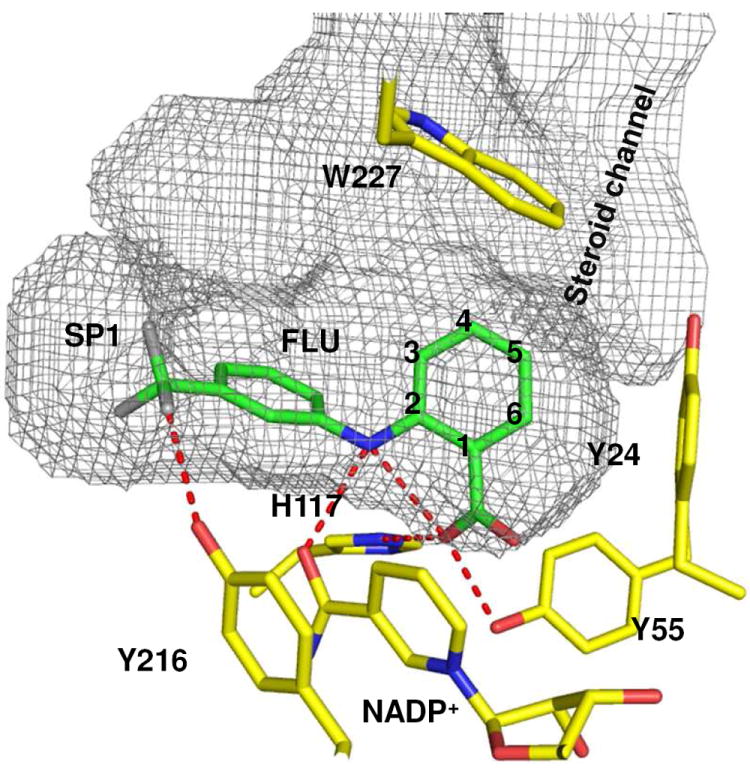
Predicted binding poses for phenylamino-benzoates in the AKR1C3 active site to account for AKR1C3 selectivity. The structures are based on the crystal structure of FLU with AKR1C3. (PDB 1S2C). The A-ring of the compound is anchored to the oxyanion site through hydrogen bonding between the carboxylate group and His117 and Tyr55. The B ring is oriented towards the SP1 pocket by the interactions between the bridge amine group and the carboxamide of the nicotinamide ring and between the trifluoromethyl group and Tyr 216. Possible positions of the bridge amine relative to the carboxylic acid are numbered 2- 6 for the various inhibitor classes. The position of the -CO2H group is invariant between the compounds in classes 1 (ortho-carboxyl) 4 (meta-carboxy) and 5 (para-carboxyl). Thus, the amine in class 4 would be at positions 3 or 5, while the amine group of class 5 compounds will be at position 4. This movement disrupts the interaction between the bridge amine and the carboxamnide of the nicotinamide ring, and results in the B-ring no longer interacting with the SP1 site. The solvent-occupied surface of the active site cavity was generated by Voidoo using probe size of 1.6 Å and was rendered as a mesh surface. Carbon atoms are colored in green for FLU and yellow for AKR1C3. All the other atoms are color coded as follows: nitrogen, blue oxygen, red fluorine, grey. Hydrogen bonds are shown as red dashes. The figure was generated with PyMol (Delano Scientific).
Comparison of class 1, 4 and 5: Movement of the A-ring Carboxylic acid
Class 1, 4 and 5 compounds are defined by the A-ring carboxylic acid being ortho, meta and para, relative to the bridge amine, respectively. Most class 1 and class 4 compounds gave IC50 values in the nanomolar range, however the highest selectivity for AKR1C3 was observed with the class 4 compounds. Loss of AKR1C3 inhibitory potency in all classes coincided with the removal of B-ring substituents, indicating that these substituents are essential for optimal interaction with amino acid residues in SP1 subpocket. This effect was not apparent when AKR1C2 inhibitory potency was examined.
The electronic effect (σ) of the B-ring substituents is a critical determinant of the AKR1C3 inhibitory potency of the class 4 compounds. Compounds with EWG on the B-ring were consistently more potent AKR1C3 inhibitors than those with EDG at all positions on the ring. Using regression analysis, a Hammet relationship was observed between the electronic effect of the B-ring substituents and AKR1C3 potency at the para (R2= 0.81, p = 0.009) and meta (R2= 0.90, p = 0.009) positions (A similar trend is obvious at the ortho position, however σ values for these substituents were unavailable to perform the correlative analysis). By contrast, no correlation was observed between the electronic effect of the B-ring substituents and AKR1C2 potency (data not shown).
B-ring substituents that alter the electron density of the aromatic system are predicted to change the pKa of the carboxylic acid and the basic amine, which could influence hydrogen bond formation within the active site and possibly alter the inhibitory potency of these compounds. Although, there was a significant correlation between the AKR1C3 potency and the pKa of the acid (R2 ≥ 0.77 at all positions) and amine (R2 ≥ 0.79 at all positions) for compounds with substitution at the ortho, meta and para positions of the B-ring, it is unlikely that AKR1C3 inhibitory potency is related to electronic effects on the acidity of the carboxylic acid and/or basicity of the amine for two reasons. First, since these compounds are anchored to the active site via the A-ring CO2H, moving this group from the ortho to the meta position would likely eliminate the intramolecular hydrogen bond formation between the carboxyl group and the bridging amine. It would also move the amine outside the hydrogen bond distance of the carboxamide group of the cofactor (Figure 4). Second, the changes in the pKa of the acid is minimal, (a range of 4.20- 4.33 for the para-substituted analogs, Supporting Information-Table S1), and are insufficient to account for the dramatic changes in the AKR1C3 potency seen with the B-ring substituents.
The dependence of AKR1C3 potency on the electronic effect of B-ring substituents was not seen with the class 1 and 5 compounds, suggesting a difference in the binding pose of these compounds. Movement of the -CO2H group from the ortho to the para position on the A-ring (class 5 analogs) causes the greatest separation between this group and the amine, and results in loss of hydrogen bond interaction of the amine with the carboxylic acid and the cofactor, Figure 4.
