

Abstract
Spinal muscular atrophy (SMA) is an autosomal-recessive disorder characterized by α-motor neuron loss in the spinal cord anterior horn. SMA results from deletion or mutation of the Survival Motor Neuron 1 gene (SMN1) and retention of SMN2. A single nucleotide difference between SMN1 and SMN2 results in exclusion of exon 7 from the majority of SMN2 transcripts, leading to decreased SMN protein levels and development of SMA. A series of splice enhancers and silencers regulate incorporation of SMN2 exon 7; these splice motifs can be blocked with antisense oligomers (ASOs) to alter SMN2 transcript splicing. We have evaluated a morpholino (MO) oligomer against ISS-N1 [HSMN2Ex7D(−10,−29)], and delivered this MO to postnatal day 0 (P0) SMA pups (Smn−/−, SMN2+/+, SMN▵7+/+) by intracerebroventricular (ICV) injection. Survival was increased markedly from 15 days to >100 days. Delayed CNS MO injection has moderate efficacy, and delayed peripheral injection has mild survival advantage, suggesting that early CNS ASO administration is essential for SMA therapy consideration. ICV treatment increased full-length SMN2 transcript as well as SMN protein in neural tissue, but only minimally in peripheral tissue. Interval analysis shows a decrease in alternative splice modification over time. We suggest that CNS increases of SMN will have a major impact on SMA, and an early increase of the SMN level results in correction of motor phenotypes. Finally, the early introduction by intrathecal delivery of MO oligomers is a potential treatment for SMA patients.
INTRODUCTION
Spinal muscular atrophy (SMA), the leading cause of hereditary infant mortality (1), is an autosomal-recessive disorder characterized by α-motor neuron loss in the anterior horn of the spinal cord (2). SMA is caused by the loss or mutation of the Survival Motor Neuron (SMN1) gene and the retention of SMN2 (3,4). The SMN1 and SMN2 genes essentially differ at a single nucleotide in exon 7 (C to T transition) which disrupt a modulator of splicing and leads to exon 7 loss in 90% of SMN2 mRNA transcripts (5–10). The SMN protein lacking exon 7 encoded amino acids does not oligomerize efficiently and is rapidly degraded, leading to low SMN levels and thus SMA (11–13). The SMN2 copy number has a major influence on the severity of the SMA phenotype with milder patients (type III–IV, juvenile/adult onset) having a higher SMN2 copy number, type II having intermediate SMN2 copy number, and severe patients (type I, infantile) having a lower SMN2 copy number (14,15). Furthermore, different SMN2 alleles that are capable of producing more SMN act as improved modifiers of SMA (16,17). Missense mutations occur in both mild and severe patients (4,18), such mutations in severe patients often disrupt the ability of SMN to oligomerize efficiently (12,18). In the case of mild missense mutations, the SMN protein is often able to interact with wild-type SMN produced by SMN2, resulting in more functional SMN complexes (19).
SMN forms a complex with a series of proteins called gemins, and the best characterized function of this complex is the assembly of Sm proteins onto snRNA, thus forming a crucial component of the splicing machinery (20–22). Thus, the complete loss of Smn in mice is embryonic lethal, as SnRNPs are required for gene splicing (23). SMN has also been found in axons and is suggested to play a role in the axonal transport of mRNA (24–27). The exact role that SMN plays in the axon is unclear (4), and motor axon growth is not disrupted in SMA mice models (28). The reduced SnRNP assembly has been reported in SMA mice (29,30), and certainly specific target genes could have disrupted splicing (4,31). Controversy remains as to whether SMA is caused by the disrupted splicing of select genes important for motor neuron circuitry, by disrupted axonal maintenance, or both (4). It is well established that SMA is caused by insufficient SMN levels (32,33) and thus current therapeutic strategies are focused on increasing the amount of SMN.
Mice possess only one Smn gene and the loss of this gene is embryonic lethal (23). Thus, the production of mice with an SMN deficiency that models human SMA required the introduction of the SMN2 gene. Two copies of human SMN2 in mice lacking Smn results in mice with severe SMA that live an average of 5 days, while eight copies of SMN2 results in complete rescue (34,35). The introduction of SMN▵7, an SMN transgene lacking exon 7, into the severe SMA mice increases the average life span to ∼14 days (36). It has recently been shown that postnatal induction of SMN can modulate SMA in SMN▵7 mice (37). The SMNΔ7 mice can therefore serve as an SMA model in which one can characterize disease-modifying pharmacologic interventions.
Using the SMNΔ7 mouse model of SMA, multiple treatment strategies to increase SMN expression have been performed. Targeting SMN production with various pharmacologic compounds to either activate the SMN promoter (38–40) or to alter exon 7-splicing patterns (41) has shown modest improvements in the phenotype of SMNΔ7 SMA mice due to a relatively weak SMN induction. Restoration of SMN using scAAV9-SMN delivered via the facial vein (FV) in newborn SMNΔ7 SMA mice results in rescue of the phenotype with survival in all cases beyond 200 days (42,43) and in one instance beyond 400 days (44). Thus, induction of SMN postnatally has a remarkable therapeutic effect.
Antisense oligomers (ASO), including 2′-O-methyl (2′-OMe) or 2′-O-methoxyethyl (MOE)-modified nucleotides on a phosphorothioate backbone, peptide nucleic acids (PNA) and phosphorodiamidate morpholino (MO) oligomers(45–47), can be used as specific splice-switching agents to alter pre-mRNA processing. ASOs can block targeted sequences, including exon splice enhancers or intron splice silencers (ISSs)(46–50). Numerous regions are important in SMN2-splicing regulation (51), and one particularly important element is the negative splice regulator ISS-N1, a 15 nucleotide-splice-silencing motif located downstream of SMN2 exon 7 (52). An 18mer MOE chemistry ASO overlapping ISS-N1 effectively increases in vivo exon 7 incorporation and SMN levels through SMN2 splice modulation (53,54). Intracerebroventricular (ICV) injection of this MOE at a dose of 0.58 mm did increase the average survival in SMNΔ7 SMA mice from 14 to 26 days (55). However, there was evidence of toxicity with this MOE at doses >1.16 mm. More recently, while this work was in review, Hua et al. (56) reported that peripheral dosing of MOEs was essential for longer survival, and propose that extra-CNS targets (i.e. peripheral) are required for rescue of the SMA phenotype. This assertion contradicts previous results showing the necessity of neuronal SMN expression using both scAAV8-SMN delivery by ICV, and transgenic approaches which limit SMN expression to CNS or peripheral tissue (57,58); moreover, the authors show CNS SMN2 splicing modulation after serial high-dose peripheral deliveries, suggesting MOE transport across the blood-brain barrier when delivered at high doses in young mice.
MOs have a low toxicity and a wide distribution of uptake as evidenced by their extensive use in target gene expression knockdown in the developing zebrafish (59). MOs have been used to induce exon skipping in Duchenne muscular dystrophy (DMD) with excellent body-wide distribution and restoration of dystrophin expression after systemic administration in the mdx mouse (49,60,61). Likewise, there have been encouraging clinical results in patients with DMD after MO administration (62–64).
In this study, we delivered a bolus ICV injection of a high concentration, highly stable MO targeted to ISS-N1 to alter SMN2 splicing and increase SMN levels. We have named this MO HSMN2Ex7D(−10,−29) (referred to as MO) based on its position relative to the exon 7 donor site. Treated SMA mice had improvement in weight gain, motor activity and increased survival from 15 to >100 days. Peripheral FV delivery at P0, combined peripheral and ICV delivery and dual ICV injection at P0 and P30 produced an equivalent survival. Delayed CNS delivery (P4) had an intermediate advantage, evidence that earlier CNS treatment yields more robust effects, while delayed peripheral delivery after blood-brain-barrier maturation had only a modest increased survival.
ICV injection yielded an increase in both SMN2 exon 7 incorporation and SMN protein levels in brain and spinal cord, while digital droplet polymerase chain reaction (PCR) (ddPCR) showed no alteration in SMN2 splicing in peripheral tissues (the kidney, liver, heart) with Smn+/− heterozygotes and small alterations with SMA animals. P0 FV delivery of MO clearly altered SMN2 splicing within the CNS, evidence that the MO ASO does cross the blood-brain barrier, but does not have effect across the cerebrospinal (CSF)–blood barrier in young mouse pups. Immunofluorescence analysis of lumbar spinal cord sections delineates increased SMN and dissemination of the MO to caudal CNS structures. These results suggest that it is essential to restore SMN levels within neurons to have a major impact in SMA, and long survival benefit can be obtained without significantly enhancing SMN levels in the periphery. It remains possible that increasing SMN expression in the ANS outside of the blood-brain barrier may be required for full correction of SMA. These results support the continued investigation of MO ASOs as promising approaches in the early treatment of SMA.
RESULTS
MO modulates exon splicing and increases SMN production
Mutation or steric block at the 5′ region of SMN2 intron 7 (ISS-N1) yields the greatest rates of exon 7 inclusion (51,52,54). We designed our MO to mask this splice-silencing motif (Fig. 1). A sequence nonspecific scramble MO (referred to as scMO) was manufactured with identical length and molecular weight to MO. To assess exon 7-splicing inclusion, we injected by ICV newborn (P0) pups of genotype Smn+/−; SMN2+/+; Δ7+/+ (‘heterozygotes’) with one of the three doses of MO (high, middle, low dose) or scMO. Heterozygotes were used due to the normal phenotype (as a result of the presence of one Smn gene), thus controlling for additional splicing changes that could accompany an SMA mouse as well as secondary disease effects (e.g. poor nutrition and muscle atrophy). The availability of human specific primers and antibody facilitated analysis. Injected animals were sacrificed at interval ages with collection of the brain, spinal cord, heart, liver and kidney tissue. The total number of mice allowed analysis of all samples in triplicate. For assessment of exon 7 incorporation, we performed reverse transcription polymerase chain reaction (RT–PCR) and then amplified the cDNA with SMN2-specific primers. Amplification yields both a full-length (1491 nucleotides, top) and exon 7-deficient product (1433 nucleotides, bottom). scMO-injected animals had a strong exon 7-deficient band, and transgenic animals with 16 copies of SMN (8SMN2+/+; Smn−/−; HiCopy or ‘HiC’) (35) also had a relative band intensity of exon 7-deficient greater than full-length. All MO-injected heterozygotes showed a strong band reversal (full-length > exon 7 deficient) at all time points (P7 → P65) in both the brain (Fig. 2A) and spinal cord (Fig. 2B).
