

Abstract
Background
Mendelian analysis of disorders of immune regulation can provide insight into molecular pathways associated with host defense and immune tolerance.
Methods
We identified three families with a dominantly inherited complex of cold-induced urticaria, antibody deficiency, and susceptibility to infection and autoimmunity. Immunophenotyping methods included flow cytometry, analysis of serum immunoglobulins and autoantibodies, lymphocyte stimulation, and enzymatic assays. Genetic studies included linkage analysis, targeted Sanger sequencing, and next-generation whole-genome sequencing.
Results
Cold urticaria occurred in all affected subjects. Other, variable manifestations included atopy, granulomatous rash, autoimmune thyroiditis, the presence of antinuclear antibodies, sinopulmonary infections, and common variable immunodeficiency. Levels of serum IgM and IgA and circulating natural killer cells and class-switched memory B cells were reduced. Linkage analysis showed a 7-Mb candidate interval on chromosome 16q in one family, overlapping by 3.5 Mb a disease-associated haplotype in a smaller family. This interval includes PLCG2, encoding phospholipase Cγ2 (PLCγ2), a signaling molecule expressed in B cells, natural killer cells, and mast cells. Sequencing of complementary DNA revealed heterozygous transcripts lacking exon 19 in two families and lacking exons 20 through 22 in a third family. Genomic sequencing identified three distinct in-frame deletions that cosegregated with disease. These deletions, located within a region encoding an autoinhibitory domain, result in protein products with constitutive phospholipase activity. PLCG2-expressing cells had diminished cellular signaling at 37°C but enhanced signaling at subphysiologic temperatures.
Conclusions
Genomic deletions in PLCG2 cause gain of PLCγ2 function, leading to signaling abnormalities in multiple leukocyte subsets and a phenotype encompassing both excessive and deficient immune function. (Funded by the National Institutes of Health Intramural Research Programs and others.)
The genetic dissection of unique inflammatory phenotypes can identify and elucidate immunologic pathways and mechanisms. Such investigations have ultimately led to findings whose significance extends beyond the monogenic diseases harboring the mutations. Examples include the recognition that FOXP3 is essential for the differentiation of regulatory T cells in Scurfy mice and in patients with profound immune dysregulation,1-4 the demonstration of a critical role for AIRE in thymic negative selection of T cells in patients with a specific autoimmune polyendocrinopathy,5 and the identification of NLRP3 as a critical regulator of interleukin-1 in families with cold-induced inflammation.6
Cold-induced urticaria is a unique inflammatory phenotype that is characterized by mast-cell degranulation and triggered by exposure to cold stimuli, a condition that can culminate in life-threatening anaphylaxis.7,8 There are various forms of cold urticaria that differ with respect to the mode of inheritance, ages at onset and resolution, and responses to a range of cold provocative testing.7 Although mast cells have been clearly implicated in the pathogenesis of cold urticaria,9 the processes leading to the degranulation of such cells are poorly understood. We describe three families with cold urticaria, the genetic causes of their disease, and the implications of these findings for the understanding of disease mechanisms.
Methods
Study Subjects
Subjects were admitted to the National Institutes of Health Clinical Center, where they were enrolled in clinical protocols approved by the institutional review board of the National Institute of Allergy and Infectious Diseases. All subjects provided written informed consent.
Genetic Analysis
We isolated genomic DNA from whole blood of members of two families and genotyped single-nucleotide polymorphisms (SNPs). We performed parametric multipoint linkage and haplotype analysis independently in each family, using Merlin software.10 We carried out whole-genome sequencing in one affected member of Family 1, as described previously.11 We screened the coding exons, splice junctions, and complementary DNA (cDNA) of PLCG2 for mutations by means of polymerase-chain-reaction (PCR) amplification of overlapping segments and then sequencing of the products using Sanger's method. We mapped genomic deletions using long-range PCR amplification and Sanger sequencing. Additional data on assays, reagents, and primer sequences are provided in the Supplementary Appendix, available with the full text of this article at NEJM.org.
Evaluation of Phospholipase cγ2 Function
We assayed the enzymatic activity of phospholipase Cγ2 (PLCγ2) in a COS-7–cell transfection system.12 We measured intracellular calcium flux in peripheral-blood lymphocytes13 and determined degranulation of natural killer cells14 using flow cytometry. We quantified B-cell class switching in cultured peripheral-blood mononuclear cells ex vivo.15 Secondary recombination in B cells was measured by sequencing the Igκ transcript of single CD19+CD10+IgMhiCD27– transitional B cells obtained by fluorescence-activated cell sorting.16 We measured the effect of temperature on the intracellular calcium concentration in negatively selected, column-sorted, FLUO-4–stained, unstimulated B cells from the study subjects, using confocal microscopy. Laboratory of Allergic Diseases 2 (LAD2) human mast cells were transfected with mutant PLCG2 cDNA fused to green fluorescent protein, and transfected cells were then assayed for degranulation by means of flow cytometry. Additional information about assays, reagents, and statistical techniques is provided in the Supplementary Appendix.