Likewise, the para arrangement would prevent the B-ring substituents from optimal interaction with the SP1 subpocket. It is predicted that the B-ring would now occupy the larger adjacent steroid cavity and that the flexible side chain of W227 now resides in the steroid channel. This would explain the loss of AKR1C3 inhibitory potency observed with these analogs and the observation that with most class 5 compounds, B-ring substitution did not produce any remarkable increase in AKR1C3 potency.
There was also a pronounced loss of AKR1C2 inhibition with the class 4 and 5 analogs relative to the class 1 compounds which indicates the existence of different binding orientations (or conformations) for these compounds in AKR1C2 relative to the o-CO2H analogs. The trends in inhibitory potency for AKR1C3 among the 3 classes was 1 ≥ 4 > 5 whereas the selectivity trend was class 4 > 5 > 1. The smaller size and narrower shape of the SP1 in AKR1C2 might preclude the binding of the class 4 and 5 compounds into this subpocket. It is likely that these compounds occupy the larger steroid channel in AKR1C2.
Comparison of class 1, 2 and 3: Substitution on the A-ring
Class 1, 2 and 3 inhibitors are defined by an ortho-carboxylic acid group on the A-ring relative to the bridge amine. Class 1 compounds have no other substituents on the A-ring while class 2 and class 3 compounds have in addition, a 4-OCH3 and 5-COCH3, respectively on the A-ring. With the three classes, increased AKR1C3 potency was observed with B-ring substitution, similar to the trend seen with the class 4 compounds. However, there were no B-ring positional effects on AKR1C3 potency and there was no correlation between the electronic effect of B-ring substituents and AKR1C3 potency. The AKR1C3 potency of these compounds suggests that several factors may be involved.
First, the replacement of a m-CF3 and p-CF3 with a CH3 led to a decrease in AKR1C3 potency in the three classes (compare 1a vs 1h, 2a vs 2g, 3a vs 3k, and 3n vs 3r), and supports the formation of interactions with polar amino acid residues in the subpocket. Second, the low IC50 values displayed by the p-t-butyl analogs 1o, 2o and 3s make them among the most potent compounds in these three classes, and suggest that the introduction of steric bulk into SP1 contributes to AKR1C3 potency. Third, the presence of the 4-OCH3 group on the A-ring was better tolerated than the 5-COCH3 as the rank order of AKR1C3 potency for the three classes was 1 ≥ 2 > 3 for most B-ring substituents, an example being the p-OMe analogs, 1m > 2m > 3q which gave IC50 values of 90 nM, 250 nM, and 610 nM, respectively. These trends are consistent with steric interference between the 5-COCH3 group of the class 3 compounds and the residues W227 and Y24, whereas the 4-OCH3 (in the class 2 structures) is farther away from these two amino acids (Supporting Information-Figure S1).
The introduction of the substituents into the N-benzoic acid A-ring did not improve AKR1C3 selectivity since compounds in classes 1-3 displayed slightly better potency on AKR1C3 than AKR1C2.
Promising Leads
Compounds with IC50 values for AKR1C3 less than 300 nM and over 100 fold selectivity for AKR1C3 over AKR1C2 were selected for further screening. The compounds were tested for activity against other AKR1C isoforms and AKR1B enzymes. AKR1C4 is a liver specific enzyme that shares 84% sequence identity with AKR1C3 and is involved in bile acid biosynthesis and metabolism of steroid hormones.14, 37, 38 AKR1B1 (aldose reductase) and AKR1B10 are involved in glucose metabolism and retinal metabolism, respectively.39, 40 We conducted counter screens on AKR1B isoforms as both are inhibited by FLU.33 All the compounds displayed remarkable selectivity for AKR1C3 over other AKR1C enzymes (AKR1C1, AKR1C2 and AKR1C4) as well as the AKR1B isoforms (AKR1B1 and AKR1B10). All the compounds were also devoid of inhibition of COX-1 and COX-2, with the exception of 4m, consistent with the reported SAR of N-PA inhibition of COX.26-28 The lack of COX inhibitory activity is important since it minimizes the risk of gastrointestinal and cardiovascular adverse effects that have been associated with chronic COX inhibition. The efficacy of the FLU isomer 4o in the LNCaP-AKR1C3 prostate cancer model as shown by the robust inhibition of testosterone formation indicates these compounds are effective in a prostate cancer cell setting.
It will be important to demonstrate that the lead compounds can inhibit prostate cancer cell proliferation driven by the AKR1C3 substrate (Δ4-AD) that they have desirable adsorption, metabolic, excretion and toxicity (ADMET) profiles, and that they are effective in xenograft models of CRPC. We predict that their ADMET properties will be favorable since they are built on NSAIDs which are well tolerated.
Diaryl inhibitors for 17β-HSD type 1 which catalyzes the NADPH dependent reduction of estrone (E1) to give the potent estrogen, estradiol (E2) have been reported. 41-43. In contrast to AKR1C3, 17β-HSD type 1 and all other known 17β-HSDs belong to the short chain dehydrogenase / reductase (SDR) superfamily of enzymes. The SDR family have different properties to the AKRs. SDRs are multimeric proteins, have a unique protein fold which includes a Rossman fold for cofactor binding and catalyze 4-pro-S hydride transfer from the nicotinamide ring while AKRs are monomeric proteins, have a triosephosphate isomerase (TIM) barrel motif and catalyze 4-pro-R hydride transfer.44 These differences are expected to confer selectivity on AKR1C3 inhibitors relative to 17β-HSD type 1.