Figure 1.

Illustration of SMN2 exon and intron 7 with highlighted ISS-N1 and the target site for morpholino HSMNEx7D(−10–29) (MO).
Figure 2.

Analysis of exon 7 incorporation in SMN2 transcript. Smn+/−; SMN2+/+; Δ7SMN+/+ mice were injected by P0 ICV with low- (27 μg), middle- (54 μg) or high- dose (81 μg) MO. RNA from brain (A) and spinal cord (B) tissue was isolated and cDNA amplified with SMN2-specific primers. All dosings show increased full-length SMN2 (top band) versus exon 7-deficient SMN2 (bottom band) at all time points (P7, P21, P45, P65) relative to scMO-injected control animals (HiC = 16 copies SMN2 (8SMN2+/+; Smn−/−); WT, wild type. (C) ddPCR for relative full-length SMN2 (FL-SMN2) relative to cyclophilin; low-, middle-, high-dose MO (dose–response curve); P7 brain and spinal cord tissue. (D) Quantitative RT–PCR for full-length SMN2 in spinal cord P7–P65 (low, middle, high dose). There is increased P7 full-length SMN2, with decay at later time points.
Quantification of full-length SMN2 transcript was performed by both real-time RT-PCR and ddPCR. The latter has many advantages for both quantification and detection of low levels of a specific transcript (65). The sample is first partitioned into droplets; the distribution of any particular molecule (such as a specific cDNA) will conform to a Poisson distribution. The number of droplets amplified by PCR is proportional to the concentration of that molecule. The final result is a count of positive droplets after PCR has gone to completion. ddPCR has the ability to detect small amounts of a particular cDNA, and does not suffer from the logarithmic error seen in real-time PCR. In our experience, this method has been highly reliable in quantification when compared with real-time PCR methods.
We performed ddPCR of P7 pup brain and spinal cord tissue injected at P0 with either MO (2 µL of 2, 4 or 6 mm) or scMO (control). Figure 2C shows the full-length SMN2 response curve with escalating MO dose. We next quantified by real-time RT–PCR full-length SMN2 transcript as a function of increasing animal age through P65 (Fig. 2D). Low, middle and high P0 MO doses yielded an increase in exon 7 incorporation at P7, with lower levels observed through P65. It should again be realized that these increases in SMN2 exon 7 incorporation are a result of a single P0 bolus dose, delineating the robust CNS stability of the MO.
We next asked whether CNS administered MO crosses the CSF–blood barrier and remains sufficiently stable in the blood stream to affect systemic structures. These experiments aimed to characterize the importance of CNS versus peripheral SMN expression. SMN2 RNA splicing analysis was performed on CNS and systemic organs (the heart, liver and kidney) 1 week after middle dose (4 mm) MO injection into mice heterozygous for mouse Smn. The analysis was done by both RT–PCR and ddPCR. RT–PCR of the heart, liver and kidney did not show subjective modulation of splicing in any visceral organ, although the spinal cord again showed strong exon 7 incorporation (Fig. 3A). ddPCR (Fig. 3B) revealed no significant splicing changes in tissue outside the nervous system after ICV delivery (the liver and heart). Furthermore, there was an increase in SMN2 exon 7 incorporation within the CNS after peripheral delivery, suggesting that high doses of MO delivered peripherally do have the ability to cross the blood-brain barrier in the physically immature neonatal mouse pup. We also injected SMNΔ7 SMA mice (n= 2) by middle dose MO ICV and compared tissue SMN2 exon 7 incorporation with control SMNΔ7 SMA animals (Fig. 3C). As was seen with mSmn+/− heterozygotes, there were large CNS increases in full-length SMN2, up to 5-fold in both the brain and spinal cord. There were also smaller (2-fold) increases in the liver, suggesting immaturity or derangement of the blood–CSF barrier in SMNΔ7 SMA mice, but again reinforcing the ‘leakiness’ of CSF barriers in neonatal pups. Taken together, these results are important for two reasons: first, the phenotypic changes seen in MO ICV-injected SMA mice are most likely a result of the large amount of exon 7 splice modulation within the CNS relative to only modest splice modulation within the periphery, and second, peripherally administered MO in the neonatal mouse profoundly alters CNS SMN2 splicing.
Figure 3.

Central versus peripheral SMN2 splice modulation. (A) RT–PCR systemic analysis of full-length SMN2 after P0 MO ICV injection. There is no increased SMN2 exon 7 incorporation in visceral structures (the heart, liver, kidney) when compared with spinal cord (n= 2). (B) ddPCR (full-length SMN2 relative to cyclophilin) of the brain, spinal cord, heart and liver after P0 MO ICV (54 µg) or P0 FV (50 µg/g) injection in Smn+/−; SMN2+/+; Δ7SMN+/+ mice. Results per organ are relative to scramble-injected control, which has been set at 1 on the y-axis. There is increased CNS splice modulation after both ICV and peripheral dosing. There is no increased splicing in the heart and liver after ICV injection, suggesting limited translocation across the CSF–blood barrier or rapid systemic degradation. (C) ddPCR (FL-SMN2 relative to cyclophilin) of the brain, spinal cord, heart and liver after P0 MO ICV (54 µg) injection in Smn−/−; SMN2+/+; Δ7SMN+/+ mice; SMA scramble-injected control per each organ. Results are displayed as absolute relative ratios on the y-axis. There are large increases in CNS splice modulation, and a more modest increase in liver full-length SMN2.
We quantified SMN protein levels after P0 ICV MO injection (Fig. 4). Primary antibody detected human SMN and did not have affinity for mouse SMN (53). At P7, all MO doses increased SMN levels (relative to β-actin) in both brain (Fig. 3A and C) and spinal cord tissue (Fig. 4B and D) when compared with scMO-injected control animals. Again, as was seen with splicing changes, there was a decrease in relative SMN levels at later time points.
Figure 4.

Analysis of SMN induction after MO delivery. Smn+/−; SMN2+/+; Δ7SMN+/+ mice were injected by P0 ICV with low- (27 μg), middle- (54 μg) or high-dose (81 μg) MO. Quantification of SMN by human specific antibody relative to actin in the brain (A) and spinal cord (B) for each dose at P7, P21, P45 and P65. All doses increased P7 SMN (strongest increase with high dose) and decay through P65 (n= 3 for each dose per time point). (C and D) Representative western blots for human SMN and actin in the brain (A) and spinal cord (B) after low-dose injection.
P0 MO was injected by ICV into SMN▵7 SMA mice which had motor neuron-specific GFP expression (Smn−/−; SMN2+/+; Δ7+/+; HB9:GFP), and animals were sacrificed at P7 (66). Staining of lumbar cord sections with human SMN-specific antibody showed gems in the nucleus and SMN staining in the cytosol of both motor neurons and spinal cord support cells (Fig. 5). There was robust evidence of SMN in the most distal segments of the spinal cord parenchyma. An ICV-injected lissamine-tagged MO also showed lumbar cord parenchymal distribution (Supplementary material, Fig. S1).
Figure 5.

Motor neuron SMN expression increases in MO-injected Δ7SMN SMA mice. (A–C) 54 μg MO (middle) dose was injected by P0 ICV. Lumbar spinal cord tissue was harvested at P7, sectioned and stained with human specific anti-SMN KH antibody. (A) Motor neurons in the ventral horn were identified by HB9:GFP transgenic expression. (B) SMN is found in the cytoplasm and in gems in the nucleus of motor neurons, as well as other cell types, throughout the spinal cord. Insert highlights the motor neuron indicated by the arrowhead. (C) The merged image of SMN expression (red) in the motor neuron (green). Insert highlights the motor neuron indicated by the arrowhead. (D–F) SMN expression in a non-injected carrier control animal. (D) Motor neurons identified by HB9:GFP expression. (B) A low level of SMN expression from SMN2 is present throughout the spinal cord. (C) The merged image of SMN expression (red) in the motor neuron (green). Scale bar = 200 µm.
MO-targeting ISS-N1 extends survival in SMA animals
SMNΔ7 SMA mice (Smn−/−; SMN2+/+; SMN▵7+/+) were injected by P0 ICV with either MO at low (n= 8), middle (n= 10) or high dose (n= 13), or with scMO (n= 8). Control mice (Smn+/+; SMN2+/+; SMN▵7+/+) were injected with scMO (n= 30). Animals were weighed daily, assessed for morbidity (peripheral necrosis, acute weight loss, bladder or bowel retention, priapism, decreased grooming) and mortality, and righting response was tested through P21. MO-injected animals had increased survival at all concentrations (Fig. 6A), with a median survival increasing from 15 ± 1.1 days (scMO) to 83 ± 37.7 days (MO, low dose), 104 ± 15.0 days (middle dose) and 112 ± 6.6 days (high dose). The survival advantage was significant for all three groups (log-rank P< 0.001) when compared with scMO control injected mice. There was no significant difference between MO doses (log rank P> 0.8); indeed, the low-dose strategy yielded our longest lived mouse (161 days).
Figure 6.
