

Abstract
MUC4 is a large transmembrane type I glycoprotein that is overexpressed in pancreatic cancer (PC) and has been shown to be associated with its progression and metastasis. However, the exact cellular and molecular mechanism(s) through which MUC4 promotes metastasis of PC cells has been sparsely studied. Here we showed that the NIDO domain of MUC4, which is similar to the G1-domain present in the nidogen or entactin (an extracellular matrix protein), contributes to the protein-protein interaction property of MUC4. By this interaction, MUC4 promotes breaching of basement membrane integrity, and spreading of cancer cells. These observations are corroborated with the data from our study using an engineered MUC4 protein without the NIDO domain, which was ectopically expressed in the MiaPaCa PC cells, lacking endogenous MUC4 and nidogen protein. The in vitro studies demonstrated an enhanced invasiveness of MiaPaCa cells expressing MUC4 (MiaPaCa-MUC4) compared to vector-transfected cells (MiaPaCa-Vec; p=0.003) or cells expressing MUC4 without the NIDO domain (MiaPaCa-MUC4-NIDOΔ; p=0.03). However, the absence of NIDO-domain has no significant role on cell growth and motility (p=0.93). In the in-vivo studies, all the mice orthotopically implanted with MiPaCa-MUC4 cells developed metastasis to the liver as compared to MiaPaCa-Vec or the MiaPaCa-MUC4-NIDOΔ group, hence, supporting our in vitro observations. Additionally, a reduced binding (p=0.0004) of MiaPaCa-MUC4-NIDOΔ cells to the fibulin-2 coated plates compared to MiaPaCa-MUC4 cells indicated a possible interaction between the MUC4-NIDO domain and fibulin-2, a nidogen-interacting protein. Furthermore, in PC tissue samples, MUC4 colocalized with the fibulin-2 present in the basement membrane. Altogether, our findings demonstrate that the MUC4-NIDO domain significantly contributes to the MUC4-mediated metastasis of PC cells. This may be partly due to the interaction between the MUC4-NIDO domain and fibulin-2.
Keywords: MUC4, pancreatic cancer, metastasis, NIDO domain, MUC4 domains
Introduction
Pancreatic cancer remains an unfortunate disease with an overall cumulative 5-year survival rate below 5%. Despite advances in therapy and surgical techniques, it represents one of the leading cause of cancer related deaths (Jemal et al. 2010; Li et al. 2004). One of the main characteristic of pancreatic cancer is its propensity to locally progress and to disseminate at an early stage (DiMagno et al. 1999). To this regard, the specific targeting of pancreatic cancer metastasis remains a real therapeutic challenge. During the last decade, various experimental and clinical evidences have helped to enrich our comprehension regarding the genetic alterations that are involved in pancreatic cancer progression. However, the underlying mechanisms that regulate the progression of pancreatic cancer through different stages of metastatic cascade are still unclear. Particularly, how the malignant cells detach from the primary tumor site and invade different host barriers is an important area to be dissected.
Metastasis to distant organs requires a series of coordinated steps. During hematogenous metastasis of tumor cells (epithelial origin) starting from the spreading of tumor cells from the primary site to the establishment of secondary tumors at the distant organs, the tumor cells have to disrupt the basement membrane integrity at three different steps (Langley and Fidler 2007). First the tumor cells have to break the epithelial basement membrane to invade the local tissues, followed by disrupting the endothelial basement membrane integrity during tumor cells intravasation and extravation process (Nguyen et al. 2009). Metastatic tumor cells can rupture the basement membrane integrity either by promoting its proteolytic degradation (protease-dependent) or by mediating a physical disruption (protease-independent transmigration) (Bacac and Stamenkovic 2008; Rowe and Weiss 2008). Protease dependent degradation of basement membrane is well explored and is mostly carried out by membrane-type matrix metalloproteinases (Hotary et al. 2006). Whereas, the exact mechanisms for a protease-independent mode of invasion are yet to be defined. In this article, we discuss the role of MUC4-NIDO domain as a mediator of protease independent invasion in pancreatic cancer metastasis.