6-(3’-hydroxyphenyl)-2-naphthols were found to be potent and selective inhibitors of 17β-HSD type 1 with limited inhibitory activity on 17β-HSD type 2.41-43 The planarity of the molecules and the presence of two hydroxyl groups, one on the 3-position (meta) of the phenyl ring and the other on the 2-position of the naphthalene ring were optimal for 17β-HSD type 1 inhibition. The distance between the hydroxyl groups (11Ǻ) on the two rings was optimal for interaction with the polar amino acids on either end of the inhibitors. Substitution on the phenyl ring and movement of the hydroxyl groups to the 4-position of the phenyl ring and 1-position of the naphthalene ring resulted in a series of potent inhibitor of 17β-HSD type 2 that were selective over 17β-HSD type 1.45, 46 One compound with a 3-trifluoromethyl group on the phenyl ring displayed weak AKR1C3 inhibition at 1 μM.46 The absence of a linker region between the two aromatic rings in these compounds make them more rigid than the substituted 3-(phenylamino)benzoates reported in our study. This lack of flexibility and the planarity would prevent the 6-(3’hydroxyphenyl)naphthalenes from having an optimal interaction with the SP1 pocket of AKR1C3, an interaction that is important to achieve highly potent inhibitors of AKR1C3. It is also unlikely that the lead compounds discovered in our study will have a significant effect on 17β-HSD type 1 as the distance between the two ends of the reported AKRC3 inhibitors is shorter (about 8Ǻ), precluding the necessary interactions needed for 17β HSD type 1 inhibition. A virtual screen was also used to identify potential inhibitors for AKR1C3. In this screen, it was necessary to consider that different ligand classes occupy different subpockets in the enzyme. One model used the AKR1C3·NADP+·FLU structure and in silico screening identified phenylamino carboxylates as inhibitors.47
Since AKR1C3 plays a pivotal role in prostate tumor androgen biosynthesis, inhibitors of this enzyme have the potential to be superior to abiraterone acetate, a CYP17/20 hydroxylase/lyase inhibitor. Abiraterone acetate is effective for the treatment of CRPC but it has to be co-administered with a glucocorticoid to prevent the buildup of the potent mineralocorticoid, desoxycorticosterone.6-8 Chronic use of glucocorticoids can cause Cushing’s syndrome, immunosuppression and diabetes. Since AKR1C3 acts further downstream in the androgen biosynthetic pathway, co-administration of AKR1C3 inhibitors with a glucocorticoid is not necessary.
Taken together, we have systematically developed potent and selective AKR1C3 inhibitors that are efficacious in a prostate cancer model and are potential therapeutic agents for the treatment of CRPC.
EXPERIMENTAL
Chemistry
Solvents used for extraction and purification were HPLC grade from Fisher. Unless otherwise indicated, all reactions were run under an inert atmosphere of Argon. Anhydrous toluene was obtained via distillation from the appropriate drying agents. Deuterated solvents were obtained from Cambridge Isotope labs. Merck pre-coated silica gel plates (250 μm, 60 F254) were used for analytical TLC. Spots were visualized using 254 nm ultraviolet light, with either anisaldehyde or potassium permanganate stains as visualizing agents. Chromatographic purifications were performed on Sorbent Technologies silica gel (particle size 32-63 microns). 1H and 13C NMR spectra were recorded at 500 MHz, 360 MHz and 125 MHz, 90 MHz, respectively in CDCl3 or (CD3)2SO on a Bruker AM-500, DRX-500, or DRX-360 spectrometer. Chemical shifts are reported relative to deuterated or 13C labeled chloroform (δ 7.27 for 1H, δ 77.23 for 13C) or deuterated or 13C labeled dimethyl sulfoxide ((δ 2.50 for 1H, δ 39.53 for 13C) as internal standards. High resolution mass spectra were obtained by Dr. Rakesh Kohli at the University of Pennsylvania Mass Spectrometry Service Center on an Autospec high resolution double-focusing electrospray ionization/chemical ionization spectrometer equipped with either DEC 11/73 or OPUS software data system.
Compound characterization
All compounds were obtained in milligram quantities. Details of the synthesis and characterization of a compound from each class (1-5) is described below. Complete details of the synthesis and physicochemical characterization of all compounds are included in the supplementary information. Structures of all reported compounds were confirmed by 1H and 13C-NMR, and their mass confirmed by HRMS. Purity was determined by HPLC analysis using a Waters 2690 Alliance system with an inline photodiode array detector. All reported compounds had ≥ 95% purity with the exception of 5l (94%).
2-((2-methoxyphenyl)amino)benzoic acid (1c)
Reaction of methyl 2-(trifluoromethanesulfonyloxy)benzoate with o-anisidine according to step A above provided the methyl ester of 1c as a yellow oil (99% yield). 1H NMR (CDCl3, 500 MHz): δ = 9.44 (s, 1H), 7.97 (d, J = 8.0 Hz, 1H), 7.44 (d, J = 7.8 Hz, 1H), 7.30-7.36 (m, 2H), 7.05 (m, 1H), 6.92-7.00 (m, 2H), 6.75 (m, 1H), 3.92 (s, 3H), 3.90 (s, 3H). 13C NMR (CDCl3, 125 MHz): δ = 169.0, 151.8, 147.5, 134.1, 131.9, 130.4, 123.5, 120.9, 120.7, 117.4, 114.6, 113.0, 111.6, 56.0, 52.0. Hydrolysis of the ester according to Step B provided 1c as a white solid (53% yield). 1H NMR (DMSO, 500 MHz): δ = 12.93 (bs, 1H), 9.57 (s, 1H), 7.90 (m, 1H), 7.31-7.42 (m, 2H), 7.19 (d, J = 8.4 Hz, 1H), 7.00-7.11 (m, 2H), 6.94 (m, 1H), 6.76 (m, 1H), 3.82 (s, 3H). 13C NMR (DMSO, 125 MHz): δ = 169.2, 150.5, 146.3, 133.6, 131.3, 128.9, 122.9, 120.1, 119.8, 116.7, 113.1, 112.4, 111.4, 55.2. HRMS (ES) Calcd. for C14H13NO3: 242.0817 (M-H-), found 242.0812 (M-H-).