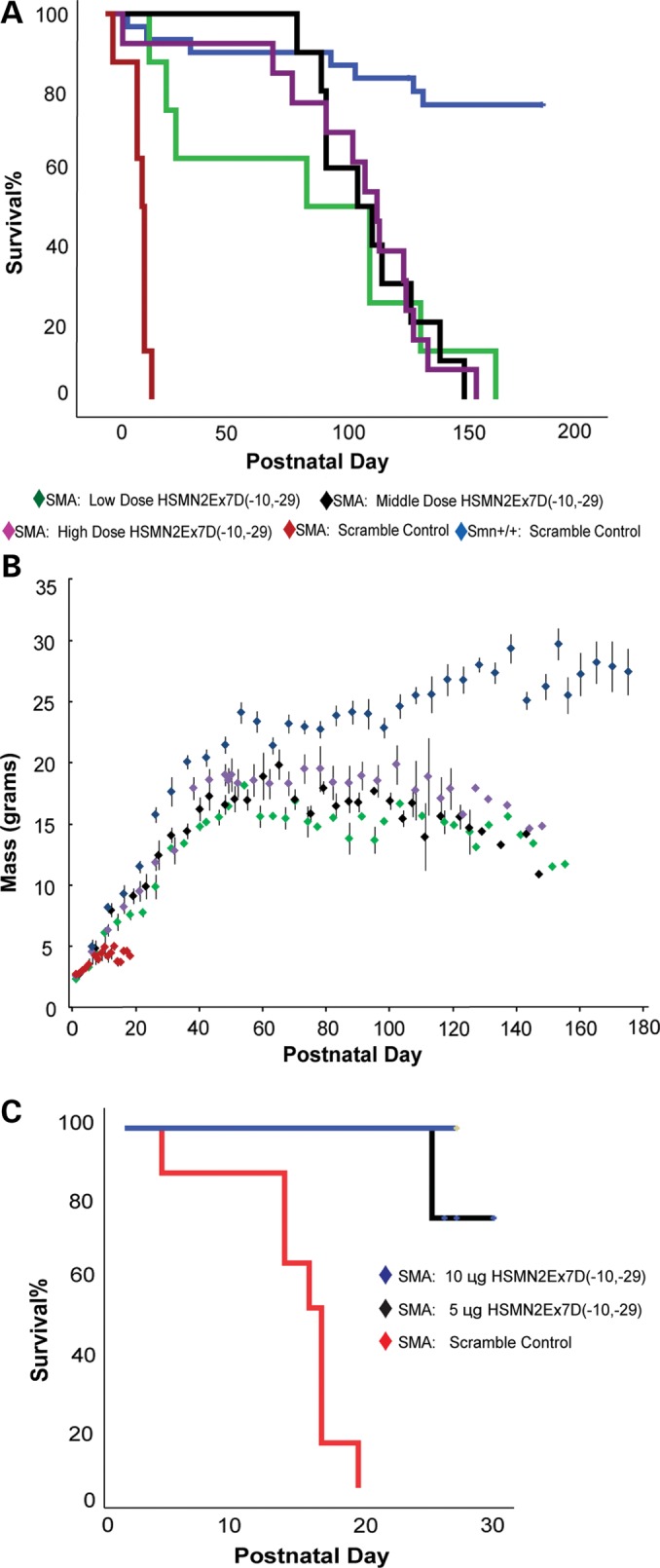
Survival and weight gain are increased in MO-treated Δ7SMN SMA animals. (A) The Kaplan–Meier survival curve for low (n= 8), middle (n= 10) and high dose (n= 13). The median survival for low (83 ± 37.7 days, maximum 161), middle (104 ± 15.0 days, maximum 148) and high dose (112 ± 6.6 days, maximum 153) were not statistically different (log-rank P> 0.8 for all pairwise comparisons). The median survival for each group was significantly extended when compared with scMO (6 mm, 81 µg)-injected Δ7SMN SMA animals (15 ± 1.1 days, log-rank P< 0.001 for all pairwise comparisons). (B) MO-treated Δ7SMN SMA mice increased mass nearly parallel to that of control animals (Smn+/+; SMN2+/+; SMNΔ7+/+, scMO injection) through ∼P30, at which point the mass curves leveled while control animals continued to gain weight. P80 mean: low dose = 16 ± 0.9 g, middle dose: 17 ± 0.6 g, high dose: 19 ± 1.9 g, control = 22 ± 1.4 g. (C) The Kaplan–Meier survival curve after 5 µg (n= 4) and 10 µg (n= 3) MO ICV P0 injection (censored, through P30).
We also injected SMNΔ7 SMA mice with 5 µg (0.74 mm, n= 4) and 10 µg (1.48 mm, n= 3) MO (Fig. 6C); the former dose more closely approximates the MOE–ASO dose (0.58 mm) that was reported to have some survival advantage (median 26-day survival) for the SMNΔ7 SMA model in a previous ICV ASO administration study by Passini et al. (55). Although it is too early in these low-dose investigations to calculate the median survival, through P26, there is a 75% survival with 5 µg injection, and 100% survival with 10 µg. Additionally, three mice were injected by ICV at P0, P1 and P2 with 135 µg MO (2 µl of 10 mm MO) per dose for a total dose of 405 µg ICV. These mice, currently 38 days old, are healthy and show no evidence of toxicity. These results clearly indicate that the long survival can be obtained with CNS delivery alone. Mortality was not increased as a result of CNS cannulation or MO injection. We did have 7 deaths among the 30 scramble control-injected mice, although four mice were sacrificed iatrogenically due to injuries from fighting, cage flooding and a grossly infected foot. No control mice died within the first week of life. Therefore, 3 of 30 control animals died from unknown causes, although none displayed declining weight before death. While it is plausible that control treatment contributed to this mortality, the lack of a pattern of animal decline and death in this group largely reinforces the safety of ICV cannulation and of the MO chemistry ASO.
Treated animals had weight gain nearly parallel to control animals through ∼P30 (Fig. 6B) at which time the mass of treated animals stabilized at ∼80% that of control animals (P80 average weight of SMA-treated animals: low = 16 g, middle = 17 g and high = 19 g, versus controls = 22 g). There was normalization of righting response within days of treatment (Fig. 7A). Forelimb and hindlimb grip strength at P30 showed equivalent force generation (per animal mass) in treated SMN▵7 SMA mice when compared with controls (Fig. 7B).
Figure 7.

Treated SMA animals have an improved motor phenotype. (A) Righting response quickly approaches 100% by P3 in ICV MO-treated animals, while scMO-injected SMNΔ7 SMA animals have a rapid decline in righting ability during the first week of life. No SMA animals injected with scMO were able to right after P6. (B) Mass-corrected forelimb and hindlimb grip strength is equivalent between control animals (Smn+/+; SMN2+/+; SMNΔ7+/+, scMO injection) and MO-treated SMNΔ7 SMA animals (grip strength assessed at P30 in Newtons/mg body mass; forelimb: control 34.5 ± 2.2 versus SMA 34.2 ± 2.1, hindlimb: control 28.2 ± 5.0 versus SMA 25.8 ± 4.4).
MO-treated mice developed an interesting phenotype of peripheral necrosis, similar to that seen in a previous study of early SMN modulation (34,38,44). All SMA mice had shorter tails during neonatal, adolescent and early adulthood (Supplementary material, Fig. S2A). Beginning at ∼P50, both pinna and tails of SMA mice necrosed until healing granulation tissue appeared at the base of these structures (Supplementary material, Fig. S2B). Three treated mice (one middle dose and two high dose) developed signs of possible autonomic nervous system (ANS) dysfunction, including priapism, urinary retention and bowel obstruction. One middle dose and three high dose adult SMA animals died unexpectedly without evident morbidity (weight loss, decreased grooming, decreased activity or urinary retention). These four mice weighed the most among all treated mice. This mortality suggests hyperacute cardiopulmonary failure as has previously been demonstrated in rescued mice of the SMNΔ7 SMA mouse model (67).
A variety of P0 injection modalities was attempted in an effort to investigate whether either a supplementary CNS dosing or targeting of peripheral structures (i.e. extra-CNS ANS) could increase the survival of animals and decrease necrosis. SMA mice were injected with a high-dose MO through the peripheral FV, as well as a combination of ICV and FV (Fig. 8). Additionally, another group received P0 ICV MO coupled with P30 stereotactic ICV injection (n= 4). All of these administration techniques produced an impressive survival advantage equivalent to but not greater than high-dose PO ICV MO (log-rank P> 0.09 for all pairwise comparisons). Necrosis was not prevented or delayed in these groups. Mice treated with a single peripheral injection (50 µg/g animal mass, n= 3) had a median survival of 35 ± 6.5 days, with a maximum survival of 112 days, suggesting either peripheral MO toxicity or inadequate full-length SMN translation. Mice injected at P0 with both peripheral (50 µg/g) and ICV (54 µg, middle dose) MO, however, had substantial increases in the median survival (93 ± 6.4 days, n= 5). These results evidence the non-toxicity of peripheral MO at this dose, as well as reinforce the central importance of CNS SMN2 splice modulation.
Figure 8.
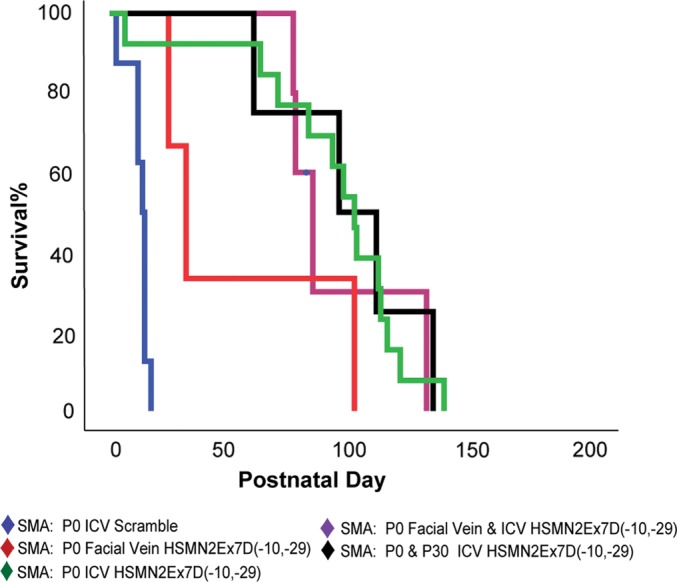
Alternative MO injection strategies do not increase the survival beyond P0 ICV MO injection. Three cohorts of mice each received an injection of MO by P0 FV (50 µg/g body mass, n= 3), P0 FV and ICV (50 µg/g peripheral, 54 µg ICV, n= 5) or P0 and P30 ICV (with stereotactic guidance, 54 µg/g P0, 18 µg/g P30, n= 4). Survival for all groups was equivalent to high-dose P0 ICV MO injection (log-rank P> 0.09).