Results
Disease Manifestations
We identified three independent families of European ancestry with cold urticaria and variable immune defects (Table 1, and Table S1 in the Supplementary Appendix). The cold urticaria developed at a very young age and was lifelong; the cold-related symptoms in one of these families have been described previously.7 The study subjects differed from patients with typical cold urticaria in that they had negative results on skin testing with ice-cube and cold-water immersion. However, they had positive results on skin testing for evaporative cooling (Fig. 1A, and Fig. S1 in the Supplementary Appendix) and generalized exposure to cold air, findings that are consistent with their reports that symptoms were often elicited by cold wind rather than contact with cold objects.
Table 1. Summary of the Clinical Manifestations of Phospholipase Cγ2–Associated Antibody Deficiency and Immune Dysregulation in the Subjects.
| Clinical Manifestation | Frequency no./total no. (%) |
|---|---|
| Cold urticaria | 27/27 (100) |
| Recurrent sinopulmonary infection | 12/27 (44) |
| Antibody deficiency* | 15/20 (75) |
| Common variable immunodeficiency | 3/27 (11) |
| Symptomatic autoimmune disease† | 7/27 (26) |
| Positive test for antinuclear antibodies‡ | 13/21 (62) |
| Symptomatic allergic disease | 15/27 (56) |
Antibody deficiency was defined as a serum IgM, IgG, or IgA level that was more than 2 SD below the age-specific mean.
Symptomatic autoimmune disease was defined as the presence of any autoimmune disease, including vitiligo, autoimmune thyroiditis, inflammatory arthritis, and undifferentiated connective-tissue disease.
The test was weakly positive (1 to 3 enzyme-linked immunosorbent assay [ELISA] units) in 12 subjects and strongly positive (>3 ELISA units) in 1 subject.
Figure 1. Clinical and Immunologic Manifestations in Patients with Phospholipase Cγ2–Associated Antibody Deficiency and Immune Dysregulation (PLAID).
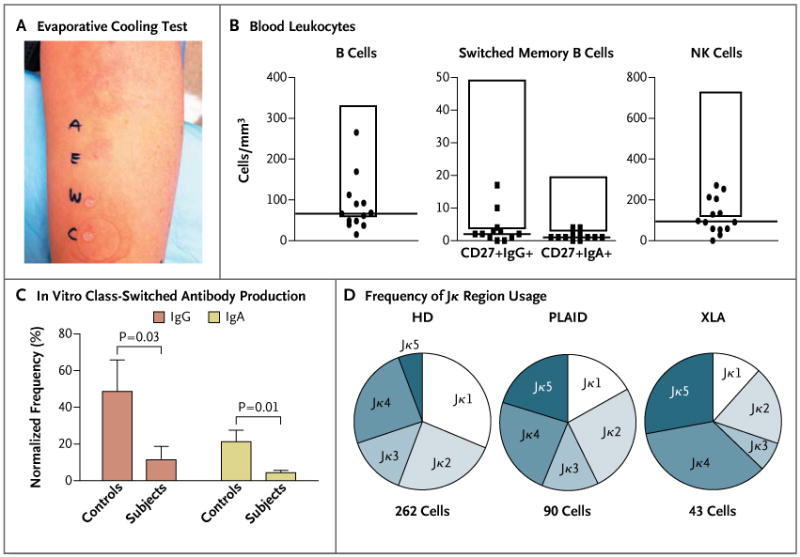
Panel A shows the results of an evaporative cooling test in one subject. Cold urticaria was provoked with droplets of ethanol (E) or air-blown water (A) but not with droplets of unblown water (W) or covered water (C). In Panel B, blood leukocyte counts are shown for 18 adult subjects with PLAID. The tops and the bottoms of the boxes represent a 2-SD range above and below the mean values in healthy control subjects, and the horizontal lines represent the median value among subjects with PLAID. NK denotes natural killer. Panel C shows the mean frequency of IgA or IgG antibody–secreting cells, measured by means of enzyme-linked immunospot assay (ELISPOT) after 4 days of expansion in culture. ELISPOT data were normalized to an equal number of B cells on the basis of the proportion of B cells in peripheral-blood mononuclear cells at the start of culture. The T bars indicate standard errors for five subjects from each group. In Panel D, Igκ secondary recombination in transitional B cells is shown for 12 healthy donors (HD), 4 subjects with PLAID, and 4 subjects with X-linked agammaglobulinemia (XLA), indicated by the frequency of Jκ region usage, as determined by single-cell Igκ sequence analysis of data from subjects who were pooled according to diagnosis. The increased usage of the terminal Jκ4 and Jκ5 genes, as seen in both PLAID and XLA, indicates impaired termination of secondary recombination.
