

Abstract
Spinal glial activation has been implicated in sustained morphine-mediated paradoxical pain sensitization. Since activation of glial CB2 cannabinoid receptors attenuates spinal glial activation in neuropathies, we hypothesized that CB2 agonists may also attenuate sustained morphine–mediated spinal glial activation and pain sensitization. Our data indicate that co-administration of a CB2-selective agonist (AM 1241) attenuates morphine (intraperitoneal; twice daily; 6 days)-mediated thermal hyperalgesia and tactile allodynia in rats. A CB2 (AM 630) but not a CB1 (AM 251) antagonist mitigated this effect. AM 1241 co-treatment also attenuated spinal astrocyte and microglial marker and pro-inflammatory mediator (IL-1β, TNFα) immunoreactivities in morphine-treated rats, suggesting that CB2 agonists may be useful to prevent the neuroinflammatory consequences of sustained morphine treatment.
Keywords: morphine, spinal glia, CB2 agonist, hyperalgesia, allodynia, pain sensitization
1. Introduction
Long-term opioid treatment frequently leads to an increased sensitivity to painful (hyperalgesia) and normally innocuous (allodynia) stimuli in humans (Arner et al., 1988; Wanigasekera et al., 2011) and in experimental animals (Vanderah et al., 2001b). It was suggested that long-term opioid-mediated paradoxical pain sensitization may play a role in apparent antinociceptive tolerance (Vanderah et al., 2001a, Ossipov et al., 2005). However, the molecular mechanisms that lead to paradoxical pain sensitization upon chronic opioid treatment are poorly understood.
Central nervous system (CNS) glia was shown to play an important role in the regulation of pain sensitivity during inflammatory and neuropathic conditions (Watkins et al., 2005). Interestingly, it was found that - similar to inflammation and nerve injury - sustained opioid agonist-treatment also activates CNS microglia and astrocytes (Watkins et al., 2005, 2007). Glial proinflammatory inhibitors were shown to attenuate sustained morphine-mediated pain sensitization and to reduce/delay the development of antinociceptive tolerance (Mika et al., 2007; Raghavendra et al., 2002, 2003) in experimental animals. The molecular mechanisms leading to sustained opioid-mediated augmentation of glial activity and glial pro-inflammatory mediator release are presently not entirely clear. Morphine may directly activate glial opioid receptors (Chang et al., 1998). Alternatively, it has been suggested that morphine may interact with Toll-like receptors (such as TLR-4) on glial cells (Hutchinson et al., 2007). Finally, sustained opioid treatment may also activate glial cells indirectly, for instance by stimulating excitatory modulator release from the sensory neurons (Wang et al., 2009).
It was suggested that spinal glial activation and glia-derived proinflammatory mediator (such as IL-1β, tumor necrosis factor-α (TNF-α), and prostaglandin E2 (PGE2)) release may be implicated in long-term opioid treatment-mediated paradoxical pain sensitization and antinociceptive tolerance (Watkins et al., 2005). Therefore, glial proinflammatory inhibitors may be useful to prevent the neuroinflammatory consequences of sustained morphine treatment. Unfortunately, the currently available “classical” glial proinflammatory inhibitors are either toxic (fluorocitrate) or are not specific for glia (minocycline and propentofylline) (Mika, 2008; Raghavendra et al., 2003). Thus, novel methods are necessary to regulate CNS glial activity during opioid agonist treatment.
Interestingly, recent data indicate that CB2 cannabinoid agonists attenuate inflammatory and neuropathic activation of CNS glia (Fernandez-Ruiz et al., 2007). Inflammation and nerve injury dramatically increase the expression of CB2 receptors in microglia and astrocytes (Stella, 2004; Maresz et al., 2005), suggesting glial CB2 receptor activation may serve as a negative feed-back mechanism to regulate glial activity. Indeed, it was found that a selective CB2 agonist, (JWH-015) attenuates glial marker expression (Ehrhart et al., 2005) and the synthesis of glial pro-inflammatory mediators in cultured rodent microglia (Eljaschewitsch et al., 2006). The molecular mechanism leading to attenuated glial activity and glial inflammatory mediator synthesis in response to CB2 receptor activation is not entirely clear, but it was suggested that CB2 agonists may activate glial MAP kinase-phosphatases, leading to attenuated MAP kinase activity (Eljaschewitsch et al., 2006, Romero-Sandoval et al., 2009).
Numerous shared characteristics between neuropathic and sustained morphine-induced pain sensitization and analgesic tolerance led to the suggestion that these processes may share common mechanisms (Ossipov et al., 2005). Similar to inflammatory and neuropathic conditions, sustained morphine treatment also induce a time-dependent upregulation of CB2 receptors within the spinal cord dorsal horn (Lim et al., 2005). Therefore, we hypothesized that systemic co-administration of a CB2-selective agonist will also be able to antagonize the pro-inflammatory consequences of sustained morphine treatment in the spinal cord. Our data show that systemic co-administration of morphine with a CB2 agonist (AM 1241) attenuates repeated morphine administration-mediated thermal hyperalgesia, tactile allodynia, spinal glial activation and upregulation of spinal pro-inflammatory mediator concentrations in rats by activation of CB2 receptors.
2. Methods
2.1 Drug administration
Adult (200–225 g) male Sprague-Dawley rats (Harlan Sprague-Dawley, Indianapolis, IN) were kept in a climate-controlled room on a 12 hr light/dark cycle with food and water available ad libitum. Handling, care, maintenance and testing of the animals were performed in accordance with the policies and recommendations of the International Association for the Study of Pain and the National Institutes of Health guidelines for the handling and use of laboratory animals. The experimental protocol was approved by the Animal Care and Use Committee of the University of Arizona. Repeated morphine administration was accomplished by intraperitoneal (i.p.) injections of morphine sulfate (NIDA; 5mg/kg/injection, 1ml/kg volume) to rats twice daily, for 6 days (vehicle-morphine group, n=12 animals). Separate groups of animals received i.p. injections of the CB2-selective agonist, AM 1241 (0.1–2.5 mg/kg/injection; 1ml/kg injection volume, Sigma, St. Louis, MO) twice daily for 6 days either together with morphine (morphine-AM 1241 group, n=6) or saline (AM 1241+saline group, n=12 animals). Separate groups of animals received i.p. injections of AM 1241 (2.5 mg/kg/injection) along with a CB2 antagonist (AM 630; 600 μg/kg/injection; Tocris, Ellisville, Missouri) or with a CB1-selective antagonist (AM 251;600 μg/kg/injection; Tocris, Ellisville, Missouri), either together with morphine (AM 1241+AM 630+morphine group, n=6) or saline (AM 630+saline group, n=6 animals) twice daily for 6 days. The doses of AM 1241, AM 630 and AM 251 were selected based on previous studies (Ibrahim et al., 2003, 2006) AM 1241, AM 630 and AM 251 were dissolved in physiological saline, containing 10% Tween 80, 10% DMSO. Control animals received the same volume of vehicle and saline (vehicle+saline group). Behavioral (thermal hyperalgesia and tactile allodynia) assays were performed prior to drug administration (naïve baseline), every day during the drug treatment period and 96 h after the last i.p. drug injection. Animals receiving saline-vehicle injections exhibited pain thresholds similar to those of naïve animals. The 96 h after drug withdrawal time point was selected in order to allow for complete drug clearance after the last drug injection, based on the withdrawal time-response studies (Largent-Milnes, unpublished observations). Fig. 1. shows a schematic representation of the experimental paradigm.