A cohort of SMA animals was injected at P4 by ICV (n= 5) to determine the extent of phenotype modification with delayed delivery. We chose delivery at P4 as this time-point approaches the latest age at which free-hand delivery can easily and reliably cannulate the lateral ventricle due to increasing cranial ossification and diminishing ventricular volume. These mice had an intermediate weight gain compared with PO-injected SMA animals (Fig. 9A). The median survival was also modestly improved (41 ± 14.2 days, maximum 79, log-rank P< 0.001) (Fig. 9B). The comparison of pairwise survival between P0- and P4-treated animals was also significant (log-rank P< 0.006). Thus, early administration of MO is more effective than late administration. This will be an important consideration when moving forward towards human clinical applications (37,68).
Figure 9.
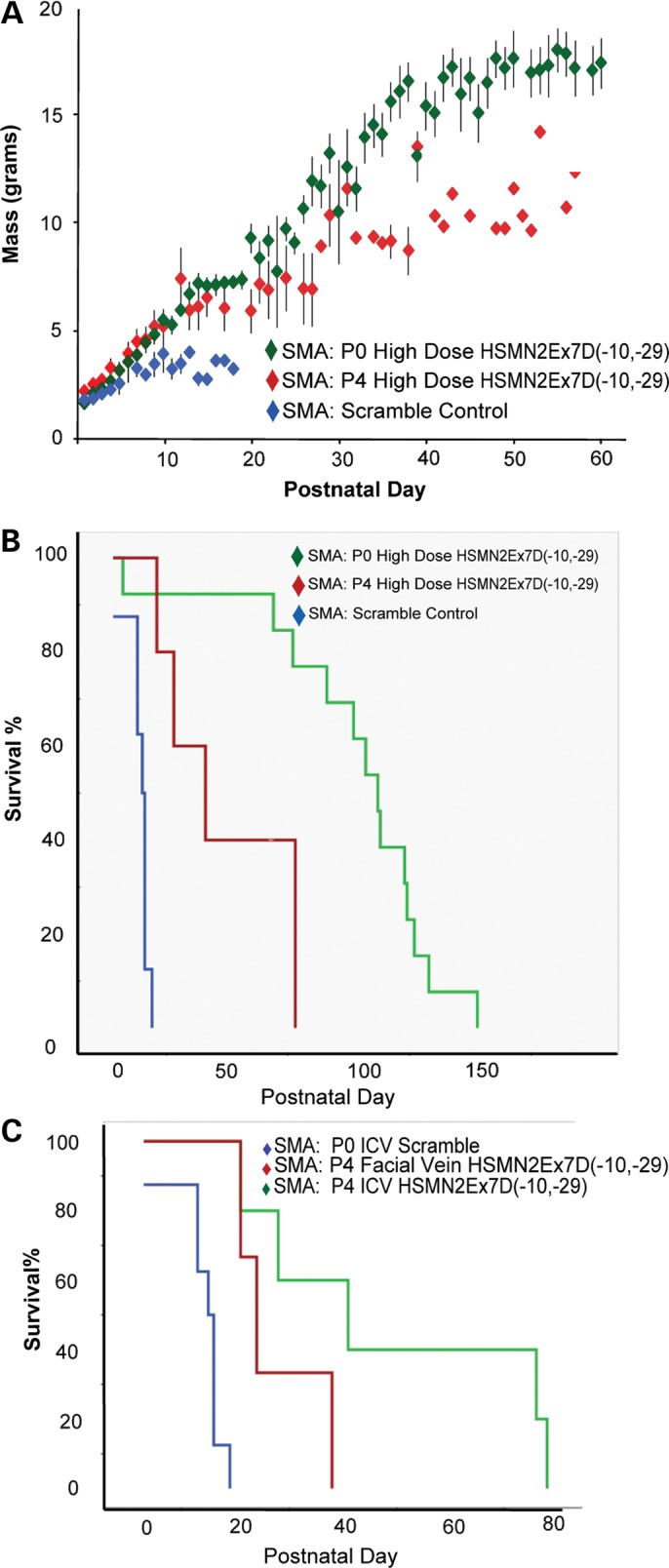
Delayed ICV injection yields intermediate weight gain and survival. (A) SMNΔ7 SMA mice treated at P4 with 54 μg MO/g body mass (‘high dose’ equivalent) had increased weight gain when compared with SMNΔ7 SMA controls, but was less than the weight gain displayed by P0 MO-treated SMNΔ7 SMA animals [P30 mean 10.6 ± 1.5 g (P4) versus 13.6 ± 1.0 g (P0)]. (B) P4 ICV MO-injected animal survival was also increased (median 41 ± 14.2 days, maximum 78 days) when compared with scMO-injected animals (15 ± 1.1 days; log-rank P< 0.001), but was decreased when compared with SMNΔ7 SMA animals treated at P0 with high-dose MO (112 ± 6.6 days, maximum 153 days, log-rank P< 0.01). (C) SMNΔ7 SMA mice were injected with P4 MO by FV. Survival was modestly extended when compared with scMO-injected animals, and there was a decreased survival when compared with animals injected by P4 ICV MO (median 21 ± 0.354 days, log-rank P< 0.014).
We also performed a P4 peripheral vascular injection (50 µg/g, FV) of MO into a group of SMA animals. This group of mice (n= 9) had decreased survival compared with the P4 ICV-injected cohort (21 ± 0.354 days, log-rank P< 0.014) (Fig. 9C). We surmise that the later age coupled with less-direct targeting of the CNS motor neurons over a shortened period of time (shorter therapeutic window) resulted in greater motor neuron morbidity. This condition argues for the early CNS delivery of bare ASOs for SMA motor neuron targeting.
DISCUSSION
A single-nucleotide alteration within exon 7 differentiates SMN1 and SMN2 capacity for full-length SMN protein production. Exon 7 exclusion, as a result of modulation of SMN2 mRNA splicing (5–10), yields a truncated SMN that does not oligomerize efficiently, is unstable and rapidly degraded (11–13). SMN functions in ensuring accurate assembly of Sm proteins onto snRNA. This function is altered in SMA animals (19,29) in direct correlation with disease severity and altered SnRNP profiles (29,30). However, assembly of other RNP complexes could also be altered, and SMN has been found in axons (4,25). The possibility exists for disruption of additional SMN functions that have not yet been elucidated, and SMA could ultimately be a result of splicing changes, RNA transport defects or a combination of both. Despite the uncertainty, it is clear that SMA is a disease of a reduced functional component of SMN, and thus increasing SMN at the right time and place is predicted to alter the disease course. Elevating SMN in the postnatal period of SMA mice has a marked impact on phenotype (38,39,41,43,44,55,56). Furthermore, we have shown using an inducible mouse that postnatal induction of SMN yields a dramatic rescue of SMA (37); Lutz et al. (68) have reported similar results using a different inducible system. The removal of SMN induction after 28 days has minimal effect on the neuromuscular system, highlighting the critical window for high SMN requirements during neonatal and juvenile mouse development (37). In agreement with these studies, we have found that SMN induction early (P0) by a single bolus dose of MO successfully modulates SMN2 splicing and results in an impressive impact on SMN▵7 SMA survival, whereas delayed delivery (P4) had a diminished impact. These results reiterate the importance of early disease diagnosis and therapy.
Many investigative SMA treatments focus on increasing SMN protein levels. Three strategies include compounds that activate the SMN2 promoter (38–40), transfection of the motor neuron with an SMN producing viral vector (42–44) and injection of short-sequence ASOs that promote modulation of splicing and inclusion of SMN2 exon 7 (53,55). There are several chemistries of ASOs, including 2′-OMe, MOE, PNA and MOs that have been used to modify RNA processing in neuromuscular disease (45–47). Multiple groups have published results of CNS ASO injection in mouse models of SMA, and all have shown SMN2 splice modulation with targeting of ISS-N1 and flanking sequences within intron 7, affirming this region as a strong modulator of exon 7 splice dynamics (45,48,53–56). The principle drawback of many of these studies is the relatively mild SMA phenotypic alteration. Williams et al. (48) used a 2′-OMe ASO and noted an increased animal mass that declined by P12 animal sacrifice, as well as improvement in righting ability. The study was complicated by multiple cranial injections on successive days, a possible confounder for animal decline. Hua et al. (53) compared 2′-OMe and MOE oligomers targeting HSMN2Ex7D(−10 to −27) and found the former as substantially less effective. 2′-OMe ASO is likely suboptimal in the treatment of SMA mice when used for negative regulator blocking, although it may work well when enhancing positive regulator binding.
Baughan et al. (45) used a bifunctional 2′-OMe ASO that targeted both an intron 6 negative regulator and attracted SR proteins, thereby enhancing exon 7 incorporation. Phenotypic advantage was again modest, although the authors used a more severe model of SMA (Smn−/−, SMN2+/+), and thus direct comparison with the current study is difficult. Hua et al. (53) showed that an ICV-injected MOE oligomer against ISS-N1 altered necrosis phenotypes in SMN-deficient mice that do not show a clear SMA motor phenotype. Recently, Passini et al. (55), using this same MOE, showed improved muscle physiology as well as survival (median 26 days, 4 μg dose) after ICV treatment in the SMNΔ7 SMA model. This study showed suspected MOE toxicity at doses >8 μg (1.16 mm). The authors reported on decay rates and longitudinal splicing changes with their most efficacious ICV bolus dose of 4 μg. The MOE decreased from an apex of 8–10 μg/g in cervical tissue to 2 μg/g at P16, and SMN2 splicing was effected at Days 3 and 16 but returned to baseline levels by Day 30. Due to little if any toxicity demonstrated by bolus CNS dosing of MO, we were able to administer up to 135 μg/day, and 405 μg from P0 to P3 without adverse effect, and measured SMN2-splicing changes after single P0 bolus of 81 μg through Day 65. Therefore, the reduced CNS toxicity of MOs may allow for higher early dosing and achievement of more prolonged splicing modulation.