4-methoxy-2-((3-nitrophenyl)amino)benzoic acid (2d)
As in 1c, methyl 4-methoxy-2-[[(trifluoromethyl)sulfonyl]oxy]benzoate and 3-nitroaniline were reacted to give the methyl ester of 2d as a yellow oil (93% yield). 1H NMR (CDCl3, 500 MHz): δ = 9.87 (s, 1H), 8.16 (s, 1H), 7.96 (d, J = 8.9 Hz, 1H), 7.88 (d, J = 8.0 Hz, 1H), 7.53 (d, J = 8.0 Hz, 1H), 7.48 (t, J = 8.0 Hz, 1H), 6.80 (d, J = 2.4 Hz, 1H), 6.43 (dd, J = 8.9, 2.4 Hz, 1H), 3.90 (s, 3H), 3.80 (s, 3H). 13C NMR (CDCl3, 125 MHz): δ = 168.8, 164.7, 149.5, 148.1, 142.6, 134.0, 130.3, 127.2, 117.6, 115.5, 106.9, 106.1, 98.9, 55.6, 51.9. The methyl ester was then hydrolyzed to give 2d as a yellow solid (79% yield). 1H NMR (DMSO, 500 MHz): δ = 12.91 (bs, 1H), 9.95 (s, 1H), 8.05 (s, 1H), 7.90 (d, J = 8.8 Hz, 1H), 7.82 (d, J = 8.1 Hz, 1H), 7.71 (d, J = 8.1 Hz, 1H), 7.59 (t, J = 8.1 Hz, 1H), 6.81 (d, J = 2.0 Hz, 1H), 6.52 (dd, J = 8.8, 2.0 Hz, 1H), 3.76 (s, 3H). 13C NMR (DMSO, 125 MHz): δ = 169.3, 163.8, 148.7, 147.0, 142.3, 134.0, 130.7, 126.4, 116.7, 113.9, 107.4, 106.0, 99.2, 55.3. HRMS (ES) Calcd. for C14H14N2O5: 287.0668 (M-H-), found 287.0656 (M-H-).
5-acetyl-2-((4-(trifluoromethyl)phenyl)amino)benzoic acid (3n)
As in 1c, methyl 5-acetyl-2-[[(trifluoromethyl)sulfonyl]oxy]benzoate and 4-aminobenzotrifluoride were reacted to yield the methyl ester of 3n as a yellow solid (89% yield). 1H NMR (CDCl3, 500 MHz): δ = 10.13 (s, 1H), 8.64 (d, J = 2.2 Hz, 1H), 7.97 (dd, J = 8.9, 2.2 Hz, 1H), 7.63 (d, J = 8.4 Hz, 2H), 7.35 (d, J = 8.6 Hz, 2H), 7.31 (d, J = 8.9 Hz, 1H), 3.97 (s, 3H), 2.57 (s, 3H). 13C NMR (CDCl3, 125 MHz): δ = 195.8, 168.7, 150.3, 142.9, 134.3, 133.7, 127.8, 127.0, 122.3, 113.8, 112.0, 52.5, 26.3. Hydrolysis of the ester gave 3n as a white solid (80% yield). 1H NMR (DMSO, 360 MHz): δ = 10.27 (s, 1H), 8.51 (d, J = 2.2 Hz, 1H), 7.99 (dd, J = 8.9, 2.2 Hz, 1H), 7.70 (d, J = 8.5 Hz, 2H), 7.49 (d, J = 8.3 Hz, 2H), 7.39 (d, J = 8.9 Hz, 1H). 13C NMR (DMSO, 90 MHz): δ = 195.3, 169.3, 148.9, 143.3, 134.0, 133.0, 127.4, 126.8, 126.7, 121.3, 114.1, 113.1, 26.2. HRMS (ES) Calcd. for C16H12F3NO3: 322.0691 (M-H-), found 322.0677 (M-H-).
3-((3, 5-bis(trifluoromethyl)phenyl)amino)benzoic acid (4zi)
As in 1c, methyl 3-bromobenzoate and 3,5-bis(trifluoromethyl)aniline were reacted to give the methyl ester of 4zi as a white solid (51% yield). 1H NMR (CDCl3, 500 MHz): δ = 7.80 (s, 1H), 7.77 (d, J = 7.7 Hz, 1H), 7.45 (t, J = 7.8 Hz, 1H), 7.41 (s, 2H), 7.34-7.39 (m, 2H), 6.21 (bs, 1H), 3.94 (s, 3H). 13C NMR (CDCl3, 125 MHz): δ = 166.9, 144.9, 141.1, 133.2, 132.9, 132.1, 130.1, 124.8, 124.1, 121.2, 116.0, 114.0, 52.6. Hydrolysis of the ester gave 4ziii as a brown solid (75% yield). 1H NMR (DMSO, 500 MHz): δ = 9.13 (s, 1H), 7.72 (s, 1H), 7.59 (d, J = 7.5 Hz, 1H), 7.52 (s, 2H), 7.47 (t, J = 7.7 Hz, 1H), 7.43 (m, 1H), 7.38 (s, 1H). 13C NMR (DMSO, 125 MHz): δ = 167.0, 145.6, 141.2, 132.4, 131.2, 129.9, 124.4, 123.2, 122.9, 122.3, 119.5, 114.7. HRMS (ES) Calcd. for C15H9F6NO2: 348.0459 (M-H-), found 348.0449 (M-H-).
4-((3-nitrophenyl)amino)benzoic acid (5d)
As in 1c, methyl 4-bromobenzoate and 3-nitroaniline were reacted to give the methyl ester of 5d as a yellow solid (57% yield). 1H NMR (DMSO, 500 MHz): δ = 9.22 (s, 1H), 7.92 (t, J = 2.1 Hz, 1H), 7.88 (d, J = 8.8 Hz, 2H), 7.74 (m, 1H), 7.54-7.63 (m, 2H), 7.19 (d, J = 8.8 Hz, 2H), 3.81 (s, 3H). 13C NMR (DMSO, 125 MHz): δ = 165.9, 148.7, 146.8, 143.1, 131.1, 130.7, 123.9, 121.0, 115.6, 115.3, 111.7, 51.7. Hydrolysis of the ester gave 5d as a yellow solid (79% yield). 1H NMR (DMSO, 500 MHz): δ = 9.19 (s, 1H), 7.91 (s, 1H), 7.87 (d, J = 8.5 Hz, 2H), 7.72 (m, 1H), 7.50-7.63 (m, 2H), 7.18 (d, J = 8.5 Hz, 2H). 13C NMR (DMSO, 125 MHz): δ = 167.0, 148.7, 146.3, 143.4, 131.2, 130.7, 123.6, 122.5, 115.7, 115.1, 111.4. HRMS (ES) Calcd. for C13H10N2O4: 257.0562 (M-H-), found 257.0567 (M-H-).
Enzyme purification
Homogenous recombinant enzymes AKR1C1-4, AKR1B1 and AKR1B10 were prepared and purified as previously described.14, 16, 48 COX-1 was purified to homogeneity from ram seminal vesicles and COX-2 was purified from baculovirus-infected SF-21 cells as described previously.49, 50 All AKR enzymes were stored at -80 °C in 20 mM potassium phosphate buffer pH 7.0 containing 30% glycerol, 1mM EDTA and 1mM β-mercaptoethanol.