Fig. 1. Experimental design.

On day 0 (A), baseline paw withdrawal latency (Radiant heat test) and paw withdrawal threshold (vonFrey filament test) values were measured for each rat. Subsequently, the animals received intraperitoneal (i.p.) injections of saline (200 μl) or morphine (5mg/kg in 200 μl) with/without AM 1241 (0.5–2.5mg/kg in 200 μl) and with/without a cannabinoid antagonist (AM 630; 600 μg/kg or AM 251 (600 μg/kg). The animals were tested for acute antinociception 30 min after drug administration (B). The injection regimen continued in each animal group for 6 days (C) twice daily (a total of 12 injections; Day 0 – Day 5). The animals were tested for paw withdrawal latencies and paw withdrawal thresholds every day in the morning (9:30) and 96 h (day 9) after the last drug treatment (D). The drug-withdrawn animals were subsequently euthanized and the spinal cords were isolated for immunohistochemistry.
2.2. Measurement of thermal hyperalgesia
The method of Hargreaves et al. (1988) was used to assess the sensitivity of rats to mildly noxious thermal stimulus, as previously described (Largent-Milnes et al., 2008). Briefly, the animals (6 individual animals in each treatment group, n=2 experiments) were placed on tempered glass in a Plexiglass container and radiant heat was directed toward the plantar surface of their hind paw. Paw withdrawal latencies were measured using an automated motion detector before (baseline) drug administration, after drug treatment on each day of the drug treatment (9:00 AM) for 6 days and 96 h after the last i.p. drug injection. The device simultaneously stops the timer and the heat source when the paw is moved. A maximal cutoff of 33 s was used to prevent tissue damage.
2.3. Measurement of mechanical allodynia
Paw withdrawal thresholds in response to mild, normally innocuous tactile stimuli were measured by probing the left hind paw of the rats with von Frey filaments (0.4–15.0 g), as previously described (Vanderah et al., 2000). The paw withdrawal thresholds were measured before (baseline) drug administration, each day during drug administration (9:30 AM) for 6 days and 96 h after the last i.p. drug injection. Paw withdrawal thresholds were measured prior to the thermal hyperalgesia assay in each animal group, by sequentially increasing and then decreasing the stimulus strength (“up-and-down” method). The data were analyzed using the Dixon nonparametric test (Dixon, 1980; Chaplan et al., 1994).
2.4. Immunohistochemistry
Immunohistochemisty was performed using a protocol modified from Gardell et al. (2002). Briefly, after the last behavioral testing, the rats were anesthetized with i.p. ketamine-xylazine (100mg/kg) injections and transcardially perfused with 0.1 M PBS (pH 7.4) until the exudate ran clear and then with 10% formalin for 15 min. Lumbar spinal cords were harvested, fixed in 10% formalin solution overnight and cryoprotected with 20% sucrose in 0.1 M PBS. The fixed tissues were saturated with 30% sucrose in 0.1 M PBS solution and embedded in Tissue-Tek Optimal Cutting Temperature Compound (Sakura, Torrance, CA) and sliced in a cryostat at −20°C. Serial lumbar spinal cord sections (20 μm each) were placed onto slides so that each slide contained an ordered series of sections from a lumbar spinal cord. The mounted sections were extensively rinsed and blocked in 0.1 M PBS containing 10% goat serum, for 2 h, at room temperature. The sections were incubated overnight at 4°C with either a mouse monoclonal anti-glial fibrillary acidic protein (GFAP) antiserum (1:2,000; Chemicon International, Temecula, CA) or a mouse monoclonal anti-OX-42 antiserum (1:2,000; Chemicon International, Temecula, CA) in 0.1 M PBS containing 2% goat serum and 0.3% Triton X-100. The slides were subsequently washed and incubated (2 h) with an Alexa Fluor 594-conjugated goat anti-mouse secondary antibody (1:1000; Molecular Probes, Eugene, OR). After the immunoreactions, the sections were rinsed and mounted in a Vectashield (Vector Laboratories) mounting medium. Fluorescence images were digitally captured using a Nikon (Tokyo, Japan) E800 fluorescence microscope, equipped with the appropriate standard filters. Images were acquired and analyzed using a Hamamatsu C5810 color CCD camera and its proprietary Image Processor Software (Hamamatsu, Bridgewater, NJ). The acquired images were processed using Adobe PhotoShop (Adobe Systems, San Jose, CA). The images were quantified by counting the number of pixels within a defined threshold fluorescence intensity using the Image J software (NIH Image, National Institutes of Health, Bethesda, MD). Immunoreactive pixel numbers were counted in randomly selected areas of the spinal cord dorsal horn for 3 rats per treatment group, using six spinal cord slices for each rat.
2.5. Measurement of IL-1β and TNFα concentrations in the lumbar spinal cord of rats
96 h after drug withdrawal, the animals (n=3 per treatment group) were euthanized and the dorsal half of their lumbar spinal cords were isolated. The tissues were flash frozen in liquid nitrogen and stored at −80°C. On the day of the experiment, the dorsal lumbar spinal cord sections were thawed, homogenized in a homogenization buffer centrifuged and the supernatants were collected as previously described (Tumati et al., 2010). Protein concentration in these supernatants was determined by the Bradford method. A rat interleukin-1 β (rat IL-1β) sandwich ELISA kit (R and D systems, Minnesota, MN) was used for the quantitative determination of rat IL-1β concentration in spinal cord superfusates. TNFα concentration in the samples was determined using a rat TNFα ELISA kit (R and D systems, Minnesota, MN). The assays were performed according to the manufacturer’s instructions. IL-1β and TNFα concentrations (pg/mg total protein) in the samples were calculated based on standard IL-1β and TNFα concentration-absorbance curves using the Graph Pad Prism 4.0 software (San Diego, CA).
2.6. Data analysis
Data were analyzed using the GraphPad Prism 4.0 software (San Diego, CA). Thermal and tactile hypersensitivity data were analyzed by two-way repeated measures ANOVA (post hoc, Newman-Keuls) while, the IL-1β and TNF-α content were analyzed by nonparametric one-way ANOVA (post hoc, Newman-Keuls). Statistical differences were considered significant at p <0.05 (*p < 0.05, **p < 0.01, and ***p < 0.001). Data were presented as mean±S.E.M, unless otherwise indicated.