While the current paper was under review, Hua et al. (56) reported the results of MOE injection targeting ISS-N1, and also explored various routes of delivery as well as elevated and intermittent dosing strategies. These experiments used a different mouse model of SMA developed by Hsieh-Li et al. (34), which is characterized by mouse Smn lacking exon 7, thus producing a truncated transcript, as well as two copies of SMN2 on a chromosome. Mice live for an average of 10–11 days and display a similar phenotype to the SMNΔ7 SMA animals. Despite many similarities, it is possible that there is unique tissue variability in SMN2 expression between these two SMA models. Nevertheless, the authors found a minor survival advantage after a single P0 MOE ICV bolus dose of 20 μg (2.9 mm, 16 versus 10 days). This dose is comparable with our 27 µg dose and the resultant median survival of 83 days. It is possible that MOs have a longer half-life within the CNS and thus a longer duration of effect.
Hua et al. reported a long survival after multiple peripheral doses (median 137 days), and conclude that peripheral SMN restoration is essential for SMA treatment. SMN2 splicing was modulated at high levels in both the periphery (the liver, heart, kidney, muscle) and in the CNS (the brain and spinal cord) after peripheral delivery. It is important to consider the pharmacokinetics of single versus multiple dosing, such that the latter would result in a more sustained therapeutic window and longer term splice modulation over the critical early period in SMA. Furthermore, it is clear that both MOEs and MOs cross the immature blood-brain barrier in neonatal mice and modulate CNS splicing, and we suspect that such translocation would not be recapitulated in human infants. We propose that sustained increased levels of CNS SMN throughout a critical early window is essential for SMA therapy, in agreement with previous findings (44,57,58).
An MO ASO directed against ISS-N1 induces a robust and sustained SMN2 exon 7 incorporation, leading to unequivocal SMN protein increases in both the brain and spinal cord tissue. These findings correlate with an impressive SMNΔ7 SMA survival advantage (maximum 161 days, median 112 days, high dose MO). When compared with scAAV9-SMN injection, our weight gain and righting paralleled with these animals, although after viral transfection the investigators had complete survival rescue (44). ICV injection was performed by bolus injection at a single time (P0) or at two time points (P0 and P30) with no difference in survival between the two groups. However, scAAV9-SMN animals were exposed to continuous SMN production due to viral cell transduction. The MO is susceptible to both dilution with animal growth and to eventual renal clearance, and multiple boluses or continuous MO dosing by intrathecal osmotic pump placed during adolescence may further prolong survival. Alternatively, ICV dosing may miss a critical target such as the extra-CNS ANS, although this seems less likely given the lack of an increased animal survival over-and-above P0 ICV when ICV was combined with FV dosing.
FV delivered MO at P0 also had long-term survival, although we surmise that the leaky blood-brain barrier in the immature mouse pup allows for translocation of ASO into the CNS, as demonstrated by ddPCR analysis. MO was also delivered by FV at P4, which resulted in only mild animal rescue, suggesting that the critical target for SMN-level modulation resides within the CNS and must be acted on early. It may still be important to increase peripheral SMN, although in a more continuous fashion so as to surmount renal, which could be accomplished with multiple intravascular administrations by tail vein injection or indwelling central venous catheter placement.
Despite both increased inclusion of exon 7 in SMN2 transcript and elevated SMN protein levels at delayed time points (P45–65), treated SMA mice developed an interesting and time-predictable phenotype of ear and tail necrosis. This necrosis pattern is not unlike that seen in less-severe SMA mouse models (34) or in mice treated with less-potent activators of SMN (38). The patterns of necrosis could represent dysfunction in cell populations outside of the CNS that are not fully or continuously exposed to the targeting MO delivered by bolus ICV or FV. We have previously shown that high levels of SMN in neurons, but not in muscle, can increase the survival of Smn−/−; SMN2+/+ mice; however, the contribution of extra-neuronal SMN has not been fully elucidated (57). The necrosis of peripheral structures, together with priapism, urinary retention and bowel obstruction, all serve as clinical evidence supporting potential ANS dysfunction in SMA. A number of groups have reported cardiac bradyarrythmias, as well as dilated cardiomyopathy coupled with decreased contractility, in SMA mouse models (67,69,70), possibly due to an SMN-deficient sympathetic nervous system leading to autonomic imbalance. Interestingly, some of our adult MO-treated mice died suddenly before showing any evidence of disease morbidity (i.e. weight loss, necrosis); we postulate that such sudden and unpredictable mortality may be due to acute cardiopulmonary failure resulting from autonomic dysfunction. There is increasing human clinical evidence of ANS imbalance in SMA. Retrospective reports describe an elevated incidence of cardiac dysrhythmia and wall-motion abnormalities in patients with type I SMA (71–73). Autonomic testing, including skin vessel vasodilatation response and circulating norepinephrine levels, also suggest autonomic dysfunction (74). Morphologic and functional cardiac evaluation of MO-injected mice will help elucidate the contribution of autonomic dysfunction on animal demise.
P0 injection of MO produced a striking increase in survival, delayed ICV injection had intermediate survival and delayed FV injection yielded only mild rescue. These results again imply the importance of early CNS elevations of SMN (37,44,57,68). Previous examination of neuronal morphology in severe SMA embryos (Smn−/−, SMN2+/+) showed decreased synapse occupation by motor axons, suggesting that SMN is a critical component for early synapse development and maturation (28). Temporal induction of SMN in SMA mice has defined a window of increased SMN production for correction of the SMA phenotype (37,68). Therefore, a period during neonatal growth in the mouse constitutes a critical time for high SMN production. It is clear that any potential SMA therapeutic will have maximum effect when given as early as possible.
Strategies for increasing SMN and altering the course of SMA are proliferating, including ASOs, viral therapy with ScAAV9-SMN and drug compounds that induce SMN. The latter modality has the simplicity of application and the ability to ensure target levels within tissue. Drawbacks include non-specific targeting and resultant potential toxicity. Viral therapy has the distinct advantage of having the longest laboratory survival increase of all interventions and the need for only a single vascular delivery (44). Disadvantages include the hurdles of large-scale production, as well as a patient subpopulation that is seropositive for the AAV9 capsid. ASOs are maturing as potential disease modifiers in SMA and other neuromuscular disease. MOs have notable efficacy through exon skipping in DMD (46,49), showing low toxicity, high specificity and excellent body-wide distribution after systemic administration (46,60,61,63,75). Disadvantages of ASO therapy for SMA include the probability of invasive repeat CNS dosing.
Our study has shown that a single CNS bolus of MO targeting ISS-N1 of SMN2 can strongly modulate splicing mechanics, increase SMN protein and increase the survival of SMNΔ7 SMA mice to a greater extent than other previously trialed ASO chemistries. Furthermore, MOs appear to have little or no toxicity when delivered at high doses within the CNS. This therapy has the potential for translation into clinical trials with early administration of MO via intrathecal delivery.
Continued refinement of dosing (when, where, how much?) should continue in order to optimize delivery for future clinical trials. A large animal model of SMA, such as the pig, could be used to both titrate the optimal dosing and to truly evaluate the efficacy of peripherally administered ASO therapy for SMA in the setting of an intact blood-brain barrier. It is clear that earlier correction of SMN protein levels has more profound effects than delayed increases, emphasizing the importance of early SMA detection and intervention in future clinical trial design. Methods of newborn SMA detection have been developed but implementation presents complications (76), including whether an SMA therapy must first demonstrate efficacy in symptomatic patients before the start of widespread newborn screening. We believe that new data showing both profound disease modification with therapy, as well as the clear advantage of pre-morbid treatment initiation, advocate for the start of SMA neonatal screening.
MATERIALS AND METHODS
Generation of SMA mice
SMN▵7 carrier breeding mice (SMN2+/+; Smn+/−; SMNΔ7+/+) were crossed to generate three types of offspring varying in mouse Smn genotype: Smn+/+, Smn+/− and Smn−/− as described previously (36,39). All breeding and subsequent use of animals in this study were approved by the IACUC of The Ohio State University, Columbus, OH, USA.
ICV injections
The P0 or P4 pup was cryo-anesthetized and hand-mounted over a back-light to visualize the intersection of the coronal and sagittal cranial sutures (bregma) (55,58). A fine-drawn capillary needle with injection assembly was inserted 1 mm lateral and 1 mm posterior to bregma, and then tunneled 1 mm deep to the skin edge (approximating) ipsilateral lateral ventricle. An opaque tracer (Evans Blue, 0.04%) was added to the reagent to visualize the borders of the lateral ventricle after injection of 2 μl of MO.
Stereotactic injections
P30 mice were anesthetized with inhalational isoflurane (3% induction, maintenance 1% mixed with high-flow 100% O2). The animal was placed into the cranial stereotactic frame (Kopf Instruments) with digital coordinate guidance (myNeuroLab), and the anesthesia nose cone was secured. The cranial apex was sterilized and a short midline incision was made with visualization of bregma and lambda. A small burr hole was drilled and cranial needle with attached Hamilton syringe was guided to preselected coordinates (A/P 0.58 mm, D/L 2.15 mm, M/L 1.10 mm) for right lateral ventricle cannulation (77); the coordinates were validated by injection of scAAV4-GFP (ependymal localization, Supplementary material, Fig. S3) in a trial P30 mouse. 18 μg/g of MO or scMO [equivalent to low dose (2 mm) injection in the P0 pups] was injected at a rate of 0.75 μl/min with digital microinjector (KD Scientific). After injection, the needle was withdrawn and skin closed with a running suture. Post-surgical care was approved by the IACUC of The Ohio State University, Columbus, OH, USA.