Of the 27 subjects who were tested, 26 had immunologic abnormalities in addition to cold urticaria (Table S1 in the Supplementary Appendix). We detected antibody deficiency in 75% of subjects who were tested, with 56% reporting a history of recurrent infection; 3 subjects had received treatment with intravenous immune globulin for severe recurrent infection and hypogammaglobulinemia. Autoantibodies or autoimmune disease was present in 56% of subjects. Granulomatous disease was observed in 7 subjects from 2 unrelated families; 3 of these subjects were born with cutaneous lesions on the fingers, nose, and ears that disappeared during infancy. The remaining 4 subjects had persistent granulomatous skin disease, and 1 of these subjects had disfiguring granulomata with soft-palate and laryngeal involvement (Fig. S2 in the Supplementary Appendix).
Common laboratory findings in affected subjects included low levels of serum IgM and IgA, depressed levels of circulating CD19+ B cells and IgA+ and IgG+ class-switched memory B cells, and low levels of natural killer cells, whereas IgE levels were elevated in a majority of the subjects (Fig. 1B, and Table S1 and Fig. S3 in the Supplementary Appendix). Neutrophil, monocyte, eosinophil, and basophil counts were normal, as were numbers of naive and memory T cells (Fig. S3 in the Supplementary Appendix).
Of the 21 subjects who were tested for the presence of antinuclear antibodies, 13 (62%) had positive results (Table 1, and Table S1 in the Supplementary Appendix). B cells were characterized by poor in vitro expansion and reduced classswitched–antibody production after stimulation with Staphylococcus aureus Cowan I (SAC) and unmethylated cytosine–guanine dinucleotide (CpG)– containing oligonucleotide (Fig. 1C, and Fig. S4 in the Supplementary Appendix). Secondary recombination in CD19+CD10highIgMhighCD27– transitional B cells was increased, as shown by the skewing of light-chain gene usage toward the terminal elements, a characteristic that is also observed in Bruton's tyrosine kinase deficiency in patients with X-linked agammaglobulinemia,17 a disease that is associated with severe impairment in B-cell signaling (Fig. 1D, and Fig. S5 in the Supplementary Appendix). Unlike patients with X-linked agammaglobulinemia, who have few peripheral-blood mature B cells, subjects with PLCG2 mutations have a substantial mature B-cell compartment, which may lead to an increased occurrence of autoimmunity.
Linkage and Mutational Analysis
Linkage analysis based on the cold urticaria phenotype in Family 1 identified a single linkage interval of 7.7 Mb on chromosome 16q21 with a logarithm of odds (LOD) score of 4.2 (Fig. 2, and Fig. S6 in the Supplementary Appendix). Whole-genome sequencing of one affected member of Family 1 did not identify any novel coding or splice-site single-nucleotide variants, insertions, or deletions within this interval. The linkage interval was narrowed by an independent analysis of Family 2 (Fig. S7 in the Supplementary Appendix), which identified a 12.6-Mb haplotype that segregated with disease status and overlapped the linkage interval in Family 1 by 3.5 Mb (Fig. 2). Among the 24 genes contained within the narrowed candidate interval, we identified PLCG2 as the primary candidate gene (Table S2 in the Supplementary Appendix). The encoded protein, PLCγ2, is a member of the phosphoinositide-specific phospholipase C family. Members of this family propagate a wide range of extracellular signals by producing diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3) through hydrolysis of a phospholipid, phosphatidylinositol 4,5-bisphosphate (PIP2).18 The IP3 that is produced is responsible for calcium release from the endoplasmic reticulum, a critical event in cellular activation. There are two members of the PLCγ family, PLCγ1 and PLCγ2. Although PLCγ1 is expressed in many cell types, the Racresponsive PLCγ2 isoform predominates and appears to have nonredundant functions in B cells19 and natural killer cells,20 both of which had abnormalities in the three families in our study.
Figure 2. Genomic Deletions in PLCG2 Encoding Phospholipase Cγ2 in the Study Subjects.

Panel A shows the pedigrees of three families with PLAID. Solid symbols indicate affected subjects, and open symbols unaffected relatives. Squares indicate male subjects, and circles female subjects. Slashes indicate deceased subjects. The starred member in Family 3 died at the age of 1 year from pneumonia. Panel B shows the candidate interval on the long arm of chromosome 16, defined by the intersection of a linkage interval in Family 1 and candidate interval in Family 2. Three distinct PLCG2 deletions were identified in the three families (red horizontal bars).
Direct sequencing of cDNA from peripheral-blood mononuclear cells from members of Family 1 identified PLCG2 transcripts lacking exon 19 on one allele, a mutation that was present only in affected family members. Amplification of the genomic segment of PLCG2 that is flanked by exons 18 and 20 revealed a heterozygous deletion that cosegregated with cold urticaria (Fig. S8 in the Supplementary Appendix). By directly sequencing PCR products from overlapping segments flanking exon 19, we identified a deletion of 5.9 kb in PLCG2 (Fig. S8 in the Supplementary Appendix).