3. Results
3.1. Co-administration of a CB2-selective agonist, AM 1241, dose-dependently attenuates repeated morphine-mediated thermal hyperalgesia in rats
Baseline paw withdrawal latencies in naïve animals was 23.9±1.2 (n=36; Fig 2A) Rats were randomly assigned into treatment groups. (n=6 in each group; 2 independent experiments). Acute paw withdrawal latencies were measured after 30 min of drug treatment. Subsequently, paw withdrawal latencies were measured once every day in the morning before drug administration (9:00 AM) for 6 days and 96h after the last drug treatment to monitor the development of thermal hypersensitivity associated with repeated morphine treatment and opioid withdrawal. Acute morphine administration significantly increased radiant heat paw-withdrawal latencies in vehicle-pre-treated rats (vehicle+morphine group; 30.1±1 s, **p < 0.01 relative to the naive baseline of 23.9±1.2 s, two-way ANOVA, n=6). Control animals treated with vehicle-saline exhibited mean paw withdrawal latencies similar to the naïve baseline value (vehicle+saline group; 20.4±1.09 s; p>0.05 relative to naïve animals, two-way ANOVA, n=6 per group) (Fig 2A). The paw withdrawal latencies of rats receiving vehicle+saline injections remained similar to the naïve baseline value throughout the treatment period (p>0.05, two-way ANOVA, n=6).
Fig. 2. A. Sustained co-administration of a CB2-selective agonist (AM 1241) attenuates morphine withdrawal-mediated thermal hyperalgesia in rats.

Male Sprague-Dawley rats (6 in each group) received intraperitoneal ▼:vehicle+saline; ◆: vehicle+morphine (5 mg/kg); ■: AM 1241 (2.5 mg/kg)+saline; ●: AM 1241 (2.5mg/kg)+morphine(5mg/kg) injections twice daily for 6 days. Thermal pain sensitivity was measured 30 min after the first injectyion (acute) on each day (at 9:00 AM) of the drug treatment and after 96h drug withdrawal using the Hargreaves test. BL represents the naïve baseline. Fig 2. B. shows the area-under-curve (AUC) values for each treatment group.
Repeated morphine treatment on the other hand, led to a gradual decrease in radiant heat paw withdrawal latencies indicating a gradual development of thermal hypersensitivity. The decrease in paw withdrawal latencies was significant starting from day 3 of morphine treatment (vehicle-morphine group, 13.1±1.9 s; *p<0.05 relative to naïve baseline of 23.9±1.2 s, two-way ANOVA, n=6). 96h after morphine withdrawal there was still a marked decrease in mean paw withdrawal latency (7.5±1.6 s, ***p<0.001 relative to both naïve and to vehicle-saline treated control, two-way ANOVA, n=6). Interestingly, co-administration of a selective CB2 agonist (AM 1241, 2.5 mg/kg) completely attenuated repeated morphine-mediated decrease in paw withdrawal latencies. Paw withdrawal latencies in animals co-treated with 1 mg/kg AM 1241 were 24.5±0.7 s on day 3 and 21.4±1.8 s at 96h withdrawal (p>0.05, two-way ANOVA, n=6). The mean paw withdrawal latencies of rats treated with AM 1241 alone (AM 1241-saline group) were not significantly different from the naïve baseline or vehicle-saline treated control either during drug treatment or after 96 drug withdrawal (23.2±1.3 s; p>0.05 relative to naïve baseline, two-way ANOVA, n=6). All treatment groups were tested for thermal pain sensitivity on each day (9:00 AM) of the treatment period.
3.2. Co-administration of a CB2-selective agonist (AM 1241) dose-dependently attenuates repeated morphine-mediated tactile allodynia in rats
Baseline mechanical sensitivity in naïve animals to VonFrey filament stimulation was 15.0±0.0 g (n=36; Fig 3A). Animals treated with vehicle-saline injections showed no significant changes in paw withdrawal thresholds throughout the treatment period relative to naïve baseline (p>0.05). Acute morphine administration (30 min) did not cause significant change in the paw withdrawal thresholds of vehicle-treated rats (15.0±0.0 g; vehicle+morphine group; p>0.05). Acute (30 min) AM 1241 administration (0.1, 1.0, 2.5 mg/kg) did not affect paw withdrawal thresholds either in the presence (AM 1241-morphine group; p>0.05 relative to the naive baseline) or absence of morphine (AM 1241 group; p>0.05 for each doses, two-way ANOVA, n=6).
Fig. 3. A. AM 1241 co-administration attenuates morphine withdrawal-mediated tactile allodynia in rats.

Male Sprague-Dawley rats (6 in each group) received intraperitoneal ▼:vehicle+saline; ◆: vehicle+morphine (5 mg/kg); ■: AM 1241 (2.5 mg/kg)+saline; ●: AM 1241 (2.5mg/kg)+morphine(5mg/kg) injections twice daily for 6 days. Thermal pain sensitivity was measured 30 min after the first injectyion (acute) on each day (at 9:30 AM) of the drug treatment and after 96h drug withdrawal using VonFrey filaments. Fig 2. B. shows the area-under-curve (AUC) values for each treatment group.
Repeated morphine treatment led to a gradual decrease in paw withdrawal thresholds. The decrease was significant starting from day 2 of morphine treatment, (10.2±2.0 s on day 5; **p<0.01, two-way ANOVA, n=6). 96h after morphine withdrawal there was still a marked decrease in the mean paw withdrawal threshold (1.0±0.5 g; ***p<0.001 relative to both naïve and to vehicle-saline treated control, two-way ANOVA, n=6). Rats receiving AM 1241 (2.5 mg/kg) together with morphine did not exhibit significant decrease in paw withdrawal latencies (12.2±0.8 g at 96 withdrawal; p>0.05 relative to naïve baseline; two-way ANOVA, n=6), indicating that AM 1241 co-treatment attenuates the development of mechanical allodynia due to repeated morphine treatment and morphine withdrawal in rats (Fig 3A). In the presence low AM 1241 dose (0.1 mg/kg), repeated morphine treatment caused a slight decrease in paw withdrawal latency (17.5±1 s; on day 5, *p<0.05, two-way ANOVA, n=6, Fig. 2B). After 96h drug withdrawal the mean paw withdrawal latency in the AM 1241 (0.1 mg/kg)+morphine group was not significantly different from the naïve baseline (19.6±1.9 s vs 23.9±1.2 s; p>0.05). Paw withdrawal latencies in animals co-treated with higher doses (1–2.5 mg/kg) of AM 1241 were not significantly different from naïve baseline (p>0.05, two-way ANOVA) either during drug treatment or after 96h withdrawal. Paw withdrawal thresholds in animals receiving 2.5 mg/kg AM 1241 alone were slightly decreased on day 5 (7.2±2.0 g, *p<0.05 relative to naïve baseline, two-way ANOVA) and at 96h withdrawal (9.0±2.5 g, *p<0.05 relative to naïve baseline, two-way ANOVA), indicating that repeated treatment with racemic (S,R)AM 1241 alone causes a slight increase in mechanical sensitivity. These data show that co-treatment with AM 1241 attenuates the development of mechanical allodynia during repeated morphine treatment and after morphine withdrawal in rats.