FV injection
FV injection on was performed as previously described (78). MO or scMO was dosed at 50 μg/g body mass.
Mouse genotyping
The SMN2, Smn knockout allele and SMNΔ7 alleles were genotyped as described previously (36,44,57). Tail snips were gathered at P0, and each pup was identified by paw tattooing. All genotyping was performed on P0 as described previously (44).
Behavioral analysis
All injected animals, as well as breeding pairs, were monitored each day for morbidity, mortality and new litters. Control and experimental animals were weighed each day, and righting response was assessed during the animals' first 21 days as described previously (79). The grip strength was recorded as described previously with correction for the weight of the animals (80).
RT–PCR and real-time RT–PCR analysis
RNA was isolated from Trizol (Invitrogen) homogenized tissue and purified with the RNeasy kit (Qiagen). RT–PCR was performed as described previously (57). The SMNΔ7 transgene lacks the terminal portion of exon 8. Primers were designed to amplify only the SMN2 transcripts that contain this region, thus distinguishing SMNΔ7 from SMN2: (hSMN2E8rev) TTATATACTTTTAAACATATAGAAGA TAG, (hSMNE6fwd) AGATTCTCTTGATGATGCTGAATG.
Real-time RT–PCR assayed for full-length SMN2 transcripts relative to cyclophilin. SMN2 amplification: (hSMNFull Fb) GTTTCAGACAAAATCAAAAAGAAGGA, (hSMNFull Rc) TCTATAACGCTTCACATTCCAGATCT, probe: (hSMNFull FAM) ATGCCAGCATTTCTCCT TAATTTAAGG. Cyclophilin: (QcycloF) GTCAACCCCACCGTGTTCTT, (QcycloR) TTGGAACTTTGTCTGCAAACA, probe: (Probecyclo NED) CTTGGGCCGCGTCT. PCR for SMN2 used 2 μl of cDNA and 0.6 μl (300 nm) of forward and reverse primers; cyclophilin, 1.8 μl (900 nm) forward and reverse primer. The transcript level was determined as described previously (81).
Digital droplet PCR
cDNA was collected as detailed above. Identical primers and probe were used for ddPCR as was used for real-time RT–PCR. PCR for SMN2 used 1.0 μl of cDNA and 0.1 μl of cyclophilin. 1.8 μl (900 nm) of forward and reverse primers was used for both SMN2 and cyclophilin. Droplet generation and reader analysis were performed on QuantaLife (www.quantalife.com/, now Bio-Rad) hardware. Ten to fifteen thousand droplets containing cDNA transcript and PCR reagents were generated before amplification. The concentration of transcripts was first calculated by the droplet reader using Poisson statistical distributions (65), and relative SMN2 levels were determined by calculating the ratio of SMN2 versus cyclophilin.
Western blot analysis
Western blot analysis was performed as described previously (36,37,57). Detection was performed using the LI-COR Odyssey Imaging System (Biosciences) and quantification was determined using Odyssey Infrared Imaging System Application Software (Biosciences).
MO ASO preparation
The MO sequence, numbered from the SMN2 exon 7 donor site (Fig. 1), was ATTCACTTTCATAATGCTGG (MWT = 6754, Gene Tools). The scMO sequence was TCCTTTAAAG TATTGTGACC (MWT = 6754, Gene Tools). MOs were resuspended in sterile 0.9% sodium chloride, aliquoted and mixed with Evans Blue (final concentration 0.04%). Three different molar concentrations were prepared (high: 6 mm = 40.5 μg/μl; middle: 4 mm = 27 μg/μl; low: 2 mm = 13.5 μg/μl). Stock solutions were stored at −20°C, and working solutions were stored at 4°C. Lissamine-tagged MO (sequence CCTCTTACCTCAGTTACAATTTATA) was resuspended to 2 mm in 0.9% NaCl. Two microliters of MO oligomer (2 μl) were injected, yielding total doses per animal of 81 μg (high), 54 μg (middle) and 27 μg (low).
SMN immunofluorescence
SMNΔ7 SMA mice (SMN2+/+; HB9:GFP; Smn−/−; SMNΔ7+/+) were injected by P0 ICV with 4 mm MO. Carrier control was not injected. Spinal cords were harvested, frozen, fixed and sectioned at P7 as described previously (37). Tissue sections were stained with anti-human SMN KH antibody 1:10 overnight and Alexa Fluor® 594 goat anti-rabbit IgG (Molecular probes) (1:1000). Endogenous lissamine and GFP fluorescence were imaged with a Nikon E800 Eclipse fluorescent microscope, Ultrapix Digital Camera (Olympus) with MagnaFIRE v2.1C software (Optronics), and further processed with Adobe Photoshop CS2.
Statistical analysis
Data are expressed as means ± standard errors. Kaplan–Meier curves were generated from the survival data and tested using the Mantel–Cox log-rank test. All statistical analyses were performed using SPSS, v.16.0.
SUPPLEMENTARY MATERIAL
AUTHORS’ ROLES
P.N.P., A.H.M.B. and C.M. designed and executed experiments. S.W. and B.K. provided conceptual experimental design. V.M. performed animal perfusion, immunohistochemistry and manuscript revision. A.B. and K.F. performed immunohistochemistry for stereotactic coordinate confirmation. P.N.P. and A.H.M.B. wrote the manuscript.
FUNDING
This work was supported by the National Institutes of Health R01 HD060586 (A.H.M.B.) and RC2 NS069476 (A.H.M.B.); The Ohio State University Department of Neurological Surgery (P.N.P.); AFM/SMA Europe (S.W.), the Spinal Muscular Atrophy Association of Australia (S.W.) and the Parry Foundation of Australia (S.W.).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Yimin Hua and Dr Adrian Krainer for the generous gift of human specific KH SMN antibody and interesting discussions, as well as Aurelie Massoni-Laporte for help on the real-time RT-PCR primer design and use.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Roberts D.F., Chavez J., Court S.D. The genetic component in child mortality. Arch. Dis. Child. 1970;45:33–38. doi: 10.1136/adc.45.239.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crawford T.O., Pardo C.A. The neurobiology of childhood spinal muscular atrophy. Neurobiol. Dis. 1996;3:97–110. doi: 10.1006/nbdi.1996.0010. [DOI] [PubMed] [Google Scholar]
- 3.Lefebvre S., Burglen L., Reboullet S., Clermont O., Burlet P., Viollet L., Benichou B., Cruaud C., Millasseau P., Zeviani M., et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 4.Burghes A.H., Beattie C.E. Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat. Rev. Neurosci. 2009;10:597–609. doi: 10.1038/nrn2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parsons D.W., McAndrew P.E., Monani U.R., Mendell J.R., Burghes A.H., Prior T.W. An 11 base pair duplication in exon 6 of the SMN gene produces a type I spinal muscular atrophy (SMA) phenotype: further evidence for SMN as the primary SMA-determining gene. Hum. Mol. Genet. 1996;5:1727–1732. doi: 10.1093/hmg/5.11.1727. [DOI] [PubMed] [Google Scholar]
- 6.Kashima T., Manley J.L. A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat. Genet. 2003;34:460–463. doi: 10.1038/ng1207. [DOI] [PubMed] [Google Scholar]
- 7.Monani U.R., Lorson C.L., Parsons D.W., Prior T.W., Androphy E.J., Burghes A.H., McPherson J.D. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 1999;8:1177–1183. doi: 10.1093/hmg/8.7.1177. [DOI] [PubMed] [Google Scholar]
- 8.Lorson C.L., Hahnen E., Androphy E.J., Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl Acad. Sci. USA. 1999;96:6307–6311. doi: 10.1073/pnas.96.11.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cartegni L., Krainer A.R. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat. Genet. 2002;30:377–384. doi: 10.1038/ng854. [DOI] [PubMed] [Google Scholar]
- 10.Gennarelli M., Lucarelli M., Capon F., Pizzuti A., Merlini L., Angelini C., Novelli G., Dallapiccola B. Survival motor neuron gene transcript analysis in muscles from spinal muscular atrophy patients. Biochem. Biophys. Res. Commun. 1995;213:342–348. doi: 10.1006/bbrc.1995.2135. [DOI] [PubMed] [Google Scholar]
- 11.Burnett B.G., Munoz E., Tandon A., Kwon D.Y., Sumner C.J., Fischbeck K.H. Regulation of SMN protein stability. Mol. Cell Biol. 2009;29:1107–1115. doi: 10.1128/MCB.01262-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lorson C.L., Strasswimmer J., Yao J.M., Baleja J.D., Hahnen E., Wirth B., Le T., Burghes A.H., Androphy E.J. SMN oligomerization defect correlates with spinal muscular atrophy severity. Nat. Genet. 1998;19:63–66. doi: 10.1038/ng0598-63. [DOI] [PubMed] [Google Scholar]
- 13.Lorson C.L., Androphy E.J. An exonic enhancer is required for inclusion of an essential exon in the SMA-determining gene SMN. Hum. Mol. Genet. 2000;9:259–265. doi: 10.1093/hmg/9.2.259. [DOI] [PubMed] [Google Scholar]
- 14.Burghes A.H. When is a deletion not a deletion? When it is converted. Am. J. Hum. Genet. 1997;61:9–15. doi: 10.1086/513913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McAndrew P.E., Parsons D.W., Simard L.R., Rochette C., Ray P.N., Mendell J.R., Prior T.W., Burghes A.H. Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am. J. Hum. Genet. 1997;60:1411–1422. doi: 10.1086/515465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prior T.W., Krainer A.R., Hua Y., Swoboda K.J., Snyder P.C., Bridgeman S.J., Burghes A.H., Kissel J.T. A positive modifier of spinal muscular atrophy in the SMN2 gene. Am. J. Hum. Genet. 2009;85:408–413. doi: 10.1016/j.ajhg.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vezain M., Saugier-Veber P., Goina E., Touraine R., Manel V., Toutain A., Fehrenbach S., Frebourg T., Pagani F., Tosi M., et al. A rare SMN2 variant in a previously unrecognized composite splicing regulatory element induces exon 7 inclusion and reduces the clinical severity of spinal muscular atrophy. Hum. Mutat. 2010;31:E1110–E1125. doi: 10.1002/humu.21173. [DOI] [PubMed] [Google Scholar]