Using this same approach, we identified additional heterozygous deletions of PLCG2 in Families 2 and 3 (Fig. S8 in the Supplementary Appendix). In Family 2, we identified PLCG2 transcripts that lacked exons 20 through 22, caused by an 8.2-kb deletion. In Family 3, we identified PLCG2 transcripts that also lacked exon 19 but were caused by a 4.8-kb deletion with breakpoints distinct from those found in Family 1. These family-specific deletions were not detected in 200 healthy subjects of Northern European ancestry. A post hoc analysis of the whole-genome sequence from 1 member of Family 1 confirmed the 5.9-kb deletion that was initially discovered on Sanger sequencing (see the Methods section in the Supplementary Appendix). We propose the term PLAID (PLCγ2-associated antibody deficiency and immune dysregulation) to describe this unique constellation of clinical, genetic, and functional findings.
Functional Studies of Mutant PLCγ2
Each of the three deletions that were identified in these families involved the C-terminal Src-homology 2 (cSH2) domain of PLCγ2 (Fig. 3A), which is autoinhibitory and normally prevents constitutive enzymatic function.21,22 Transfection of COS-7 cells with PLCG2 expression constructs with a deletion of the full cSH2 domain, a deletion of exon 19 (Δ19), and a deletion of exons 20 through 22 (Δ20–22) produced equivalent PLCγ2 protein expression (Fig. S9 in the Supplementary Appendix) but elevated basal and Rac-activated phospholipase activity, as compared with a nonmutant PLCγ2 expression construct (Fig. 3A).
Figure 3. Effects of PLAID-Associated PLCG2 Deletions, Including Increased Phospholipase Activity, Deceased Cellular Activation at Physiologic Temperatures, and Enhanced Cellular Activation at Subphysiologic Temperatures.
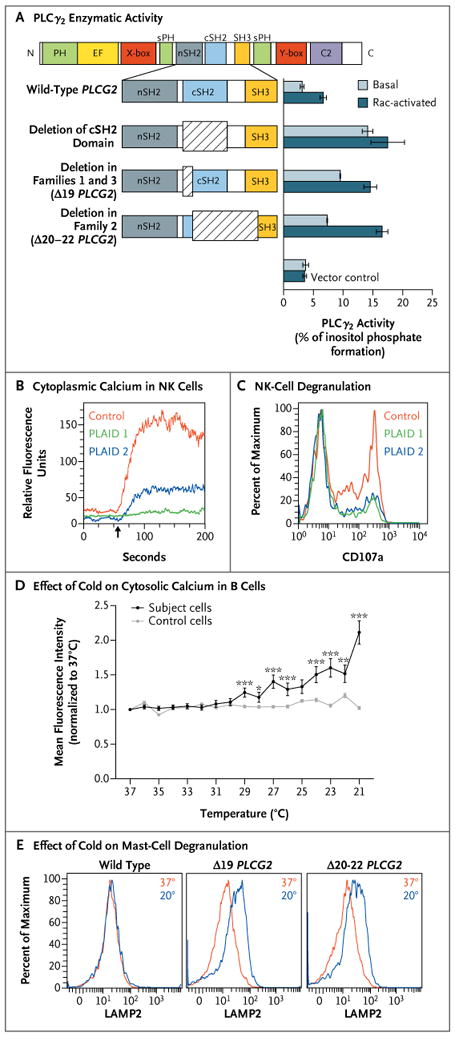
Panel A shows enzymatic activity of phospholipase Cγ2 (PLCγ2) mutants. COS-7 cells were transfected with DNA encoding either wild-type PLCG2 or PLCG2 with deletions of the C-terminal Src homology 2 (cSH2) domain, exon 19 (Δ19), and exons 20 through 22 (Δ20–22) (at left). PLCγ2 activity was assayed by quantification of [3H] inositol phosphates generated by the respective enzymes (at right). Basal activity was quantified in COS-7 cells transfected with PLCG2 alone, whereas the Rac-activated condition included cotransfection with a Rac2 V12 expression construct. The I bars indicate 1 SD above and below the mean release of inositol phosphate for each condition. Data are representative of three independent experiments. C2 denotes calcium-binding C2 domain, EF EF hand motif, nSH2 N-terminal SH2 domain, PH pleckstrin homology domain, SH3 SH3 domain, sPH split pleckstrin homology domain, X-box × catalytic domain, and Y-box Y catalytic domain. Panel B shows cytoplasmic calcium content (as measured by FLUO-4 staining) in natural killer (NK) cells before and after surface-receptor cross-linking (arrow indicates time of administration) in two subjects with PLAID and one control subject. Panel C shows degranulation of NK cells after incubation with sensitive target cells, as measured by cell-surface expression of CD107a as a percent of the maximum CD107a expression observed in control NK cells. Panel D shows the effect of temperature reduction, in the absence of surface stimulation, on cytosolic calcium content in B cells sorted from a case subject and a control subject, as measured on confocal microscopy. Data points reflect the average mean fluorescence intensity of 100 individually imaged cells, and I bars indicate standard errors. Data are representative of three independent experiments. One asterisk indicates a P value of 0.01 to 0.05, two asterisks indicate a P value of 0.001 to less than 0.01, and three asterisks indicate a P value of less than 0.001. Panel E shows mast-cell degranulation, as indicated by the surface expression of lysosomal-associated membrane protein 2 (LAMP2) and measured by means of flow cytometry at 37°C (red) and 20°C (blue) in Laboratory of Allergic Diseases 2 (LAD2) human mast cells transfected with wild-type and deleted forms of PLCG2.