Basement membranes are specialized extracellular matrices composed of many proteins. Many of the basement membrane proteins like collagenV, laminin, entactin/nidogen, and proteoglycans have specific protein-protein interaction motifs in their domain structures. These proteins interact with each other in a coordinated fashion and maintaining the supramolecular structural organization. Likewise, some proteins present on the surface of the normal or neoplastic transformed cells also specifically interact with these basement membrane proteins (Casey and Skubitz 2000). Nidogen (entactin) is a ubiquitous constituent of the basement membrane. It has three globular structures (G1, G2 and G3). It was shown that the G3 and G2 domains of nidogen interact with laminin and collagen IV respectively, and thus, bridges laminin and collagen IV in the basement membrane (Aumailley et al. 1993; Fox et al. 1991). Furthermore, a recent study has also shown that the G1 domain of nidogen interacts with fibulin-2 (Ries et al. 2001).
MUC4 is a type I glycoprotein overexpressed in pancreatic cancer cells, which promotes metastasis of pancreatic cancer cells (Chaturvedi et al. 2007a; Singh et al. 2004; Singh et al. 2007b; Singh et al. 2007b). MUC4 apomucin is a 930 kDa protein, composed of a 850 kDa mucin-type subunit (MUC4α) and an 80 kDa membrane-tethered growth factor-like subunit (MUC4β). MUC4α possesses three important domains [TR (tandem-repeat), NIDO (nidogen-like) and AMOP (adhesion-associated domain in MUC4 and other proteins)], while MUC4β has three EGF-like domains and a short cytoplasmic tail (Chaturvedi et al. 2008a; Singh et al. 2007a; Duraisamy et al. 2006). The MUC4α-subunit is thought to participate in adhesion and anti-adhesion mechanisms, while the role of MUC4β is proposed in cell signaling. In our previous studies, using ‘loss’ and ‘gain’ of function approach, we have shown a direct association of the MUC4 mucin with the metastatic PC phenotype and provided experimental evidence for a functional role of MUC4 in altered growth and invasive properties of tumor cells (Chaturvedi et al. 2007a; Singh et al. 2004).
Out of all the functional domains of MUC4, the MUC4-NIDO domain is considered to be playing a role in rupturing the integrity of basement membrane and helping in the metastasis of pancreatic cancer cells, due to its similarity with the G1 (NIDO) domain of nidogen protein. As mentioned above, the G1-domain of nidogen interacts with fibulin-2 and is thought to contribute to the protein-protein crosslinking property of nidogen (Aumailley et al. 1993), which is essential to maintain the basement membrane integrity. Previous studies have also shown that the NIDO domain of the endogenous nidogen protein has a potential binding site for fubulin-2 (Aumailley et al. 1993). Also, fibulin-2 plays a major role in the maintenance of epithelial and endothelial basement membrane integrity (Yi et al. 2007). Therefore, it is possible that MUC4-NIDO domain may also interact with fibulin-2, and thus, inhibit normal interaction between nidogen and fibulin-2 proteins. By doing so, the MUC4-NIDO domain may promote the metastasis of MUC4-expressing cancer cells.
In this report, we have investigated whether the deletion of the NIDO domain from the MUC4 protein alters the MUC4-mediated metastasis of pancreatic cancer cells. Expression of modified MUC4 protein without the NIDO domain showed a reduced invasive property of PC cells compared to cells expressing MUC4 with the NIDO domain. Further, the MUC4-NIDO domain also promoted the extravasion of pancreatic cancer cells in vitro. Importantly, the MUC4-NIDO domain interacts with fibulin-2, and this may be a possible mechanism through which MUC4 promotes pancreatic cancer metastasis.
Results
Characterization of MiaPaCa-derived MUC4 or MUC4-NIDOΔ transfected sublines
To investigate if MUC4 NIDO domain is associated with some of the functional properties of MUC4, in-frame deletion of NIDO-domain was done within the previously generated engineered MUC4 (Moniaux et al. 2007) (Figure 1A). The MUC4, MUC4-NIDOΔ construct and empty vector were transfected into MiaPaCa cells. This cell line was chosen as it expressed neither MUC4 nor nidogen protein (Figure 1B; Supplementary figure 1). Multiple stable clones were selected upon growing the cells in zeocin containing media. Cells expressing MUC4 (MiaPaCa-MUC4) or the MUC4 without the NIDO domain (MiaPaCa- MUC4-NIDOΔ) proteins were examined via Western blot analysis (Figure 1B). The selected sub-lines were monitored over a period of two to three months for the stable overexpression of the proteins. Further, the cell surface localization of these proteins was confirmed by flow cytometric analysis (Figure 1C). A pooled population of the sub-lines was selected for additional in vitro and in vivo studies.