3.3. The anti-hyperalgesic effect of AM 1241 is attenuated by a CB2 (AM 630) but not by a CB1 receptor antagonist (AM 251)
Mean baseline paw withdrawal latency in naïve animals was 24.3±1.5 s (n=48; Fig 4). Control animals treated with vehicle+saline exhibited mean paw withdrawal latencies similar to the mean baseline value (p>0.05 relative to baseline, one-way ANOVA, n=6). 96h withdrawal after repeated morphine treatment (vehicle+morphine group) led to a marked decrease in mean paw withdrawal latency (***p<0.001 relative to the baseline, one-way ANOVA, n=6). Rats receiving i.p. AM 1241 (2.5 mg/kg) concurrent with i.p. morphine (AM 1241+morphine) exhibited paw withdrawal latencies similar to the baseline 96h after drug withdrawal (p>0.05 relative to baseline, one-way ANOVA, n=6). Co-administration of a CB2 receptor-selective antagonist AM 630 (600 μg/kg, i.p.) together with AM 1241 (2.5 mg/kg) and morphine significantly attenuated AM 1241-mediated anti-thermal hypersensitive effects 96h after drug withdrawal (AM 1241+AM 630+morphine; **p<0.01 relative to the baseline and p>0.05 relative to vehicle+morphine, one-way ANOVA, n=6). On the other hand, co-administration of a CB1 receptor antagonist AM 251 (600 μg/kg, i.p.) together with AM 1241 (2.5 mg/kg) and morphine did not produce any significant changes in the anti-thermal hyperalgesic effects mediated by AM 1241 (AM 1241+AM 251+morphine; p>0.05 relative to AM 1241+morphine or baseline, **p<0.01 relative to vehicle+morphine, one-way ANOVA, n=6). AM 1241 or AM 630 or AM 251 treatment alone had no significant effect on paw withdrawal latencies (p>0.05 relative to baseline, one-way ANOVA, n=6 per group) (Fig 4). These data indicate that the anti-thermal hyperalgesic effects observed upon treatment with AM 1241 are mediated by activation of CB2 receptors.
Fig. 4. The anti-hyperalgesic effect of AM 1241 in morphine withdrawn rats is attenuated by CB2- (AM 630) but not CB1- (AM 251) antagonist.

Male Sprague-Dawley rats (6 in each group) received drug treatment twice daily for 6 days. Mean baseline paw withdrawal latencies were measure in naïve animals (dotted line; n=48) and 96 h after the last injection using the Hargraeves test. (*=p<0.05; **= p<0.01 ***= p<0.001 relative to the baseline, one-way ANOVA).
3.4. The anti-mechanical hypersensitivity effects of AM 1241 are attenuated by a CB2 (AM 630) but not by a CB1 receptor antagonist (AM 251)
Mean baseline paw withdrawal thresholds in naïve animals was 15.0±0.0 g (n=48; Fig 5). Control animals treated with vehicle+saline exhibited mean paw withdrawal thresholds similar to the mean baseline value (p>0.05 relative to baseline, one-way ANOVA, n=6). Withdrawal (96 h) after repeated morphine treatment (vehicle+morphine group) led to a marked decrease in mean paw withdrawal threshold (***p<0.001 relative to the baseline, one-way ANOVA, n=6). Rats receiving i.p. AM 1241 (2.5 mg/kg) concurrent with i.p. morphine (AM 1241+morphine) exhibited paw withdrawal thresholds that were not significantly different from the baseline (p>0.05, one-way ANOVA, n=6). Co-administration of a CB2 receptor-selective antagonist (AM 630, 600 μg/kg, i.p.) together with AM 1241 and morphine significantly attenuated AM 1241-mediated anti-mechanical hypersensitive effects (***p<0.01 relative to the baseline and p>0.05 relative to vehicle+morphine, one-way ANOVA, n=6). Co-administration of a CB1 receptor antagonist (AM 251,600 μg/kg, i.p.) did not produce any significant changes in the anti-allodynic effects mediated by AM 1241 (AM 1241+AM 251+morphine; p>0.05 relative to AM 1241+morphine, **p<0.01 relative to vehicle+morphine, one-way ANOVA, n=6). Treatment with AM 1241 or AM 630 or AM 251 alone had no significant effect (p>0.05 relative to baseline, one-way ANOVA) on paw withdrawal thresholds (Fig 5). These data indicate that the anti-allodynic effect of AM 1241 co-treatment is mediated by activation of the CB2 receptors.
Fig. 5. The antiallodynic effect of AM 1241 in morphine withdrawn rats is attenuated by CB2- (AM 630) but not CB1- (AM 251) antagonist.

Male Sprague-Dawley rats (6 in each group) received drug treatment twice daily for 6 days. Mean baseline paw withdrawal thresholds were measured in naïve animals (dotted line; n=48) and 96 h after the last injection using Von Frey filaments.
3.5. AM 1241 co-treatment reduces microglia (OX-42) and astrocyte (GFAP) marker immunoreactivities in the dorsal horn of the lumbar spinal cord of morphine-withdrawn rats
After the behavioral assays, the animals were perfused as described in the methods section and lumbar spinal cords were isolated from three animals in each treatment group. Fluorescent immunohistochemistry in lumbar spinal cord slices indicated that morphine treatment increased in both OX-42- and GFAP-like immunoreactivities in the dorsal horn of the spinal cord (Figs. 6b, 7b). AM 1241 co-administration completely attenuated sustained morphine-mediated augmentation of both OX-42 and GFAP-like immunoreactivities in the lumbar dorsal horn of morphine-withdrawn animals (Figs 6c, 7c). Each figure is a representative of similar immunohistochemistry data obtained from 3 animals in each treatment group.
Fig. 6. AM 1241 co-administration attenuates spinal astrocyte marker (GFAP) immunoreactivity in morphine-withdrawn rats.

Lumbar spinal cord slices from saline-saline (a), morphine-saline (b) and morphine-AM 1241(c) -treated animals (3 animals from each group) were incubated with a rabbit anti-GFAP primary antibody followed by incubation with an Alexa Fluor 594-conjugated goat anti-rabbit secondary antibody. Fluorescent images were collected using a Nikon E800 fluorescence microscope. The images have been collected from matching dorsal horn areas in each treatment group. Scale bar represents 100 μm. (d) the number of pixels occupied by GFAP immunoreactive cells within a defined threshold in a randomly selected area of the spinal cord dorsal horn, measured by Image J software (NIH). The data plotted is mean+SEM obtained from 3 independent rats; 6 spinal cord sections per rat per treatment group.
Fig. 7. AM 1241 co-administration attenuates spinal microglia marker (OX-42) immunoreactivity in morphine-withdrawn rats.