Functional Lymphocyte Studies
Despite the gain of enzymatic function caused by disruption of the autoinhibitory domain, distal signaling and PLCγ2-dependent functions were diminished in the affected family members. B cells showed defective calcium flux and phosphorylation of extracellular signal-regulated kinase (ERK) in response to surface IgM cross-linking (Fig. S10 in the Supplementary Appendix). Transfection of A20 cells with Δ19 or Δ20–22 PLCG2 resulted in reduced ERK phosphorylation stimulated by B-cell– receptor ligation, as compared with untransfected or nonmutant PLCG2-transfected cells, indicating a dominant effect of these mutations on B-cell receptor signaling (Fig. S11 in the Supplementary Appendix). In addition, natural killer cells from affected patients showed reduced calcium flux after cross-linking of activating receptors NKG2D and 2B4 (Fig. 3B) and reduced degranulation after incubation with sensitive target cells (Fig. 3C). In contrast, T-cell calcium flux triggered by CD3 cross-linking was normal (Fig. S12 in the Supplementary Appendix).
Effect of Cold Exposure on Mutant Cells
Cytosolic calcium levels in unstimulated, purified primary B cells from subjects with PLAID steadily rose with decreasing temperature but were unchanged with cooling in nonmutant B cells (Fig. 3D). Likewise, PLAID B cells that were stimulated by B-cell–receptor cross-linking had increased ERK phosphorylation with decreasing temperatures, as compared with nonmutant B cells (Fig. S13 in the Supplementary Appendix). Mutant PLCG2-transfected A20 cells had spontaneous ERK phosphorylation at 20°C but not at 37°C (Fig. S11 in the Supplementary Appendix). Moreover, transfection with mutated PLCG2 into LAD2 mast cells, which endogenously express PLCγ2, led to spontaneous degranulation at 20°C, which was not observed in LAD2 cells transfected with nonmutated PLCG2. The degranulation was equivalent to that seen after treatment with thapsigargin (Fig. S14 in the Supplementary Appendix) but not in LAD2 cells transfected with nonmutated PLCG2 (Fig. 3E).
Discussion
In three families with a dominantly inherited syndrome of cold urticaria and pleiotropic immune dysregulation, we found that genomic deletions in PLCG2 were responsible for this unique phenotype. Diminished receptor-mediated signaling in PLAID B cells resulted in abnormal activation, class-switch recombination, and receptor editing, which have been associated with antibody deficiency, recurrent infection, and impaired central tolerance. Furthermore, the signaling defect in PLAID was temperature-sensitive, as shown by the enhanced signaling and cellular activation observed at subphysiologic temperatures, even in the absence of surface-receptor ligation in B cells and mast cells expressing mutant PLCγ2.
The PLAID-associated PLCG2 deletions affected the regulatory region, including the autoinhibitory cSH2 domain, which normally couples the enzymatic activity of PLCγ2 to upstream pathways.21 PLAID-associated deletions resulted in the failure of autoinhibition and constitutive phospholipase activity, as measured by standard enzymatic assays. These data are consistent with the hypothesis that, at rest, cSH2 obstructs the PLCγ2 catalytic site and that this inhibitory interaction is relieved by phosphorylation of critical tyrosine residues, which facilitates a conformational change in PLCγ2, thereby exposing the catalytic site.21-23
Nonetheless, B cells and natural killer cells from subjects with PLAID clearly had decreased PLCγ2-dependent signaling and function, with responses similar to those observed in Plcg2-deficient mice.19 This apparently paradoxical loss of downstream function may be the direct result of chronic signaling, just as chronic B-cell–receptor stimulation results in calcium currents of diminished amplitude, alteration of signaling cascades, and ultimately, proliferative anergy.24-26 Although the specific mechanisms that underlie the reduction in PLCγ2-mediated signal transduction remain to be elucidated, there are several possible explanations. Increased phospholipase activity may lead to depletion of the substrate PIP2 proximate to PLCγ2, resulting in impaired IP3 production and IP3-mediated calcium release.27 Alternatively, excessively high concentrations of the products of PLCγ2 may induce feedback-mediated down-regulation of the distal signaling pathways.28 Both these mechanisms suggest that phospholipase inhibitors29 might be used to treat PLAID. It is also possible that the deleted regions have other functions that are critical for PLCγ2-mediated signaling, in addition to the phospholipase activity.30
Cold urticaria in patients with PLAID is characterized by mast-cell degranulation after cold stimulation, which is similar to the lesions reported in other types of cold urticaria.7,9 Previous serum transfer experiments have suggested that causative autoantibodies may underlie some cases of cold urticaria.9,31 However, in our study, B cells and mast cells expressing the mutant forms of PLCγ2 had increased cellular activation and effector function at subphysiologic temperatures without receptor stimulation, indicating that the altered function of mutant PLCγ2 was responsible for cold urticaria in the subjects in a dominant, cell-intrinsic fashion.