Figure 1. Generation of MUC4-NIDO domain deletion construct and its stable-expression in MiaPaCa cells.
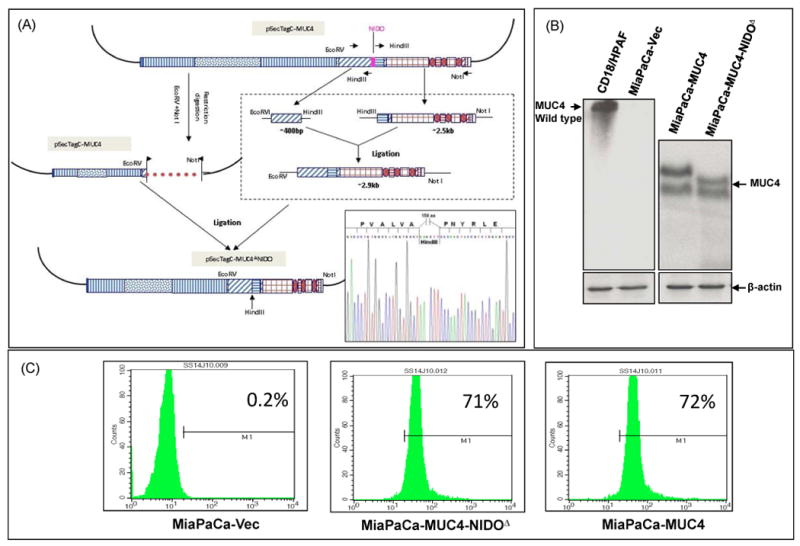
(A) The construction of the MUC4-NIDO domain deletion construct was performed in three steps. First, using Expand Long PCR, and generating two fragments were generated just 5′ and 3′ to the NIDO domain. Then the MUC4-NIDO domain deletion fragment was successfully inserted into the first generation MUC4 c-DNA construct using restriction digestion and cloning strategies (Moniaux et al. 2007). Further, sequencing of the newly generated construct was performed, showing an in-frame deletion of 474 nucleotides that code for the MUC4-NIDO domain (of 158 amino acids). (B) MUC4 expression was analyzed by immunoblot in different MiaPaCa-derived sublines (pooled population obtained from three different clones). Cell lysate from CD18/HPAC pancreatic cancer cells was taken as a positive control. A total of 20 μg protein from cell extracts was electrophoretically resolved on a 2% agarose gel. Resolved proteins were transferred onto PVDF membrane and probed with MUC4 MAb (8G7). β-actin was probed as an internal control.
(C) Flow cytometric analysis showed the cell surface localization of MUC4 and MUC4-NIDOΔ proteins on MiaPaCa-MUC4 (72%) and MiaPaCa-MUC4-NIDOΔ (71%) cells, respectively. The MiaPaCa cell lines transfected with the empty vector (MiaPaCa-Vect) was taken as a negative control (0.2%).
The MUC4-NIDO domain does not alter the motility of pancreatic cancer cells; however promotes invasion and extravasations of pancreatic cancer cells
In order to examine the role of the MUC4-NIDO domain in cell motility, a motility assay was performed by using uncoated porous membranes of 8.0 mm pore diameter (Boyden chamber). The number of cells that migrated to the lower surface of the Boyden chamber was almost equal in both MUC4 and MUC4 without NIDO domain expressing MiaPaCa cells (Figure 2A, p=0.933). However, as observed before (Moniaux et al. 2007), an overexpression of either MUC4 or MUC4-NIDOΔ significantly (∼2 fold) increased the number of cells that migrated through the Boyden chamber when compared to the untransfected/vector-transfected cells (Figure 2A; p=0.02).
Figure 2. Effect of MUC4-NIDO domain deletion on cell motility, invasion and extravasations.