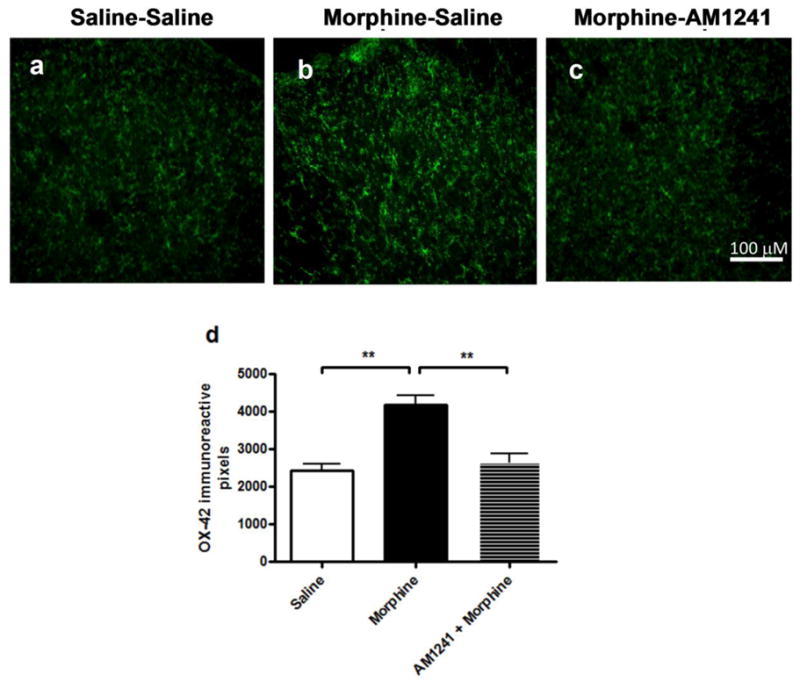
Lumbar spinal cord slices from saline-saline (a), morphine-saline (b) and morphine-AM 1241 (c) -treated animals (3 animals from each group) were incubated with a rabbit anti-OX-42 primary antibody followed by incubation with an Alexa Fluor 594-conjugated goat anti-rabbit secondary antibody. Fluorescent images were collected using a Nikon E800 fluorescence microscope. The images correspond approximately to the same area of the dorsal horn in the spinal cord of the rats. Scale bar represents 100 μm. (d) the number of pixels occupied by GFAP immunoreactive cells within a defined threshold in a randomly selected area of the spinal cord dorsal horn, measured by Image J software (NIH). The data plotted is mean+SEM obtained from 3 independent rats; 6 spinal cord sections per rat per treatment group.
3.6. Co-administration of AM 1241 attenuates spinal pro-inflammatory mediator immunoreactivities in morphine-withdrawn rats
In lumbar dorsal spinal cord tissues collected after 96h drug withdrawal from the vehicle+morphine-treated animal group, there was a significant increase in spinal IL-1β (Fig 8A, 101±9 pg/mg total protein; *p<0.05 relative to vehicle-saline-treated control, one-way ANOVA, n=3 per group) and TNFα (Fig 8B, 65±4 pg/mg total protein; *p<0.05 relative to the vehicle+saline group, one-way ANOVA, n=3 per group) concentration. AM 1241 (2.5 mg/mg) co-administration significantly reduced IL-1β (59±10 pg/mg protein; p>0.05 relative to vehicle+saline, *p<0.05 relative to vehicle+morphine; one-way ANOVA, n=3 per group, Fig 8A) and TNFα (36±6 pg/mg protein; p>0.05 relative to vehicle+saline, *p<0.05 relative to vehicle+morphine group; Fig 8B) immunoreactivities in the lumbar spinal cord of morphine-withdrawn rats. Inflammatory mediator concentrations in the spinal cord of animals that received AM 1241 (2.5 mg/kg) alone were not significantly different from vehicle+saline group (p>0.05 relative to vehicle+saline group, one-way ANOVA, n=3 per group, Figs 8A, 8B). These data indicate that AM 1241 might mediate its effects by reducing the release of pro-inflammatory mediators in the spinal cord.
Fig. 8. AM 1241 co-administration attenuates proinflammatory cytokine immunoreactivity in the lumbar spinal cord of morphine-withdrawn rats.

After the behavioral tests, the animals were sacrificed and the lumbar spinal cords were isolated and flash frozen in liquid nitrogen. The spinal cords were homogenized on ice as described in the methods sections and the concentrations of IL-1β and TNFα proteins were measured by ELISA assays.
2. Discussion
The present study demonstrates that a) co-treatment of rats with AM 1241, a CB2-selective cannabinoid agonist attenuates sustained morphine-mediated hyperalgesia and allodynia (Figs 2,3); b) AM 1241 co-treatment attenuates sustained morphine-mediated paradoxical pain sensitization by activation of CB2 cannabinoid receptors c) co-treatment of rats with AM 1241 attenuates spinal microglia and astrocyte marker immunoreactivities in morphine withdrawn rats (Figs 6,7); and e) co-treatment of rats with AM 1241 attenuates up-regulation of pro-inflammatory mediators in the lumbar spinal cord of morphine-withdrawn rats (Fig 8). Taken together, these data suggest that CB2 agonist co-administration may attenuate the neuroinflammatory consequences of long-term opioid agonist therapy.
CNS glia play an important role in regulation of the “gain” (or amplification) of neuronal pain transmission. Activated glial cells release multiple pro-inflammatory neuromodulators into the spinal cord (DeLeo and Yazierski, 2001). Augmented spinal pro-inflammatory mediator levels increase the excitability of the primary sensory neurons (Ji and Strichartz, 2004), leading to augmented pain neurotransmitter release in response to external stimuli (Gardell et al., 2002). Indeed, glial proinflammatory inhibitors (such as fluorocitrate, propentophylline and minocycline) were shown to attenuate inflammatory and neuropathic pain sensitization (Cichewicz and Welch, 2003; Romero-Sandoval and Eisenach, 2007).
Interestingly, similar to inflammatory and neuropathic conditions, sustained morphine treatment also elevates pro-inflammatory neuromodulator concentrations (such as IL-1β, IL-6, TNF-α) (Raghavendra et al., 2003) and microglia and astrocyte marker immunoreactivities (Mika et al., 2007; Raghavendra et al., 2002, 2003; Song and Zhao, 2001; Watkins et al., 2005, 2007) and in the spinal cord of rodents. Spinal glial activation may play an important role in sustained morphine-mediated paradoxical pain sensitization and antinociceptive tolerance, since glial proinflammatory inhibitors were found to attenuate sustained morphine mediated paradoxical pain and to diminish analgesic tolerance in rodents (Cui et al., 2006, Johnston et al., 2004; Li et al., 2001; Mao et al., 1994; Raghavendra et al., 2002, 2003; Trang et al., 2004; Watkins et al., 2007).