There are two gain-of-function Plcg2 mouse models, but they differ from PLAID in that these mice lack constitutively activated PLCγ2. In addition, the mouse cells are subject to abnormally high calcium flux after surface-receptor ligation, and all the mice have severe inflammatory arthritis.32,33 Furthermore, the autoimmunity in subjects with PLAID and in one of these mouse models (Plcg2Ali5 mice) appears to develop through different mechanisms, given that the induction of intracellular signaling by receptor stimulation is reduced in PLAID B cells but increased in Plcg2Ali5 B cells. Although cold urticaria is not manifested in either of these mouse models, Plcg2Ali5 mice have inflammatory dermatitis on the coolest body parts (the ears and distal extremities), resembling the distribution of cutaneous granulomatous lesions in several subjects with PLAID in our study.
Common variable immunodeficiency (CVID) is a prototypic antibody-deficiency disorder, which is also associated with autoimmunity. Although CVID was present in only three subjects in our study, there is substantial phenotypic overlap between PLAID and CVID. In addition to antibody deficiency and autoimmunity, common features of the two disorders include granulomatous disease,12 diminished class-switched memory B cells,17 impaired B-cell calcium flux,34 and low numbers of natural killer cells.35 Like PLAID, CVID is associated with mutations that affect proteins involved in B-cell activation.36-40 Given the overlap of pathways and phenotypes between the two diseases, it seems likely that an understanding of PLAID will provide mechanistic insights into the pathogenesis of CVID and CVID-associated symptoms.
The deletions that we discovered in the three families occur in a region of PLCG2 that is rich in repetitive elements known to facilitate aberrant recombination events. Indeed, five of the six deletion breakpoints occurred within repetitive elements, suggesting that sporadic cases of PLAID may be caused by similar de novo or even somatic mutations in PLCG2.
PLAID-associated PLCG2 mutations and the resulting phenotypes provide a rare example of a monogenic disease defined by an atopic phenotype. The effects of the mutant forms of PLCγ2 in this complex disorder illuminate vital elements of phospholipase-mediated signaling, including its role in leukocyte function, host defense, and self-tolerance. In addition, such effects illuminate how novel genetic variants can cause atypical temperature responses by altering the balance among complex biologic pathways.
Supplementary Material
Acknowledgments
Supported by the Intramural Research Programs of the National Human Genome Research Institute, the National Institute of Allergy and Infectious Diseases (NIAID), and the National Institute of Arthritis and Musculoskeletal and Skin Diseases; grants from NIAID (AI061093, AI071087, and AI082713, to Dr. Meffre); the University of California, San Diego (UCSD) Department of Pediatrics; a National Institutes of Health training grant (T32 AI07469); and the UCSD Clinical Translational Research Institute, which has funding from awards issued by the National Center for Research Resources (UL1RR031980, to Dr. Hoffman).
We thank Ms. Colleen Satorius for SNP genotyping and sequencing support; Ms. Julia Fekecs for her assistance in preparing the original versions of the illustrations; and the subjects and their families for their cooperation and participation in this study.
Appendix
The authors' affiliations are as follows: the Inflammatory Disease Section, National Human Genome Research Institute (M.J.O., E.F.R., I.A., D.L.K.); the Laboratories of Allergic Diseases (G.S., S.D., H.K., G.C., M.-Y.J., A.M.G., D.D.M., C.N., M.O., L.W., K.S., J.D.M.), Clinical Infectious Diseases (A.F.F., S.M.H.), Systems Biology (P.T.-P., N.S.), Immunogenetics (H.S.K., E.O.L.), and Immunoregulation (J.H., S.M.) and the Vaccine Research Center (D.C.D.), National Institute of Allergy and Infectious Diseases; and the Critical Care Medicine Department, Clinical Center (P.T.-P.) — all at the National Institutes of Health, Bethesda, MD; the Department of Structural and Molecular Biology, Division of Biosciences, University College London, London (T.D.B., R.W.B., M.S.M., M.K.); the Department of Immunobiology, Yale University School of Medicine, New Haven, CT (N.R., E.M.); the Department of Medicine, Graduate School, Medical Research Center, University of Ulsan, Ulsan, South Korea (H.S.K.); the Division of Allergy, Immunology, and Rheumatology, Department of Pediatrics and Medicine, University of California San Diego (C.G., H.M.H.), and Rady Children's Hospital of San Diego and Ludwig Institute of Cancer Research (H.M.H.) — all in San Diego; the University of Colorado Health Sciences Center, Aurora (A.A.W.); the Department of Human Genetics, University of California Los Angeles, Los Angeles (H.L., S.F.N.); and the Center for Human Genome Variation, Duke University School of Medicine, Durham, NC (K.V.S., E.T.C., D.B.G.).