Enzyme Activity Assays
AKR1C Isoforms
The potency of the compounds was determined by measuring their ability to inhibit the NADP+ dependent oxidation of S-(+)-1,2,3,4-tetrahydro-1-naphthol (S-tetralol) catalyzed by AKR1C isoforms. The continuous assay was conducted using a 96-well plate format and the reaction measured the appearance of NADPH fluorimetrically (Exc / Emi, 340/460nm) production on a BIOTEK Synergy 2 Multimode plate reader at 37 °C. The assay mixture consisted of S-tetralol (in DMSO), inhibitor (in DMSO), 100 mM phosphate buffer, pH 7.0, 200 μM NADP+, and purified recombinant enzyme to give a total volume of 200 μl and 4% DMSO. The concentration of S-tetralol used in the assays for each AKR1C isoform was equal to the KM value for the respective enzyme. This enabled direct comparison of the IC50 values of the compounds on each enzyme isoform assuming competitive enzyme inhibition, which is observed with all other N-PA analogs studied to date.23 The KM value obtained for S-tetralol for AKR1C1, AKR1C2, AKR1C3 and AKR1C4 under the same experimental conditions are 8 μM, 22.5 μM, 165 μM and 25 μM, respectively.
AKR1B1 Isoforms
The ability of compounds to inhibit AKR1B1 and AKR1B10 was determined by measuring the inhibition of the enzyme catalyzed NADPH dependent reduction of DL-Glyceraldehyde in the presence of increasing concentrations of inhibitor. The assay monitored the fluorescence change associated with NADPH consumption (Exc / Emi, 340/460nm) on a BIOTEK Synergy 2 Multimode plate reader at 37 °C. The assay mixture consisted of DL-Glyceraldehyde, inhibitor (in DMSO), 100 mM phosphate buffer, pH 7.4, 20 μM NADPH, and the purified recombinant enzyme to give a total volume of 200 μl and 1% DMSO. The concentration of DL-Glyceraldehyde used in the assays was 20 μM for AKR1B1 and 2.5 mM for AKR1B10 respectively, and equal to the KM values obtained for the individual enzymes under the same experimental conditions. Zopolrestat, an inhibitor of AKR1B1 with activity on AKR1B10 was used as a positive control.
COX 1 and 2
The ability of compounds to inhibit COX activity was determined by a continuous colorimetric assay that monitored the oxidation of N, N, N, N- tetramethyl-1, 4-phenylenediamine (TMPD) when it is coupled to the formation of PGH2 from PGG2 using arachidonic acid as substrate. The assays were conducted in a 96 well plate format and absorbance of the oxidized TMPD was monitored at 610 nm on BIOTEK Synergy 2 Multimode plate reader at 25 °C. The assay mixture consisted of 100 mM Tris-HCl pH 8.0, 2 μM Fe3+ protophoryrin IX, 80 μM TMPD and 20 μM arachidonic acid in a total reaction volume of 200 μl with 5% DMSO as cosolvent. The enzyme was pre-incubated with the compound for 5 min before the reaction was initiated by the addition of a mixture of TMPD and arachidonic acid. The pre-incubation step is required since several NSAIDs form a tight binding enzyme inhibitor complex that is time-dependent.28, 51
IC50 Determination
Initial velocities of an enzyme reaction measured in the presence of varying concentrations of inhibitor were compared to a solvent control to give percent inhibition values. IC50 values of compounds was determined by fitting the inhibition data using Grafit 5.0 software[y _ (range)/[1 _ (I/IC50)S] _ background] and obtained from a single experiment with each inhibitor concentration run in quadruplicate with the exception of COX assays that were conducted in duplicate. Substrate, cofactor and enzyme were titrated for each experiment. Selectivity of a compound for AKR1C3 relative to another tested enzyme is defined as the ratio of the IC50 values for the tested enzyme: IC50 value for AKR1C3. The higher the ratio, the greater the selectivity for AKR1C3.
LNCaP-AKR1C3 Mediated Formation of Testosterone
Androgen dependent prostate cancer cells (LNCaP) were genetically engineered to stably express AKR1C3 (LNCaP-AKR1C3).52 The cell line was used to determine the ability of lead compounds to block the conversion of Δ4-AD to testosterone. LNCaP-AKR1C3 cells were plated in RPMI-1640 supplemented with 5% charcoal stripped serum, 1% Pen/strep, 2 mM L-glutamine and 500 μg/ml G418 (CSS media). After 24h incubation, the media was aspirated and fresh CSS media containing 0.1 μM 14C radiolabeled 4-androstene-3,17-dione ([14C]-Δ4-AD) with or without inhibitor was added. The media was collected after 48 h treatment and analyzed for testosterone levels. Briefly, the collected media was extracted with ethyl acetate. The aqueous fraction was then treated with β-glucuronidase (B-glucuronidase type VII-A from E. coli (Sigma)-200 units/ml at pH 6.6) for 24 h at 37 °C to liberate conjugated testosterone and extracted with ethyl acetate. The organic extracts were pooled and dried under vacuo. Steroid reference standards and extracts were re-dissolved in 100 μl ethyl acetate were applied to LK6D Silica TLC plates (Whatman Inc., Clifton, NJ). TLC plates were developed using a dichloromethane/ethyl acetate (80:20 v/v) solution and were counted with a Bioscan System 200 plate reader (Washington, DC).
Supplementary Material
Acknowledgments
Supported by R01-CA90744, P30-ES013508, Prostate Cancer Foundation Challenge grant and Grant Number UL1RR024134 from the National Center for Research Resources awarded to T.M.P. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institute of Health. We thank Ms. Ling Duan for help with the metabolism studies.
Abbreviation
- ACTH
adrenocorticotropic hormone
- ADMET
absorption, distribution, metabolism, excretion and toxicity 5α-Adione, 5α-androstane-3 17-dione; Δ4-AD, 4-androstene-3-17dione
- AKR
aldo-keto reductase
- AR
androgen receptor
- COX
cyclooxygenases
- CRPC
castrate resistant prostate cancer
- DHEA
dehydroepiandrosterone
- DHT
5α-dihydrotestosterone
- FLU
flufenamic acid
- N-PA
N-Phenylanthranilates
- NSAIDs
non-steroidal anti-inflammatory drugs
- PC
prostate cancer
Footnotes
Supporting Information Available: Details of the synthesis and physicochemical characterization of reported compounds is available as supporting information. This material is available free of charge on the internet at http://pubs.acs.org.