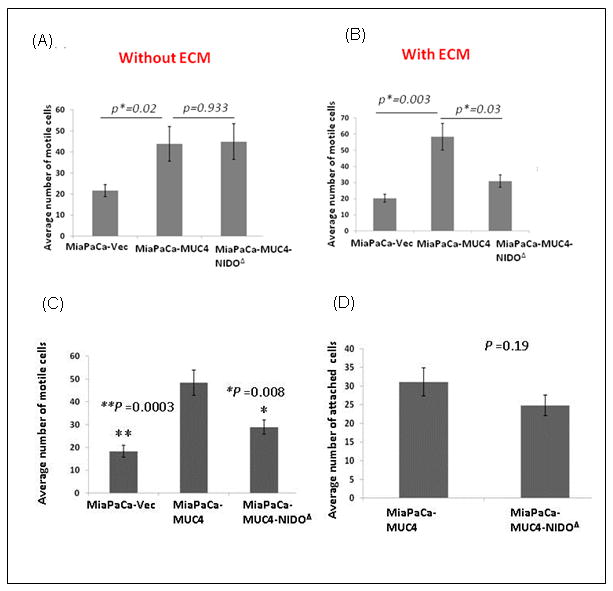
(A) Cell motility assay: 1×106 cells were plated in the top chamber of non-coated polyethylene teraphthalate membranes and incubated for 22 hrs. Cells that transversed the membranes were stained with a Diff-Quick cell staining kit. The number of cells transversing the membrane were determined by averaging 10 random fields of view at 10× and expressed as the average number of cells/field of view and as the average of three independent experiments. Mean+/- SE p<0.05. No significant difference was found between MiaPaCa-MUC4 and MiaPaCa-MUC4-NIDOΔ cells (p= 0.933). (B) The invasion assay was carried out in a similar fashion; except at the place of non-coated trans-well inserts, ECM/Matrigel-coated transwell inserts (BD Biosciences, Bedford, MA) were used. The MiaPaCa-MUC4 cells showed a significantly higher number of invasive cells than MiaPaCa-MUC4-NIDOΔ cells (p=0.03). In both the assays, the MiaPaCa-MUC4 cells showed a significantly higher number of motile (p=0.02) and invasive (p=0.003) cells than the vector control ones. (C) The MiaPaCa-MUC4-NIDOΔ cells show less trans-HUVEC invasion (extravasation) compared to MiaPaCa-MUC4 cells. The number of cells transversing the membrane were determined by averaging 10 random fields of view at 10× and expressed as the average number of cells/field of view and as the average of three independent experiments. Mean+/- SE; p<0.008. (D) The cell-cell adhesion assay showed no significant difference between the number of MiaPaCa-MUC4 and MiaPaCa-MUC4-NIDOΔ cells that adhered to HUVECs (p=0.19).
Tumor cell invasion through the extracellular matrix (ECM) and tissue barriers require the combined effects of increased cell motility and proteolytic and/or non-proteolytic degradation of ECM. Therefore, to check the role of the MUC4-NIDO domain in the MUC4-induced invasive property of pancreatic cancer cells, the above mentioned trans-well migration assay was carried out in presence of extracellular matrix proteins (Matrigel). Interestingly, MUC4-overexpressing cells showed a significantly higher number of cells migrating through the ECM proteins than cells without the MUC4 expression (∼2.4 fold; p=0.003) or cells expressing MUC4 without the NIDO domain (∼1.6 fold; p= 0.03) (Figure 2B).
Additionally, in the hematogenous metastasis of pancreatic cancer cells, during intravasation and extravasation steps, the tumor cells interact with the endothelial basement membrane. To investigate the role of the MUC4-NIDO domain in these processes, an in vitro trans endothelial invasion assay was carried out. In this experiment, MiaPaCa-derived cells were subjected to migration through a tight monolayer of Human umbilical vein endothelial cells (HUVECs), which were cultured on the surface of trans well inserts coated with matrigel (8 micron pore filters; BD Falcon). Statistically highly significant number of tumor cells expressing MUC4 (∼ 1.7 fold; p=0.008) migrated through the ECM layer covered with the endothelial cells, compared to the MiaPaCa- MUC4-NIDOΔ cells (Figure 2C). However, in a parallel experiment, NIDO-domain deletion did not have any effect on the number of cancer cells that adhered to the endothelial cells (p=0.19; Figure 2D).