The molecular mechanisms leading to sustained morphine-mediated augmentation of glial pro-inflammatory mediator synthesis are presently not entirely clear. Since morphine was shown to elicit functional responses (such as membrane ruffling and chemotaxis and enhanced BDNF gene expression (Takayama and Ueda, 2005);); augmented TNFα and IL-1 release (Mahajan et al., 2005) and increased p38 MAP kinase phosphorylation (Mouledos et al., 2004) in cultured glial cells, opioids may directly interact with glial cell membrane receptors. However, irregularities in the stereoselectivity of the opioid actions indicate that these specific glial binding sites may not correspond to classical opioid receptors. Thus, recently it was suggested that morphine may activate glial cells by interacting with Toll-like receptors (such as TLR-4) in a (+) naloxone-sensitive manner (Hutchinson et al., 2007). Alternatively, sustained opioids may also activate glial cells indirectly, for instance by stimulating excitatory modulator release from the central termini of the primary sensory neurons and/or from second order spinal neurons (again, either directly or by descending facilitatory mechanisms (Ossipov et al., 2005; Wang et al., 2009).
Regulation of spinal glial activity may be a useful novel method to increase the efficacy and duration of opioid antinociception. However, the “classical” glial proinflammatory inhibitors are either toxic (fluorocitrate) or are not specific for glia (minocycline and propentofylline) (Mika, 2008; Raghavendra et al., 2003). CNS microglia and astrocytes express numerous G protein coupled receptors (such as the CB2 cannabinoid receptor) offering new, potentially more selective methods to regulate glial activity.
The CB2 receptor is a member of the cannabinoid receptor family of G-protein coupled receptors. CB2 receptors are predominantly present in peripheral tissues, most remarkably, in the immune system. Interestingly, it was found that despite the predominantly peripheral localization of the CB2 receptors, CB2 agonists mediate remarkable pain relief in rodents (Ibrahim et al., 2003, 2006). CB2 agonists are especially efficient antinociceptive agents in hard-to-treat chronic pain conditions, such as inflammatory and neuropathic hyperalgesia and cancer pain (Beltramo et al., 2009;Lozano-Ondoua et al., 2010). Since CB2 receptors are expressed in very low concentration on CNS neurons, CB2-selective drugs may be devoid of the well-known centrally-mediated side effects of the classical cannabinoids. Consequently, CB2-selective cannabinoid agonists are promising new candidates for pain management. It was suggested earlier that the antinociceptive effect of the CB2-selective agonists may be due to stimulation of endogenous opioid release in peripheral tissues (Ibrahim et al., 2006). On the other hand, the mechanism(s) involved in the unexpectedly high efficacy of CB2-selective agonists against chronic inflammatory and neuropathic pain are presently not entirely clear.
CNS microglia and astrocytes express multiple functionally active G protein coupled receptors including the CB2 cannabinoid receptor type (Pocock and Kettenmann, 2007). Recent data suggest that CB2 agonist-mediated inhibition of glial activation may play an important role in their efficacy in inflammatory and neuropathic pain. Inflammation and nerve injury dramatically increases the expression of CB2 receptors in cultured microglia and astrocytes in vitro and in the spinal cord of rodents in vivo (Maresz et al., 2005; Stella, 2004, Fernandez-Ruiz et al., 2007). Augmented spinal glial CB2 receptor expression may play a role in regulation of microglia and astrocyte activity and glial excitatory neuromodulator synthesis (Ehrhart et al., 2006; Fernandez-Ruiz et al., 2007). Recent data indicate that glial CB2 cannabinoid receptors regulate the synthesis of excitatory immunomodulators by augmenting MAP kinase phosphatase expression in cultured microglia, leading to the attenuation of MAP kinase signaling in these cells (Elijatschewitsch et al., 2006).
Numerous shared characteristics between neuropathic and sustained morphine-induced pain sensitization and analgesic tolerance led to the suggestion that these processes may share common mechanisms (Ossipov et al, 2005). Thus, similar to inflammatory and neuropathic conditions, sustained morphine treatment induced MAP kinase phosphorylation (Cui et al., 2006) elevated excitatory neuromodulator levels (Raghavendra et al., 2002) and induced a time-dependent upregulation of CB2 receptors (Lim et al., 2006) within the spinal cord of rodents. Therefore, we hypothesized that by activation of glial CB2 receptors, CB2 agonists may be able to attenuate sustained morphine-mediated spinal glial activation and thus, prevent paradoxical pain sensitization as observed in case of inflammatory and neuropathic pain states. Our data demonstrate that indeed, co-administration of a selective CB2 agonist (AM 1241) attenuates repeated morphine-mediated pain sensitization and prevents withdrawal hyperalgesia and allodynia in rats (Figs 2, 3). Our results also show that the process is mediated by activation of the CB2 receptor type, since a CB2-selective antagonist (AM 630) attenuated the protective effects of AM 1241 while a CB1-selective antagonist (AM 251) had no effect (Figs 4,5). Co-treatment with AM 1241 also reduced astrocyte and microglia marker immunoreactivities in the lumbar dorsal horn of morphine withdrawn rats (Figs 7,8), indicating that activation of CB2 receptors reduces spinal glial proliferation.
Activation of spinal glia was shown to augment the concentration of pro-inflammatory mediators (gliotransmitters) in the spinal cord (Hulsebosch CE, 2008, Hutchinson et al., 2007) leading to sensitized basal and/or evoked pain neurotransmitter release from the primary sensory neurons (Hou and Wang, 2003; Tumati et al., 2010). Accordingly, glial proinflammatory inhibitors reduced spinal inflammatory mediator concentrations and attenuated pain sensitization in neuropathic and inflammatory conditions (DeLeo et al., 2001; Watkins et al., 2007; Hulsebosch et al., 2008) Our data indicates that activation of CB2 receptors by AM 1241 also completely attenuates the upregulation of IL-1β and TNF-α immunoreactivities in the lumbar spinal cord of morphine-withdrawn rats (Figs 6A, 6B), indicating that CB2 agonist co-administration may be an effective method to reduce the pro-inflammatory consequences of sustained opioid analgesic treatment.
It was suggested that sustained opioid analgesic-mediated paradoxical pain sensitization may contribute to the need for increased opioid doses during the clinical treatment of chronic pain (Vanderah et al., 2001a). Higher doses on the other hand aggravate the dangerous side effects of opioids (such as constipation, respiratory depression and addiction liability). Our current data provides a novel pharmacological approach, the use of CB2 agonists - devoid of the well-known CNS side effects - to prevent the neuroinflammatory consequences of sustained opioid analgesic treatment and thus increase the efficacy and prolong the duration of action of the opioid analgesics in chronic pain treatment.
Acknowledgments
The work was supported by grants from the National Institutes of Health (DA027786 and DA006284).