A provisional patent application based on these compounds has been submitted to the US patent office. U.S. Provisional Patent Application no. 61/4754,091 filed April 13, 2011.
References
- 1.Altekruse SF, K C, Krapcho M, Neyman N, Aminou R, Waldron W, Ruhl J, Howlader N, Tatalovich Z, Cho H, Mariotto A, Eisner MP, Lewis DR, Cronin K, Chen HS, Feuer EJ, Stinchcomb DG, Edwards BK. SEER Cancer Statistics Review, 1975-2007, based on November 2009 SEER data submission, posted to the SEER web site, 2010. National Cancer Institute; Bethesda, MD: 2010. [Google Scholar]
- 2.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 3.Knudsen KE, Penning TM. Partners in crime: deregulation of AR activity and androgen synthesis in prostate cancer. Trends Endocrinol Metab. 2010;21:315–324. doi: 10.1016/j.tem.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Knudsen KE, Scher HI. Starving the addiction: new opportunities for durable suppression of AR signaling in prostate cancer. Clin Cancer Res. 2009;15:4792–4798. doi: 10.1158/1078-0432.CCR-08-2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Locke JA, Guns ES, Lubik AA, Adomat HH, Hendy SC, Wood CA, Ettinger SL, Gleave ME, Nelson CC. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Res. 2008;68:6407–6415. doi: 10.1158/0008-5472.CAN-07-5997. [DOI] [PubMed] [Google Scholar]
- 6.Attard G, Reid AH, A’Hern R, Parker C, Oommen NB, Folkerd E, Messiou C, Molife LR, Maier G, Thompson E, Olmos D, Sinha R, Lee G, Dowsett M, Kaye SB, Dearnaley D, Kheoh T, Molina A, de Bono JS. Selective inhibition of CYP17 with abiraterone acetate is highly active in the treatment of castration-resistant prostate cancer. J Clin Oncol. 2009;27:3742–3748. doi: 10.1200/JCO.2008.20.0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O’Donnell A, Judson I, Dowsett M, Raynaud F, Dearnaley D, Mason M, Harland S, Robbins A, Halbert G, Nutley B, Jarman M. Hormonal impact of the 17α-hydroxylase/CYP(17,20)-lyase inhibitor abiraterone acetate (CB7630) in patients with prostate cancer. Br J Cancer. 2004;90:2317–2325. doi: 10.1038/sj.bjc.6601879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reid AH, Attard G, Danila DC, Oommen NB, Olmos D, Fong PC, Molife LR, Hunt J, Messiou C, Parker C, Dearnaley D, Swennenhuis JF, Terstappen LW, Lee G, Kheoh T, Molina A, Ryan CJ, Small E, Scher HI, de Bono JS. Significant and sustained antitumor activity in post-docetaxel, castration-resistant prostate cancer with the CYP17 inhibitor abiraterone acetate. J Clin Oncol. 2010;28:1489–1495. doi: 10.1200/JCO.2009.24.6819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, Wongvipat J, Smith-Jones PM, Yoo D, Kwon A, Wasielewska T, Welsbie D, Chen CD, Higano CS, Beer TM, Hung DT, Scher HI, Jung ME, Sawyers CL. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787–790. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jung ME, Ouk S, Yoo D, Sawyers CL, Chen C, Tran C, Wongvipat J. Structure-activity relationship for thiohydantoin androgen receptor antagonists for castration-resistant prostate cancer (CRPC) J Med Chem. 2010;53:2779–2796. doi: 10.1021/jm901488g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stanbrough M, Bubley GJ, Ross K, Golub TR, Rubin MA, Penning TM, Febbo PG, Balk SP. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006;66:2815–2825. doi: 10.1158/0008-5472.CAN-05-4000. [DOI] [PubMed] [Google Scholar]
- 12.Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, True LD, Nelson PS. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res. 2008;68:4447–4454. doi: 10.1158/0008-5472.CAN-08-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin HK, Jez JM, Schlegel BP, Peehl DM, Pachter JA, Penning TM. Expression and characterization of recombinant type 2 3α-hydroxysteroid dehydrogenase (HSD) from human prostate: demonstration of bifunctional 3α/17β-HSD activity and cellular distribution. Mol Endocrinol. 1997;11:1971–1984. doi: 10.1210/mend.11.13.0026. [DOI] [PubMed] [Google Scholar]
- 14.Penning TM, Burczynski ME, Jez JM, Hung CF, Lin HK, Ma H, Moore M, Palackal N, Ratnam K. Human 3α-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the aldo-keto reductase superfamily: functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. Biochem J. 2000;351:67–77. doi: 10.1042/0264-6021:3510067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chang KH, Li R, Papari-Zareei M, Watumull L, Zhao YD, Auchus RJ, Sharifi N. Dihydrotestosterone synthesis bypasses testosterone to drive castration-resistant prostate cancer. Proc Natl Acad Sci U S A. 2011;108:13728–13733. doi: 10.1073/pnas.1107898108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burczynski ME, Harvey RG, Penning TM. Expression and characterization of four recombinant human dihydrodiol dehydrogenase isoforms: oxidation of trans-7, 8-dihydroxy-7,8-dihydrobenzo[a]pyrene to the activated o-quinone metabolite benzo[a]pyrene-7,8-dione. Biochemistry. 1998;37:6781–6790. doi: 10.1021/bi972725u. [DOI] [PubMed] [Google Scholar]
- 17.Steckelbroeck S, Jin Y, Gopishetty S, Oyesanmi B, Penning TM. Human cytosolic 3α-hydroxysteroid dehydrogenases of the aldo-keto reductase superfamily display significant 3β-hydroxysteroid dehydrogenase activity: implications for steroid hormone metabolism and action. J Biol Chem. 2004;279:10784–10795. doi: 10.1074/jbc.M313308200. [DOI] [PubMed] [Google Scholar]