The MUC4-NIDO domain promotes metastasis of pancreatic cancer cells in vivo
To investigate the role of the MUC4-NIDO domain in vivo, we carried out the orthotopic implantation of MiaPaCa cells expressing MUC4 or MUC4 without the NIDO domain. In parallel, we also injected vector transfected MiaPaCa cells as a control (MiaPaCa-Vec). For this experiment, two millions of MiaPaCa-derived cells were injected into the head of the pancreas, and animals were sacrificed after 65days of the cancer cells injection. During autopsy of these mice, we did not find a significant difference in the mean tumor weight between MiaPaCa-MUC4 and MiaPaCa- MUC4-NIDOΔ groups (Figure 3B). However, animals in both the groups have a significantly higher mean tumor weight than the vector control animals (Figure 3B; p<0.005). Importantly, we found that the only animal group that developed massive metastasis to the liver was the one implanted with MUC4 expressing MiaPaCa cells. In this particular group of animals, out of eight animals, seven animals had liver metastasis and the average number of metastatic nodules per animal was ∼3. Incidence of metastasis to other organs like spleen, mesentery, peritoneum and diaphragm was similar in all the three groups (Figure 3A, B, and Table 1). Furthermore, histopathological evaluation of primary and metastatic tumors obtained from MiaPaCa-MUC4 and MiaPaCa- MUC4-NIDOΔ groups did not show any gross difference in the pattern of local tissue invasiveness of the cancer cells (Figure 4).
Figure 3. Macroscopic examinations of orthotopic pancreatic tumors and organs with metastatic lesions.
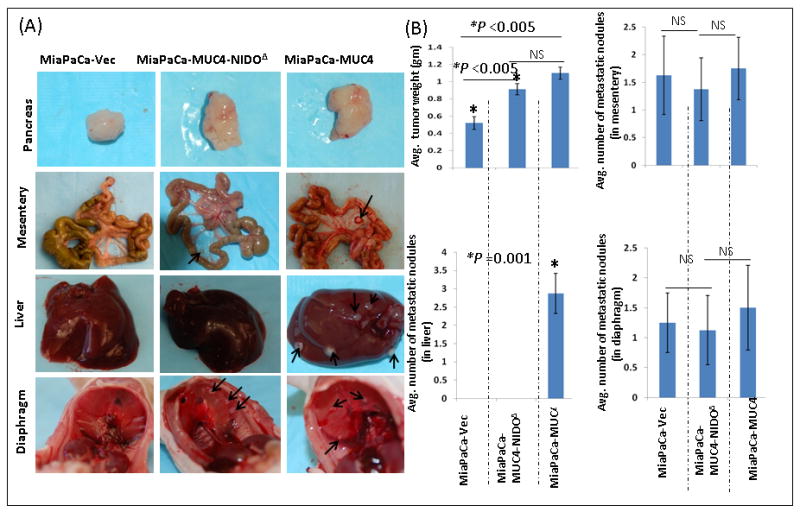
(A). Macroscopic appearance of primary pancreatic tumors from animals implanted with MiaPaCa-MUC4, MiaPaCa-MUC4-NIDOΔ and MiaPaCa-Vect cells. There was no difference in the gross appearance of tumors derived from MiaPaCa-MUC4 and MiaPaCa-MUC4-NIDOΔ groups. However, tumors of the MiaPaCa-Vect group were smaller than the other two groups. Further, metastasis to organs such as the diaphragm and mesentery was same in all the three groups excluding liver metastasis, which was observed only in animals injected with MiaPaCa-MUC4 cells. (B) Quantitative analysis showed a significantly smaller tumor generated from MiaPaCa-Vect cells than MiaPaCa-MUC4 and MiaPaCa-MUC4-NIDOΔ. Further analysis of metastatic lesions among the three groups showed no significant difference in the number of metastatic nodules that were detected in mesentery and the diaphragm. However, there was a significant difference (p=0.001) in the number of metastatic nodules present in the liver.