- 18.Sun Y., Grimmler M., Schwarzer V., Schoenen F., Fischer U., Wirth B. Molecular and functional analysis of intragenic SMN1 mutations in patients with spinal muscular atrophy. Hum. Mutat. 2005;25:64–71. doi: 10.1002/humu.20111. [DOI] [PubMed] [Google Scholar]
- 19.Workman E., Saieva L., Carrel T.L., Crawford T.O., Liu D., Lutz C., Beattie C.E., Pellizzoni L., Burghes A.H. A SMN missense mutation complements SMN2 restoring snRNPs and rescuing SMA mice. Hum. Mol. Genet. 2009;18:2215–2229. doi: 10.1093/hmg/ddp157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meister G., Buhler D., Pillai R., Lottspeich F., Fischer U. A multiprotein complex mediates the ATP-dependent assembly of spliceosomal U snRNPs. Nat. Cell Biol. 2001;3:945–949. doi: 10.1038/ncb1101-945. [DOI] [PubMed] [Google Scholar]
- 21.Pellizzoni L., Yong J., Dreyfuss G. Essential role for the SMN complex in the specificity of snRNP assembly. Science. 2002;298:1775–1779. doi: 10.1126/science.1074962. [DOI] [PubMed] [Google Scholar]
- 22.Pellizzoni L. Chaperoning ribonucleoprotein biogenesis in health and disease. EMBO Rep. 2007;8:340–345. doi: 10.1038/sj.embor.7400941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schrank B., Gotz R., Gunnersen J.M., Ure J.M., Toyka K.V., Smith A.G., Sendtner M. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc. Natl Acad. Sci. USA. 1997;94:9920–9925. doi: 10.1073/pnas.94.18.9920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rossoll W., Kroning A.K., Ohndorf U.M., Steegborn C., Jablonka S., Sendtner M. Specific interaction of Smn, the spinal muscular atrophy determining gene product, with hnRNP-R and gry-rbp/hnRNP-Q: a role for Smn in RNA processing in motor axons? Hum. Mol. Genet. 2002;11:93–105. doi: 10.1093/hmg/11.1.93. [DOI] [PubMed] [Google Scholar]
- 25.Rossoll W., Jablonka S., Andreassi C., Kroning A.K., Karle K., Monani U.R., Sendtner M. Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of beta-actin mRNA in growth cones of motoneurons. J. Cell Biol. 2003;163:801–812. doi: 10.1083/jcb.200304128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McWhorter M.L., Monani U.R., Burghes A.H., Beattie C.E. Knockdown of the survival motor neuron (Smn) protein in zebrafish causes defects in motor axon outgrowth and pathfinding. J. Cell Biol. 2003;162:919–931. doi: 10.1083/jcb.200303168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carrel T.L., McWhorter M.L., Workman E., Zhang H., Wolstencroft E.C., Lorson C., Bassell G.J., Burghes A.H., Beattie C.E. Survival motor neuron function in motor axons is independent of functions required for small nuclear ribonucleoprotein biogenesis. J. Neurosci. 2006;26:11014–11022. doi: 10.1523/JNEUROSCI.1637-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McGovern V.L., Gavrilina T.O., Beattie C.E., Burghes A.H. Embryonic motor axon development in the severe SMA mouse. Hum. Mol. Genet. 2008;17:2900–2909. doi: 10.1093/hmg/ddn189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gabanella F., Butchbach M.E., Saieva L., Carissimi C., Burghes A.H., Pellizzoni L. Ribonucleoprotein assembly defects correlate with spinal muscular atrophy severity and preferentially affect a subset of spliceosomal snRNPs. PLoS One. 2007;2:e921. doi: 10.1371/journal.pone.0000921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Z., Lotti F., Dittmar K., Younis I., Wan L., Kasim M., Dreyfuss G. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell. 2008;133:585–600. doi: 10.1016/j.cell.2008.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baumer D., Lee S., Nicholson G., Davies J.L., Parkinson N.J., Murray L.M., Gillingwater T.H., Ansorge O., Davies K.E., Talbot K. Alternative splicing events are a late feature of pathology in a mouse model of spinal muscular atrophy. PLoS Genet. 2009;5:e1000773. doi: 10.1371/journal.pgen.1000773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coovert D.D., Le T.T., McAndrew P.E., Strasswimmer J., Crawford T.O., Mendell J.R., Coulson S.E., Androphy E.J., Prior T.W., Burghes A.H. The survival motor neuron protein in spinal muscular atrophy. Hum. Mol. Genet. 1997;6:1205–1214. doi: 10.1093/hmg/6.8.1205. [DOI] [PubMed] [Google Scholar]
- 33.Lefebvre S., Burlet P., Liu Q., Bertrandy S., Clermont O., Munnich A., Dreyfuss G., Melki J. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat. Genet. 1997;16:265–269. doi: 10.1038/ng0797-265. [DOI] [PubMed] [Google Scholar]
- 34.Hsieh-Li H.M., Chang J.G., Jong Y.J., Wu M.H., Wang N.M., Tsai C.H., Li H. A mouse model for spinal muscular atrophy. Nat. Genet. 2000;24:66–70. doi: 10.1038/71709. [DOI] [PubMed] [Google Scholar]
- 35.Monani U.R., Sendtner M., Coovert D.D., Parsons D.W., Andreassi C., Le T.T., Jablonka S., Schrank B., Rossoll W., Prior T.W., et al. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(−/−) mice and results in a mouse with spinal muscular atrophy. Hum. Mol. Genet. 2000;9:333–339. doi: 10.1093/hmg/9.3.333. [DOI] [PubMed] [Google Scholar]
- 36.Le T.T., Pham L.T., Butchbach M.E., Zhang H.L., Monani U.R., Coovert D.D., Gavrilina T.O., Xing L., Bassell G.J., Burghes A.H. SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum. Mol. Genet. 2005;14:845–857. doi: 10.1093/hmg/ddi078. [DOI] [PubMed] [Google Scholar]
- 37.Le T.T., McGovern V.L., Alwine I.E., Wang X., Massoni-Laporte A., Rich M.M., Burghes A.H. Temporal requirement for high SMN expression in SMA mice. Hum. Mol. Genet. 2011;20:3578–3591. doi: 10.1093/hmg/ddr275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Avila A.M., Burnett B.G., Taye A.A., Gabanella F., Knight M.A., Hartenstein P., Cizman Z., Di Prospero N.A., Pellizzoni L., Fischbeck K.H., et al. Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy. J. Clin. Invest. 2007;117:659–671. doi: 10.1172/JCI29562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Butchbach M.E., Singh J., Thorsteinsdottir M., Saieva L., Slominski E., Thurmond J., Andresson T., Zhang J., Edwards J.D., Simard L.R., et al. Effects of 2,4-diaminoquinazoline derivatives on SMN expression and phenotype in a mouse model for spinal muscular atrophy. Hum. Mol. Genet. 2010;19:454–467. doi: 10.1093/hmg/ddp510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Farooq F., Molina F.A., Hadwen J., Mackenzie D., Witherspoon L., Osmond M., Holcik M., Mackenzie A. Prolactin increases SMN expression and survival in a mouse model of severe spinal muscular atrophy via the STAT5 pathway. J. Clin. Invest. 2011;121:3042–3050. doi: 10.1172/JCI46276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hastings M.L., Berniac J., Liu Y.H., Abato P., Jodelka F.M., Barthel L., Kumar S., Dudley C., Nelson M., Larson K., et al. Tetracyclines that promote SMN2 exon 7 splicing as therapeutics for spinal muscular atrophy. Sci. Transl. Med. 2009;1:5ra12. doi: 10.1126/scitranslmed.3000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dominguez E., Marais T., Chatauret N., Benkhelifa-Ziyyat S., Duque S., Ravassard P., Carcenac R., Astord S., Pereira de Moura A., Voit T., et al. Intravenous scAAV9 delivery of a codon-optimized SMN1 sequence rescues SMA mice. Hum. Mol. Genet. 2009;20:681–693. doi: 10.1093/hmg/ddq514. [DOI] [PubMed] [Google Scholar]
- 43.Valori C.F., Ning K., Wyles M., Mead R.J., Grierson A.J., Shaw P.J., Azzouz M. Systemic delivery of scAAV9 expressing SMN prolongs survival in a model of spinal muscular atrophy. Sci. Transl. Med. 2010;2:35ra42. doi: 10.1126/scitranslmed.3000830. [DOI] [PubMed] [Google Scholar]
- 44.Foust K.D., Wang X., McGovern V.L., Braun L., Bevan A.K., Haidet A.M., Le T.T., Morales P.R., Rich M.M., Burghes A.H., et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat. Biotechnol. 2010;28:271–274. doi: 10.1038/nbt.1610. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 45.Baughan T.D., Dickson A., Osman E.Y., Lorson C.L. Delivery of bifunctional RNAs that target an intronic repressor and increase SMN levels in an animal model of spinal muscular atrophy. Hum. Mol. Genet. 2009;18:1600–1611. doi: 10.1093/hmg/ddp076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wilton S.D., Lloyd F., Carville K., Fletcher S., Honeyman K., Agrawal S., Kole R. Specific removal of the nonsense mutation from the mdx dystrophin mRNA using antisense oligonucleotides. Neuromuscul. Disord. 1999;9:330–338. doi: 10.1016/s0960-8966(99)00010-3. [DOI] [PubMed] [Google Scholar]
- 47.Burghes A.H., McGovern V.L. Antisense oligonucleotides and spinal muscular atrophy: skipping along. Genes Dev. 2010;24:1574–1579. doi: 10.1101/gad.1961710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Williams J.H., Schray R.C., Patterson C.A., Ayitey S.O., Tallent M.K., Lutz G.J. Oligonucleotide-mediated survival of motor neuron protein expression in CNS improves phenotype in a mouse model of spinal muscular atrophy. J. Neurosci. 2009;29:7633–7638. doi: 10.1523/JNEUROSCI.0950-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alter J., Lou F., Rabinowitz A., Yin H., Rosenfeld J., Wilton S.D., Partridge T.A., Lu Q.L. Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nat. Med. 2006;12:175–177. doi: 10.1038/nm1345. [DOI] [PubMed] [Google Scholar]