Abbreviations
- AM 1241
(3-iodo-5-nitrophenyl)-[1-[(1-methylpiperidin-2-yl)methyl]indol-3-yl]methanone
- i.p
intraperitoneal
- Isoflurane
2-chloro-2-(difluoromethoxy)-1,1,1-trifluoro-ethane
- AM 251
1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-(1-piperidyl)pyrazole-3-carboxamide)
- AM 630
(6-iodo-2-methyl-1-[2-(4-morpholinyl)ethyl]-1H-indol-3-yl](4-methoxyphenyl) methanone)
- i.t
intrathecal
- Ketamine
(RS)-2-(2-Chlorophenyl)-2-(methylamino)-cyclohexan-1-one
- Morphine
(5α,6α)-7,8-didehydro-4,5-epoxy-17-methylmorphinan-3,6-diol
- Xylazine
N-(2,6-dimethylphenyl)-5,6-dihydro-4H-1,3-thiazin-2-amine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arner S, Rawal N, Gustafsson LL. Clinical experience of long-term treatment with epidural and intrathecal opioids - a nationwide survey. Acta Anaesthesiol Scand. 1988;32:253–259. doi: 10.1111/j.1399-6576.1988.tb02725.x. [DOI] [PubMed] [Google Scholar]
- Beltramo M. Cannabinoid type 2 receptor as a target for chronic pain. Mini Rev Med Chem. 2009;9:11–25. doi: 10.2174/138955709787001785. [DOI] [PubMed] [Google Scholar]
- Chang SL, Sharp BM, Madden JJ. Cellular mechanisms involved in the modulation of the immune system by drugs of abuse. Adv Exp Med Biol. 1998;437:1–12. doi: 10.1007/978-1-4615-5347-2_1. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55– 63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Cichewicz DL, Welch SP. Modulation of oral morphine antinociceptive tolerance and naloxone-precipitated withdrawal signs by oral Delta 9-tetrahydrocannabinol. J Pharmacol Exp Ther. 2003;305:812–817. doi: 10.1124/jpet.102.046870. [DOI] [PubMed] [Google Scholar]
- Cui YY, Zhi JL, Guo RX, Feng JQ, Chen PX. Activation of p38 mitogen-activated protein kinase in spinal microglia mediates morphine antinociceptive tolerance. Brain Res. 2006;1069:235–243. doi: 10.1016/j.brainres.2005.11.066. [DOI] [PubMed] [Google Scholar]
- DeLeo JA, Yezierski RP. The role of neuroinflammation and neuroimmune activation in persistent pain. Pain. 2001;90:1–6. doi: 10.1016/s0304-3959(00)00490-5. [DOI] [PubMed] [Google Scholar]
- Dixon WJ. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol. 1980;20:441–462. doi: 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- Ehrhart J, Obregon D, Mori T, Hou H, Sun N, Bai Y, Klein T, Fernandez F, Tan J, Shytle RD. Stimulation of cannabinoid receptor 2 (CB2) suppresses microglial activation. J Neuroinflammation. 2005;2:29–35. doi: 10.1186/1742-2094-2-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eljaschewitsch E, Witting A, Mawrin C, Lee T, Schmidt PM, Wolf S, Hoertnagl H, Raine CS, Schneider-Stock R, Nitsch R, Ullrich O. The Endocannabinoid Anandamide Protects Neurons during CNS Inflammation by Induction of MKP-1 in Microglial Cells. Neuron. 2006;49:67–79. doi: 10.1016/j.neuron.2005.11.027. [DOI] [PubMed] [Google Scholar]
- Fernández-Ruiz J, Romero J, Velasco G, Tolón RM, Ramos JA, Guzmán M. Cannabinoid CB2 receptor: a new target for controlling neural cell survival? Trends Pharmacol Sci. 2007;28:39–45. doi: 10.1016/j.tips.2006.11.001. [DOI] [PubMed] [Google Scholar]
- Gardell LR, Wang R, Burgess SE, Ossipov MH, Vanderah TW, Malan TP, Jr, Lai J, Porreca F. Sustained morphine exposure induces a spinal dynorphin-dependent enhancement of excitatory transmitter release from primary afferent fibers. J Neurosci. 2002;22:6747–6755. doi: 10.1523/JNEUROSCI.22-15-06747.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Hou L, Li W, Wang X. Mechanism of IL-1β-induced CGRP peptide production from DRG neurons of neonatal rats. J Neurosci Res. 2003;73:188–197. doi: 10.1002/jnr.10651. [DOI] [PubMed] [Google Scholar]
- Hulsebosch CE. Gliopathy ensures persistent inflammation and chronic pain after spinal cord injury. Exp Neurol. 2008;214:6–9. doi: 10.1016/j.expneurol.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Bland ST, Johnson KW, Rice KC, Maier SF, Watkins LR. Opioid-induced glial activation: mechanisms of activation and implications for opioid analgesia, dependence, and reward. Sci World J. 2007;7:98–111. doi: 10.1100/tsw.2007.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim MM, Deng H, Zvonok A, Cockayne DA, Kwan J, Mata HP, Vanderah TW, Lai J, Porreca F, Makriyannis A, Malan TP., Jr Activation of CB2 cannabinoid receptors by AM 1241 inhibits experimental neuropathic pain: pain inhibition by receptors not present in the CNS. Proc Natl Acad Sci USA. 2003;100:10529–10533. doi: 10.1073/pnas.1834309100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim MM, Rude ML, Stagg NJ, Mata HP, Lai J, Vanderah TW, Porreca F, Buckley NE, Makriyannis A, Malan TP., Jr CB2 cannabinoid receptor mediation of antinociception. Pain. 2006;122:36–42. doi: 10.1016/j.pain.2005.12.018. [DOI] [PubMed] [Google Scholar]
- Ji RR, Strichartz G. Cell signaling and the genesis of neuropathic pain. Sci STKE. 2004;21:E14. doi: 10.1126/stke.2522004re14. [DOI] [PubMed] [Google Scholar]
- Johnston IN, Milligan ED, Wieseler-Frank J, Frank MG, Zapata V, Campisi J, Langer S, Martin D, Green P, Fleshner M, Leinwand L, Maier SF, Watkins LR. A role for proinflammatory cytokines and fractalkine in analgesia, tolerance, and subsequent pain facilitation induced by chronic intrathecal morphine. J Neurosci. 2004;24:7353–7365. doi: 10.1523/JNEUROSCI.1850-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Angst MS, Clark JD. A murine model of opioid-induced hyperalgesia. Mol Brain Res. 2001;86:56–62. doi: 10.1016/s0169-328x(00)00260-6. [DOI] [PubMed] [Google Scholar]