Table 1. Incidence of organ specific tumor nodule formation by Miapaca Paca MUC4, vector control and MUC4 NidoΔ.
| Group | Organ | ||||
|---|---|---|---|---|---|
| Liver | Mesentery | Diaphragm | Spleen | Peritoneum | |
| MiaPaCa-MUC4 | 7/8 (87%) | 5/8 (62.5%) | 4/8 (50%) | 8/8 (100 %) | 2/8 (25%) |
| MiaPaCa-MUC4-NIDOΔ | 0/8 (0%) | 4/8 (50%) | 3/8 (37.5%) | 8/8 (100%) | 2/8 (25%) |
| MiaPaCa-Vec | 0/8 (0%) | 4/8 (50%) | 4/8 (50%) | 8/8 (100%) | 2/8 (25%) |
Figure 4. Microscopic examinations of orthotopic pancreatic tumors and organs with metastasis lesions.
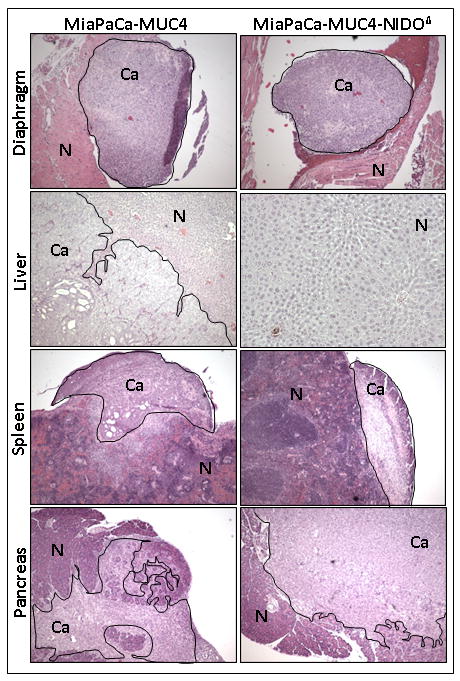
Histological appearance of pancreas, spleen, liver and diaphragm showing prominent metastasis in the mice implanted with MiaPaCa-MUC4 or MiaPaCa-MUC4-NIDOΔ cells. Between these two groups, there was no gross difference in the local tissue invasion pattern. Normal (N) and cancer cells (Ca) in different organs are separately indicated.
The MUC4 and the Fibulin-2 colocalises in pancreatic cancer tissue and MUC4 NIDO-domain interacts with Fibulin-2 in vitro
The similarity of the MUC4-NIDO domain with the G1/NIDO domain of endogenous nidogen protein indicates that MUC4-NIDO domain may interact with nidogen-G1 domain interacting proteins. In this regard, a previous study has shown an interaction between fibulin-2 and the nidogen-G1 domain thus proving that fibulin-2 is one of the interacting proteins for the nidogen protein (Ries et al. 2001). The immunohistofluorescence analysis showed the colocalization of MUC4 and fibulin-2 in the exocrine glands of the adenocarcinoma tissues. However, both the proteins' expression was absent in the endocrine glands of the same tissue section (Figure 5A). Further, to test the possible interaction between the MUC4-NIDO domain with fibulin-2, we carried out a cell adhesion assay on the surface of fibulin-2 coated plates. We observed a significantly higher number of MiaPaCa-MUC4 cells binding to the fibulin-2 coated surface than MiaPaCa-MUC4-NIDOΔ or MiaPaCa-Vec cells (Figure 5B).
Figure 5. MUC4 NIDO domain interactions with Fibulin-2 in vitro, and colocalisation in pancreatic tissue.