- 18.Rizner TL, Lin HK, Peehl DM, Steckelbroeck S, Bauman DR, Penning TM. Human type 3 3α-hydroxysteroid dehydrogenase (aldo-keto reductase 1C2) and androgen metabolism in prostate cells. Endocrinology. 2003;144:2922–2932. doi: 10.1210/en.2002-0032. [DOI] [PubMed] [Google Scholar]
- 19.Guerini V, Sau D, Scaccianoce E, Rusmini P, Ciana P, Maggi A, Martini PG, Katzenellenbogen BS, Martini L, Motta M, Poletti A. The androgen derivative 5α-androstane-3β,17β-diol inhibits prostate cancer cell migration through activation of the estrogen receptor β subtype. Cancer Res. 2005;65:5445–5453. doi: 10.1158/0008-5472.CAN-04-1941. [DOI] [PubMed] [Google Scholar]
- 20.Penning TM, Talalay P. Inhibition of a major NAD(P)-linked oxidoreductase from rat liver cytosol by steroidal and nonsteroidal anti-inflammatory agents and by prostaglandins. Proc Natl Acad Sci U S A. 1983;80:4504–4508. doi: 10.1073/pnas.80.14.4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsuura K, Shiraishi H, Hara A, Sato K, Deyashiki Y, Ninomiya M, Sakai S. Identification of a principal mRNA species for human 3α-hydroxysteroid dehydrogenase isoform (AKR1C3) that exhibits high prostaglandin D2 11-ketoreductase activity. J Biochem. 1998;124:940–946. doi: 10.1093/oxfordjournals.jbchem.a022211. [DOI] [PubMed] [Google Scholar]
- 22.Pawlowski J, Huizinga M, Penning TM. Isolation and partial characterization of a full-length cDNA clone for 3α-hydroxysteroid dehydrogenase: a potential target enzyme for nonsteroidal anti-inflammatory drugs. Agents Actions. 1991;34:289–293. doi: 10.1007/BF01993305. [DOI] [PubMed] [Google Scholar]
- 23.Bauman DR, Rudnick SI, Szewczuk LM, Jin Y, Gopishetty S, Penning TM. Development of nonsteroidal anti-inflammatory drug analogs and steroid carboxylates selective for human aldo-keto reductase isoforms: potential antineoplastic agents that work independently of cyclooxygenase isozymes. Mol Pharmacol. 2005;67:60–68. doi: 10.1124/mol.104.006569. [DOI] [PubMed] [Google Scholar]
- 24.Byrns MC, Steckelbroeck S, Penning TM. An indomethacin analogue, N-(4-chlorobenzoyl)-melatonin, is a selective inhibitor of aldo-keto reductase 1C3 (type 2 3α-HSD, type 5 17β-HSD, and prostaglandin F synthase), a potential target for the treatment of hormone dependent and hormone independent malignancies. Biochem Pharmacol. 2008;75:484–493. doi: 10.1016/j.bcp.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Desmond JC, Mountford JC, Drayson MT, Walker EA, Hewison M, Ride JP, Luong QT, Hayden RE, Vanin EF, Bunce CM. The aldo-keto reductase AKR1C3 is a novel suppressor of cell differentiation that provides a plausible target for the non-cyclooxygenase-dependent antineoplastic actions of nonsteroidal anti-inflammatory drugs. Cancer Res. 2003;63:505–512. [PubMed] [Google Scholar]
- 26.Lombardino JG. Nonsteroidal antiinflammatory drugs. Wiley Interscience; New York, NY: 1985. [Google Scholar]
- 27.Scherrer AR. Antiinflammatory Agents: Chemistry and Pharmacology. New York, Academic Press; New York, NY: 1974. [Google Scholar]
- 28.Selinsky BS, Gupta K, Sharkey CT, Loll PJ. Structural analysis of NSAID binding by prostaglandin H2 synthase: time-dependent and time-independent inhibitors elicit identical enzyme conformations. Biochemistry. 2001;40:5172–5180. doi: 10.1021/bi010045s. [DOI] [PubMed] [Google Scholar]
- 29.Csuk R, Barthel A, Raschke C. Convenient access to substituted acridines by a Buchwald-Hartwig amination. Tetrahedron. 2004;60:5737–5750. [Google Scholar]
- 30.Hamann BC, Hartwig JF. Systematic variation of bidentate ligands used in aryl halide amination. Unexpected effects of steric, electronic, and geometric perturbations. Journal of the American Society. 1998;120:3694–3703. [Google Scholar]
- 31.Old DW, Wolfe JP, Buchwald SL. A highly active catalyst for palladium-catalyzed cross-coupling reactions: Room-temperature Suzuki couplings and amination of unactivated aryl chlorides. Journal of The American Society. 1998;120:9722–9723. [Google Scholar]
- 32.Jin Y, Stayrook SE, Albert RH, Palackal NT, Penning TM, Lewis M. Crystal structure of human type III 3α-hydroxysteroid dehydrogenase/bile acid binding protein complexed with NADP(+) and ursodeoxycholate. Biochemistry. 2001;40:10161–10168. doi: 10.1021/bi010919a. [DOI] [PubMed] [Google Scholar]
- 33.Endo S, Matsunaga T, Soda M, Tajima K, Zhao HT, El-Kabbani O, Hara A. Selective inhibition of the tumor marker AKR1B10 by antiinflammatory N-phenylanthranilic acids and glycyrrhetic acid. Biol Pharm Bull. 2010;33:886–890. doi: 10.1248/bpb.33.886. [DOI] [PubMed] [Google Scholar]
- 34.Barnett J, Chow J, Ives D, Chiou M, Mackenzie R, Osen E, Nguyen B, Tsing S, Bach C, Freire J, Chan H, Sigal E, Ramesha C. Purification, characterization and selective inhibition of human prostaglandin G/H synthase 1 and 2 expressed in the baculovirus system. Biochim Biophys Acta. 1994;1209:130–139. doi: 10.1016/0167-4838(94)90148-1. [DOI] [PubMed] [Google Scholar]
- 35.Lovering AL, Ride JP, Bunce CM, Desmond JC, Cummings SM, White SA. Crystal structures of prostaglandin D(2) 11-ketoreductase (AKR1C3) in complex with the nonsteroidal anti-inflammatory drugs flufenamic acid and indomethacin. Cancer Res. 2004;64:1802–1810. doi: 10.1158/0008-5472.can-03-2847. [DOI] [PubMed] [Google Scholar]