Footnotes
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
References
- 1.Bennett CL, Christie J, Ramsdell F, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27:20–1. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- 2.Brunkow ME, Jeffery EW, Hjerrild KA, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. 2001;27:68–73. doi: 10.1038/83784. [DOI] [PubMed] [Google Scholar]
- 3.Chatila TA, Blaeser F, Ho N, et al. JM2, encoding a fork head-related protein, is mutated in X-linked autoimmunity-allergic disregulation syndrome. J Clin Invest. 2000;106:R75–R81. doi: 10.1172/JCI11679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wildin RS, Ramsdell F, Peake J, et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet. 2001;27:18–20. doi: 10.1038/83707. [DOI] [PubMed] [Google Scholar]
- 5.Nagamine K, Peterson P, Scott HS, et al. Positional cloning of the APECED gene. Nat Genet. 1997;17:393–8. doi: 10.1038/ng1297-393. [DOI] [PubMed] [Google Scholar]
- 6.Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet. 2001;29:301–5. doi: 10.1038/ng756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gandhi C, Healy C, Wanderer AA, Hoffman HM. Familial atypical cold urticaria: description of a new hereditary disease. J Allergy Clin Immunol. 2009;124:1245–50. doi: 10.1016/j.jaci.2009.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wanderer AA, Hoffman HM. The spectrum of acquired and familial cold-induced urticaria/urticaria-like syndromes. Immunol Allergy Clin North Am. 2004;24:259–86. doi: 10.1016/j.iac.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 9.Kaplan AP, Garofalo J, Sigler R, Hauber T. Idiopathic cold urticaria: in vitro demonstration of histamine release upon challenge of skin biopsies. N Engl J Med. 1981;305:1074–7. doi: 10.1056/NEJM198110293051808. [DOI] [PubMed] [Google Scholar]
- 10.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin — rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 11.Pelak K, Shianna KV, Ge D, et al. The characterization of twenty sequenced human genomes. PLoS Genet. 2010;6(9):pii–e1001111. doi: 10.1371/journal.pgen.1001111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Everett KL, Bunney TD, Yoon Y, et al. Characterization of phospholipase C gamma enzymes with gain-of-function mutations. J Biol Chem. 2009;284:23083–93. doi: 10.1074/jbc.M109.019265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim HS, Das A, Gross CC, Bryceson YT, Long EO. Synergistic signals for natural cytotoxicity are required to overcome inhibition by c-Cbl ubiquitin ligase. Immunity. 2010;32:175–86. doi: 10.1016/j.immuni.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bryceson YT, Rudd E, Zheng C, et al. Defective cytotoxic lymphocyte degranulation in syntaxin-11 deficient familial hemophagocytic lymphohistiocytosis 4 (FHL4) patients. Blood. 2007;110:1906–15. doi: 10.1182/blood-2007-02-074468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ho J, Moir S, Wang W, et al. Enhancing effects of adjuvanted 2009 pandemic H1N1 influenza A vaccine on memory B-cell responses in HIV-infected individuals. AIDS. 2011;25:295–302. doi: 10.1097/QAD.0b013e328342328b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human B cell precursors. Science. 2003;301:1374–7. doi: 10.1126/science.1086907. [DOI] [PubMed] [Google Scholar]
- 17.Ng YS, Wardemann H, Chelnis J, Cunningham-Rundles C, Meffre E. Bruton's tyrosine kinase is essential for human B cell tolerance. J Exp Med. 2004;200:927–34. doi: 10.1084/jem.20040920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bunney TD, Katan M. PLC regulation: emerging pictures for molecular mechanisms. Trends Biochem Sci. 2011;36:88–96. doi: 10.1016/j.tibs.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 19.Wang D, Feng J, Wen R, et al. Phospholipase Cgamma2 is essential in the functions of B cell and several Fc receptors. Immunity. 2000;13:25–35. doi: 10.1016/s1074-7613(00)00005-4. [DOI] [PubMed] [Google Scholar]
- 20.Tassi I, Presti R, Kim S, Yokoyama WM, Gilfillan S, Colonna M. Phospholipase C-gamma 2 is a critical signaling mediator for murine NK cell activating receptors. J Immunol. 2005;175:749–54. doi: 10.4049/jimmunol.175.2.749. [DOI] [PubMed] [Google Scholar]