- Lim G, Wang S, Mao J. Central glucocorticoid receptors modulate the expression of spinal cannabinoid receptors induced by chronic morphine exposure. Brain Res. 2005;1059:20–27. doi: 10.1016/j.brainres.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Lozano-Ondoua AN, Wright C, Vardanyan A, King T, Largent-Milnes TM, Nelson M, Jimenez-Andrade JM, Mantyh PW, Vanderah TW. A Cannabinoid 2 receptor agonist attenuates bone cancer-induced pain and bone loss. Life Sci. 2010;86:646–653. doi: 10.1016/j.lfs.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan SD, Schwartz SA, Aalinkeel R, Chawda RP, Sykes DE, Nair MP. Morphine modulates chemokine gene regulation in normal human astrocytes. Clin Immunol. 2005;115:323–32. doi: 10.1016/j.clim.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Mao J, Price DD, Mayer DJ. Thermal hyperalgesia in association with the development of morphine tolerance in rats: roles of excitatory amino acid receptors and protein kinase C. J Neurosci. 1994;14:2301–2312. doi: 10.1523/JNEUROSCI.14-04-02301.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maresz K, Carrier EJ, Ponomarev ED, Hillard CJ, Dittel BN. Modulation of the cannabinoid CB2 receptor in microglial cells in response to inflammatory stimuli. J Neurochem. 2005;95:437–445. doi: 10.1111/j.1471-4159.2005.03380.x. [DOI] [PubMed] [Google Scholar]
- Mika J. Modulation of microglia can attenuate neuropathic pain symptoms and enhance morphine effectiveness. Pharmacological Reports. 2008;60:297–307. [PubMed] [Google Scholar]
- Mika J, Osikowicz M, Makuch W, Przewlocka B. Minocycline and pentoxifylline attenuate allodynia and hyperalgesia and potentiate the effects of morphine in rat and mouse models of neuropathic pain. Eur J Pharmacol. 2007;560:142–149. doi: 10.1016/j.ejphar.2007.01.013. [DOI] [PubMed] [Google Scholar]
- Moulédous L, Diaz MF, Gutstein HB. Modulation of extracellular signal-regulated kinase (ERK) activity by acute and chronic opioid treatment in neuronal and glial cell lines. J Neurochem. 2004;90:1371–1377. doi: 10.1111/j.1471-4159.2004.02610.x. [DOI] [PubMed] [Google Scholar]
- Ossipov MH, Lai J, King T, Vanderah TW, Porreca F. Underlying mechanisms of pronociceptive consequences of prolonged morphine exposure. Biopolymers. 2005;80:319–324. doi: 10.1002/bip.20254. [DOI] [PubMed] [Google Scholar]
- Pocock JM, Kettenmann H. Neurotransmitter receptors on microglia. Trends Neurosci. 2007;30:527–535. doi: 10.1016/j.tins.2007.07.007. [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Rutkowski MD, DeLeo JA. The role of spinal neuroimmune activation in morphine tolerance/hyperalgesia in neuropathic and sham-operated rats. J Neurosci. 2002;22:9980–9989. doi: 10.1523/JNEUROSCI.22-22-09980.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavendra V, Tanga F, DeLeo JA. Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J Pharmacol ExpTher. 2003;306:624–630. doi: 10.1124/jpet.103.052407. [DOI] [PubMed] [Google Scholar]
- Rodriguez Parkitna J, Korostynski M, Kaminska-Chowaniec D, Obara I, Mika J, Przewlocka B, Przewlocki R. Comparison of gene expression profiles in neuropathic and inflammatory pain. J Physiol Pharmacol. 2006;57:401–414. [PubMed] [Google Scholar]
- Romero-Sandoval A, Eisenach JC. Spinal cannabinoid receptor type 2 activation reduces hypersensitivity and spinal cord glial activation after paw incision. Anesthesiology. 2007;106:787–794. doi: 10.1097/01.anes.0000264765.33673.6c. [DOI] [PubMed] [Google Scholar]
- Romero-Sandoval EA, Horvath R, Landry RP, DeLeo JA. Cannabinoid receptor type 2 activation induces a microglial anti-inflammatory phenotype and reduces migration via MKP induction and ERK dephosphorylation. Mol Pain. 2009;5:25. doi: 10.1186/1744-8069-5-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song P, Zhao ZQ. The involvement of glial cells in the development of morphine tolerance. Neurosci Res. 2001;39:281–286. doi: 10.1016/s0168-0102(00)00226-1. [DOI] [PubMed] [Google Scholar]
- Stella N. Cannabinoid signaling in glial cells. Glia. 2004;48:267–277. doi: 10.1002/glia.20084. [DOI] [PubMed] [Google Scholar]
- Takayama N, Ueda H. Morphine-induced chemotaxis and brain-derived neurotrophic factor expression in microglia. J Neurosci. 2005;25:430–5. doi: 10.1523/JNEUROSCI.3170-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trang T, McNaull B, Quirion R, Jhamandas K. Involvement of spinal lipoxygenase metabolites in hyperalgesia and opioid tolerance. Eur J Pharmacol. 2004;491:21–30. doi: 10.1016/j.ejphar.2004.03.022. [DOI] [PubMed] [Google Scholar]
- Tumati S, Roeske WR, Vanderah TW, Varga EV. Sustained morphine treatment augments prostaglandin E2-evoked calcitonin gene-related peptide release from primary sensory neurons in a PKA-dependent manner. Eur J Pharmacol. 2010;648:95–101. doi: 10.1016/j.ejphar.2010.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderah TW, Gardell LR, Burgess SE, Ibrahim M, Dogrul A, Zhong CM, Zhang ET, Malan TP, Jr, Ossipov MH, Lai J, Porreca F. Dynorphin promotes abnormal pain and spinal opioid antinociceptive tolerance. J Neurosci. 2000;20:7074–7079. doi: 10.1523/JNEUROSCI.20-18-07074.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderah TW, Ossipov MH, Lai J, Malan TP, Jr, Porreca F. Mechanisms of opioid-induced pain and antinociceptive tolerance: Descending facilitation and spinal dynorphin. Pain. 2001a;92:5–9. doi: 10.1016/s0304-3959(01)00311-6. [DOI] [PubMed] [Google Scholar]
- Vanderah TW, Suenaga NM, Ossipov MH, Malan TP, Jr, Lai J, Porreca F. Tonic descending facilitation from the rostral ventromedial medulla mediates opioid-induced abnormal pain and antinociceptive tolerance. J Neurosci. 2001b;21:279–286. doi: 10.1523/JNEUROSCI.21-01-00279.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Ma W, Chabot JG, Quirion R. Cell-type specific activation of p38 and ERK mediates calcitonin gene-related peptide involvement in tolerance to morphine-induced analgesia. FASEB J. 2009;23:2576–2586. doi: 10.1096/fj.08-128348. [DOI] [PubMed] [Google Scholar]
- Wanigasekera V, Lee MCH, Rogers R, Hu P, Tracey I. Neural correlates of an injury-free model of central sensitization induced by opioid withdrawal in humans. J Neurosci. 2011;31:2835–2842. doi: 10.1523/JNEUROSCI.5412-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins LR, Hutchinson HR, Johnston JN, Maier SF. Glia: novel counter-regulators of opioid analgesia. Trends in Neurosci. 2005;28:661–669. doi: 10.1016/j.tins.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Hutchinson MR, Ledeboer A, Wieseler-Frank J, Milligan ED, Maier SF. Glia as the “bad guys”: implications for improving clinical pain control and the clinical utility of opioids. Brain Behav Immun. 2007;21:131–146. doi: 10.1016/j.bbi.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]