- 36.Byrns MC, Jin Y, Penning TM. Inhibitors of type 5 17β-hydroxysteroid dehydrogenase (AKR1C3): overview and structural insights. J Steroid Biochem Mol Biol. 2010;125:95–104. doi: 10.1016/j.jsbmb.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Penning TM, Ma H, Jez JM. Engineering steroid hormone specificity into aldo-keto reductases. Chem Biol Interact. 2001;130-132:659–671. doi: 10.1016/s0009-2797(00)00257-x. [DOI] [PubMed] [Google Scholar]
- 38.Jin Y. Activities of aldo-keto reductase 1 enzymes on two inhaled corticosteroids: implications for the pharmacological effects of inhaled corticosteroids. Chem Biol Interact. 2011;191:234–238. doi: 10.1016/j.cbi.2011.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yabe-Nishimura C. Aldose reductase in glucose toxicity: a potential target for the prevention of diabetic complications. Pharmacol Rev. 1998;50:21–33. [PubMed] [Google Scholar]
- 40.Crosas B, Hyndman DJ, Gallego O, Martras S, Pares X, Flynn TG, Farres J. Human aldose reductase and human small intestine aldose reductase are efficient retinal reductases: consequences for retinoid metabolism. Biochem J. 2003;373:973–979. doi: 10.1042/BJ20021818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frotscher M, Ziegler E, Marchais-Oberwinkler S, Kruchten P, Neugebauer A, Fetzer L, Scherer C, Muller-Vieira U, Messinger J, Thole H, Hartmann RW. Design, synthesis, and biological evaluation of (hydroxyphenyl)naphthalene and -quinoline derivatives: potent and selective nonsteroidal inhibitors of 17β-hydroxysteroid dehydrogenase type 1 (17β-HSD1) for the treatment of estrogen-dependent diseases. J Med Chem. 2008;51:2158–2169. doi: 10.1021/jm701447v. [DOI] [PubMed] [Google Scholar]
- 42.Marchais-Oberwinkler S, Frotscher M, Ziegler E, Werth R, Kruchten P, Messinger J, Thole H, Hartmann RW. Structure-activity study in the class of 6-(3’-hydroxyphenyl)naphthalenes leading to an optimization of a pharmacophore model for 17β-hydroxysteroid dehydrogenase type 1 (17β-HSD1) inhibitors. Mol Cell Endocrinol. 2009;301:205–211. doi: 10.1016/j.mce.2008.09.024. [DOI] [PubMed] [Google Scholar]
- 43.Marchais-Oberwinkler S, Kruchten P, Frotscher M, Ziegler E, Neugebauer A, Bhoga U, Bey E, Muller-Vieira U, Messinger J, Thole H, Hartmann RW. Substituted 6-phenyl-2-naphthols. Potent and selective nonsteroidal inhibitors of 17β-hydroxysteroid dehydrogenase type 1 (17β-HSD1): design, synthesis, biological evaluation, and pharmacokinetics. J Med Chem. 2008;51:4685–4698. doi: 10.1021/jm800367k. [DOI] [PubMed] [Google Scholar]
- 44.Penning TM, Burczynski ME, Jez JM, Lin HK, Ma H, Moore M, Ratnam K, Palackal N. Structure-function aspects and inhibitor design of type 5 17β-hydroxysteroid dehydrogenase (AKR1C3) Mol Cell Endocrinol. 2001;171:137–149. doi: 10.1016/s0303-7207(00)00426-3. [DOI] [PubMed] [Google Scholar]
- 45.Wetzel M, Marchais-Oberwinkler S, Hartmann RW. 17β-HSD2 inhibitors for the treatment of osteoporosis: Identification of a promising scaffold. Bioorg Med Chem. 19:807–15. doi: 10.1016/j.bmc.2010.12.013. [DOI] [PubMed] [Google Scholar]
- 46.Wetzel M, Marchais-Oberwinkler S, Perspicace E, Moller G, Adamski J, Hartmann RW. Introduction of an electron withdrawing group on the hydroxyphenylnaphthol scaffold improves the potency of 17β-hydroxysteroid dehydrogenase type 2 (17β-HSD2) inhibitors. J Med Chem. 54:7547–57. doi: 10.1021/jm2008453. [DOI] [PubMed] [Google Scholar]
- 47.Schuster D, Kowalik D, Kirchmair J, Laggner C, Markt P, Aebischer-Gumy C, Strohle F, Moller G, Wolber G, Wilckens T, Langer T, Odermatt A, Adamski J. Identification of chemically diverse, novel inhibitors of 17β-hydroxysteroid dehydrogenase type 3 and 5 by pharmacophore-based virtual screening. J Steroid Biochem Mol Biol. 125:148–61. doi: 10.1016/j.jsbmb.2011.01.016. [DOI] [PubMed] [Google Scholar]
- 48.Quinn AM, Harvey RG, Penning TM. Oxidation of PAH trans-dihydrodiols by human aldo-keto reductase AKR1B10. Chem Res Toxicol. 2008;21:2207–2215. doi: 10.1021/tx8002005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.George HJ, Van Dyk DE, Straney RA, Trzaskos JM, Copeland RA. Expression purification and characterization of recombinant human inducible prostaglandin G/H synthase from baculovirus-infected insect cells. Protein Expr Purif. 1996;7:19–26. doi: 10.1006/prep.1996.0003. [DOI] [PubMed] [Google Scholar]
- 50.Smith T, Leipprandt J, DeWitt D. Purification and characterization of the human recombinant histidine-tagged prostaglandin endoperoxide H synthases-1 and -2. Arch Biochem Biophys. 2000;375:195–200. doi: 10.1006/abbi.1999.1659. [DOI] [PubMed] [Google Scholar]
- 51.Gierse JK, Koboldt CM, Walker MC, Seibert K, Isakson PC. Kinetic basis for selective inhibition of cyclo-oxygenases. Biochem J. 1999;339(Pt 3):607–614. [PMC free article] [PubMed] [Google Scholar]
- 52.Byrns MCMR, Duan L, Penning TM. Overexpression of aldo-keto reductase 1C3 (AKR1C3) in LNCaP cells diverts androgen metabolism towards testosterone resulting in resistance to the 5α-reductase inhibitor finasteride. JSBMB. 2012 doi: 10.1016/j.jsbmb.2011.12.012. Article in Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.