- 50.Morcos P.A. Achieving targeted and quantifiable alteration of mRNA splicing with morpholino oligos. Biochem. Biophys. Res. Commun. 2007;358:521–527. doi: 10.1016/j.bbrc.2007.04.172. [DOI] [PubMed] [Google Scholar]
- 51.Bebee T.W., Gladman J.T., Chandler D.S. Splicing regulation of the survival motor neuron genes and implications for treatment of spinal muscular atrophy. Front Biosci. 2011;15:1191–1204. doi: 10.2741/3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Singh N.K., Singh N.N., Androphy E.J., Singh R.N. Splicing of a critical exon of human survival motor neuron is regulated by a unique silencer element located in the last intron. Mol. Cell Biol. 2006;26:1333–1346. doi: 10.1128/MCB.26.4.1333-1346.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hua Y., Sahashi K., Hung G., Rigo F., Passini M.A., Bennett C.F., Krainer A.R. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 2010;24:1634–1644. doi: 10.1101/gad.1941310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hua Y., Vickers T.A., Okunola H.L., Bennett C.F., Krainer A.R. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am. J. Hum. Genet. 2008;82:834–848. doi: 10.1016/j.ajhg.2008.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Passini M.A., Bu J., Richards A.M., Kinnecom C., Sardi S.P., Stanek L.M., Hua Y., Rigo F., Matson J., Hung G., et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci. Transl. Med. 2011;3:72ra18. doi: 10.1126/scitranslmed.3001777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hua Y., Sahashi K., Rigo F., Hung G., Horev G., Bennett C.F., Krainer A.R. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature. 2011;478:123–126. doi: 10.1038/nature10485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gavrilina T.O., McGovern V.L., Workman E., Crawford T.O., Gogliotti R.G., DiDonato C.J., Monani U.R., Morris G.E., Burghes A.H. Neuronal SMN expression corrects spinal muscular atrophy in severe SMA mice while muscle-specific SMN expression has no phenotypic effect. Hum. Mol. Genet. 2008;17:1063–1075. doi: 10.1093/hmg/ddm379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Passini M.A., Bu J., Roskelley E.M., Richards A.M., Sardi S.P., O'Riordan C.R., Klinger K.W., Shihabuddin L.S., Cheng S.H. CNS-targeted gene therapy improves survival and motor function in a mouse model of spinal muscular atrophy. J. Clin. Invest. 2010;120:1253–1264. doi: 10.1172/JCI41615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ellett F., Lieschke G.J. Zebrafish as a model for vertebrate hematopoiesis. Curr. Opin. Pharmacol. 2010;10:563–570. doi: 10.1016/j.coph.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 60.Wu B., Xiao B., Cloer C., Shaban M., Sali A., Lu P., Li J., Nagaraju K., Xiao X., Lu Q.L. One-year treatment of morpholino antisense oligomer improves skeletal and cardiac muscle functions in dystrophic mdx mice. Mol. Ther. 2011;19:576–583. doi: 10.1038/mt.2010.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yokota T., Lu Q.L., Partridge T., Kobayashi M., Nakamura A., Takeda S., Hoffman E. Efficacy of systemic morpholino exon-skipping in Duchenne dystrophy dogs. Ann Neurol. 2009;65:667–676. doi: 10.1002/ana.21627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kinali M., Arechavala-Gomeza V., Feng L., Cirak S., Hunt D., Adkin C., Guglieri M., Ashton E., Abbs S., Nihoyannopoulos P., et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009;8:918–928. doi: 10.1016/S1474-4422(09)70211-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cirak S., Arechavala-Gomeza V., Guglieri M., Feng L., Torelli S., Anthony K., Abbs S., Garralda M.E., Bourke J., Wells D.J., et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet. 2011;378:595–605. doi: 10.1016/S0140-6736(11)60756-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sazani P., Van Ness K.P., Weller D.L., Poage D.W., Palyada K., Shrewsbury S.B. Repeat-Dose Toxicology Evaluation in Cynomolgus Monkeys of AVI-4658, a Phosphorodiamidate Morpholino Oligomer (PMO) Drug for the Treatment of Duchenne Muscular Dystrophy. Int. J. Toxicol. 2011;30:313–321. doi: 10.1177/1091581811403505. [DOI] [PubMed] [Google Scholar]
- 65.Zhong Q., Bhattacharya S., Kotsopoulos S., Olson J., Taly V., Griffiths A.D., Link D.R., Larson J.W. Multiplex digital PCR: breaking the one target per color barrier of quantitative PCR. Lab Chip. 2011;11:2167–2174. doi: 10.1039/c1lc20126c. [DOI] [PubMed] [Google Scholar]
- 66.Wichterle H., Lieberam I., Porter J.A., Jessell T.M. Directed differentiation of embryonic stem cells into motor neurons. Cell. 2002;110:385–397. doi: 10.1016/s0092-8674(02)00835-8. [DOI] [PubMed] [Google Scholar]
- 67.Bevan A.K., Hutchinson K.R., Foust K.D., Braun L., McGovern V.L., Schmelzer L., Ward J.G., Petruska J.C., Lucchesi P.A., Burghes A.H., et al. Early heart failure in the SMN{Delta}7 model of spinal muscular atrophy and correction by postnatal scAAV9-SMN delivery. Hum. Mol. Genet. 2010;19:3895–3905. doi: 10.1093/hmg/ddq300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lutz C.M., Kariya S., Patruni S., Osborne M.A., Liu D., Henderson C.E., Li D.K., Pellizzoni L., Rojas J., Valenzuela D.M., et al. Postsymptomatic restoration of SMN rescues the disease phenotype in a mouse model of severe spinal muscular atrophy. J. Clin. Invest. 2011;121:3029–3041. doi: 10.1172/JCI57291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Heier C.R., Satta R., Lutz C., DiDonato C.J. Arrhythmia and cardiac defects are a feature of spinal muscular atrophy model mice. Hum. Mol. Genet. 2010;19:3906–3918. doi: 10.1093/hmg/ddq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shababi M., Habibi J., Yang H.T., Vale S.M., Sewell W.A., Lorson C.L. Cardiac defects contribute to the pathology of spinal muscular atrophy models. Hum. Mol. Genet. 2010;19:4059–4071. doi: 10.1093/hmg/ddq329. [DOI] [PubMed] [Google Scholar]
- 71.Bach J.R. Medical considerations of long-term survival of Werdnig–Hoffmann disease. Am. J. Phys. Med. Rehabil. 2007;86:349–355. doi: 10.1097/PHM.0b013e31804b1d66. [DOI] [PubMed] [Google Scholar]
- 72.Hachiya Y., Arai H., Hayashi M., Kumada S., Furushima W., Ohtsuka E., Ito Y., Uchiyama A., Kurata K. Autonomic dysfunction in cases of spinal muscular atrophy type 1 with long survival. Brain Dev. 2005;27:574–578. doi: 10.1016/j.braindev.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 73.Finsterer J., Stollberger C. Cardiac involvement in Werdnig–Hoffmann's spinal muscular atrophy. Cardiology. 1999;92:178–182. doi: 10.1159/000006968. [DOI] [PubMed] [Google Scholar]
- 74.Arai H., Tanabe Y., Hachiya Y., Otsuka E., Kumada S., Furushima W., Kohyama J., Yamashita S., Takanashi J., Kohno Y. Finger cold-induced vasodilatation, sympathetic skin response, and R-R interval variation in patients with progressive spinal muscular atrophy. J. Child Neurol. 2005;20:871–875. doi: 10.1177/08830738050200110301. [DOI] [PubMed] [Google Scholar]
- 75.Wu B., Benrashid E., Lu P., Cloer C., Zillmer A., Shaban M., Lu Q.L. Targeted skipping of human dystrophin exons in transgenic mouse model systemically for antisense drug development. PLoS ONE. 2011;6:e19906. doi: 10.1371/journal.pone.0019906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Prior T.W., Snyder P.J., Rink B.D., Pearl D.K., Pyatt R.E., Mihal D.C., Conlan T., Schmalz B., Montgomery L., Ziegler K., et al. Newborn and carrier screening for spinal muscular atrophy. Am. J. Med. Genet. A. 2010;152A:1608–1616. doi: 10.1002/ajmg.a.33474. [DOI] [PubMed] [Google Scholar]
- 77.Paxinos G., Franklin K.B.J. The Mouse Brain in Stereotactic Coordinates: Compact Second Edition. 2nd edn. San Diego, CA, USA: Elsevier Science; 2004. [Google Scholar]
- 78.Foust K.D., Nurre E., Montgomery C.L., Hernandez A., Chan C.M., Kaspar B.K. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol. 2009;27:59–65. doi: 10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Butchbach M.E., Edwards J.D., Burghes A.H. Abnormal motor phenotype in the SMNDelta7 mouse model of spinal muscular atrophy. Neurobiol. Dis. 2007;27:207–219. doi: 10.1016/j.nbd.2007.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Monani U.R., Pastore M.T., Gavrilina T.O., Jablonka S., Le T.T., Andreassi C., DiCocco J.M., Lorson C., Androphy E.J., Sendtner M., et al. A transgene carrying an A2G missense mutation in the SMN gene modulates phenotypic severity in mice with severe (type I) spinal muscular atrophy. J. Cell Biol. 2003;160:41–52. doi: 10.1083/jcb.200208079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Heier C.R., Gogliotti R.G., DiDonato C.J. SMN transcript stability: could modulation of messenger RNA degradation provide a novel therapy for spinal muscular atrophy? J. Child Neurol. 2007;22:1013–1018. doi: 10.1177/0883073807305669. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.