- 21.Rodriguez R, Matsuda M, Storey A, Katan M. Requirements for distinct steps of phospholipase Cgamma2 regulation, membrane-raft-dependent targeting and subsequent enzyme activation in B-cell signalling. Biochem J. 2003;374:269–80. doi: 10.1042/BJ20021778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gresset A, Hicks SN, Harden TK, Sondek J. Mechanism of phosphorylation-induced activation of phospholipase C-gamma isozymes. J Biol Chem. 2010;285:35836–47. doi: 10.1074/jbc.M110.166512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Poulin B, Sekiya F, Rhee SG. Intramolecular interaction between phosphorylated tyrosine-783 and the C-terminal Src homology 2 domain activates phospholipase C-gamma1. Proc Natl Acad Sci U S A. 2005;102:4276–81. doi: 10.1073/pnas.0409590102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cooke MP, Heath AW, Shokat KM, et al. Immunoglobulin signal transduction guides the specificity of B cell-T cell interactions and is blocked in tolerant self-reactive B cells. J Exp Med. 1994;179:425–38. doi: 10.1084/jem.179.2.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Healy JI, Dolmetsch RE, Timmerman LA, et al. Different nuclear signals are activated by the B cell receptor during positive versus negative signaling. Immunity. 1997;6:419–28. doi: 10.1016/s1074-7613(00)80285-x. [DOI] [PubMed] [Google Scholar]
- 26.Dolmetsch RE, Lewis RS, Goodnow CC, Healy JI. Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature. 1997;386:855–8. doi: 10.1038/386855a0. Erratum, Nature 1997;388:308. [DOI] [PubMed] [Google Scholar]
- 27.Daniels RL, Takashima Y, McKemy DD. Activity of the neuronal cold sensor TRPM8 is regulated by phospholipase C via the phospholipid phosphoinositol 4,5-bisphosphate. J Biol Chem. 2009;284:1570–82. doi: 10.1074/jbc.M807270200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dufour JF, Arias IM, Turner TJ. Inositol 1,4,5-trisphosphate and calcium regulate the calcium channel function of the hepatic inositol 1,4,5-trisphosphate receptor. J Biol Chem. 1997;272:2675–81. doi: 10.1074/jbc.272.5.2675. [DOI] [PubMed] [Google Scholar]
- 29.van Blitterswijk WJ, Verheij M. Anti-cancer alkylphospholipids: mechanisms of action, cellular sensitivity and resistance, and clinical prospects. Curr Pharm Des. 2008;14:2061–74. doi: 10.2174/138161208785294636. [DOI] [PubMed] [Google Scholar]
- 30.van Rossum DB, Patterson RL, Sharma S, et al. Phospholipase Cgamma1 controls surface expression of TRPC3 through an intermolecular PH domain. Nature. 2005;434:99–104. doi: 10.1038/nature03340. [DOI] [PubMed] [Google Scholar]
- 31.Sherman WB, Seebohm PM. Passive transfer of cold urticaria. J Allergy. 1950;21:414–24. doi: 10.1016/0021-8707(50)90017-7. [DOI] [PubMed] [Google Scholar]
- 32.Yu P, Constien R, Dear N, et al. Autoimmunity and inflammation due to a gain-of-function mutation in phospholipase C gamma 2 that specifically increases external Ca2+ entry. Immunity. 2005;22:451–65. doi: 10.1016/j.immuni.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 33.Abe K, Fuchs H, Boersma A, et al. A novel N-ethyl-N-nitrosourea-induced mutation in phospholipase Cgamma2 causes inflammatory arthritis, metabolic defects, and male infertility in vitro in a murine model. Arthritis Rheum. 2011;63:1301–11. doi: 10.1002/art.30280. [DOI] [PubMed] [Google Scholar]
- 34.Foerster C, Voelxen N, Rakhmanov M, et al. B cell receptor-mediated calcium signaling is impaired in B lymphocytes of type Ia patients with common variable immunodeficiency. J Immunol. 2010;184:7305–13. doi: 10.4049/jimmunol.1000434. [DOI] [PubMed] [Google Scholar]
- 35.Aspalter RM, Sewell WA, Dolman K, Farrant J, Webster AD. Deficiency in circulating natural killer (NK) cell subsets in common variable immunodeficiency and X-linked agammaglobulinaemia. Clin Exp Immunol. 2000;121:506–14. doi: 10.1046/j.1365-2249.2000.01317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Warnatz K, Salzer U, Rizzi M, et al. B-cell activating factor receptor deficiency is associated with an adult-onset antibody deficiency syndrome in humans. Proc Natl Acad Sci U S A. 2009;106:13945–50. doi: 10.1073/pnas.0903543106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Losi CG, Silini A, Fiorini C, et al. Mutational analysis of human BAFF receptor TNFRSF13C (BAFF-R) in patients with common variable immunodeficiency. J Clin Immunol. 2005;25:496–502. doi: 10.1007/s10875-005-5637-2. [DOI] [PubMed] [Google Scholar]
- 38.Pan-Hammarström Q, Salzer U, Du L, et al. Reexamining the role of TACI coding variants in common variable immunodeficiency and selective IgA deficiency. Nat Genet. 2007;39:429–30. doi: 10.1038/ng0407-429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van Zelm MC, Reisli I, van der Burg M, et al. An antibody-deficiency syndrome due to mutations in the CD19 gene. N Engl J Med. 2006;354:1901–12. doi: 10.1056/NEJMoa051568. [DOI] [PubMed] [Google Scholar]
- 40.Grimbacher B, Hutloff A, Schlesier M, et al. Homozygous loss of ICOS is associated with adult-onset common variable immunodeficiency. Nat Immunol. 2003;4:261–8. doi: 10.1038/ni902. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.