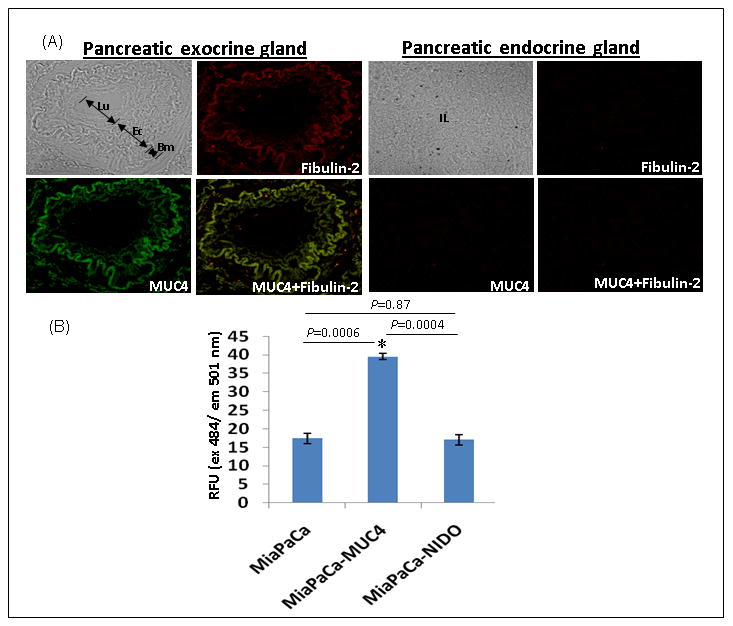
(A) Immunohistofluorescence analysis was carried out on formalin-fixed and paraffin-embedded pancreatic tumor tissue sections after rehydration and antigen-retrieval. Digital merging of the immunostained tissue section clearly showed the colocalization of MUC4 (green) and fibulin-2 (red) in an adenocarcinoma lesion. Absence of MUC4 and fibulin-2 staining in the endocrine glands of the same tissue section acted as a negative control. (B) Binding Assay Cells (25 × 103) were seeded in 96-well plate coated with fibulin-2. Wells coated with bovine serum albumin and poly-L-lysine served as negative and positive controls, respectively. A total of 100 μl of the cell suspension was seeded in triplicates onto the fibulin-2-coated 96 well plates and incubated for 1 h at 37°C. Next the adhered cells were labeled with DIO dye for 1 h at 37°C. The fluorescence of the samples was measured by using the fluorescence plate reader at an excitation wavelength of 484 nm and an emission wavelength of 501 nm. The fluorescence intensity obtained in the negative control (BSA) was subtracted from the values from fibulin-2 coated plates. A relative fluorescence (RFU) was calculated for all treatments with respect to the intensity value obtained with the positive control (poly-L-lysine-coated plates). The significance of each binding assay was evaluated using the t test assuming unequal variances. P-values lower than 0.05 were considered statistically significant.
Discussion
The MUC4 mucin, which is overexpressed in the pancreatic cancer cells, has been shown to be functionally associated with pancreatic cancer metastasis (Chaturvedi et al. 2007a; Singh et al. 2004). The length of MUC4 is estimated to range between 1.1 and 2.1 μm, and its primary structure harbors many functional domains (Chaturvedi et al. 2008a; Singh et al. 2007a). Current theory holds that, MUC4 can contribute to the metastasis of cancer cells by anti-adhesion, adhesion and signal transduction process (Singh et al. 2007a; Senapati et al. 2010). Several lines of evidence demonstrate the contribution of MUC4 in various signaling events that can regulate motility and invasion of cancer cells (Singh et al. 2007a; Senapati et al. 2008; Chaturvedi et al. 2007b; Ponnusamy et al. 2010). Furthermore, due to the large size of MUC4, overexpression of Muc4 or MUC4 has also been shown to sterically hinder integrin-mediated cell-cell and cell-ECM adhesions in vitro (Chaturvedi et al. 2007b; Komatsu et al. 2000). Domain analysis of MUC4 has revealed the presence of NIDO and AMOP domains along with others, that might play an important role in cell-cell interaction via adhesion to extracellular matrix (Chaturvedi et al. 2008a). However, due to the technical restrictions in cloning and modifying the large cDNA of MUC4, a direct experimental proof for the role of individual MUC4 extracellular domains in cancer cell metastasis has been difficult to obtain. In the present study, by expressing the engineered MUC4 without the NIDO-domain proteins, we provide the first evidence that the NIDO-domain of MUC4 significantly contributes to the MUC4-mediated metastasis of pancreatic cancer cells to organs like liver. We initially determined that the MUC4-NIDO domain has no role in cell motility, but significantly promotes the invasive property of pancreatic cancer cells in vitro. In accordance with the in vitro observations, further, we have shown that the loss of the MUC4-NIDO domain decreases the liver metastasis of pancreatic cancer cells in vivo.
In the current study, we postulated that under cancer conditions, through the NIDO-domain, MUC4 present at the basal surface of the epithelial cells may competitively inhibit the normal interaction between fibulin-2 and nidogen protein of the basement membrane (BM) (Ries et al. 2001). During this interaction MUC4 may breach the basement membrane integrity, thus promoting the metastasis of PC cells (Figure 6). Indeed, the effect of MUC4-NIDO domain in enhancing the invasiveness of PC cells, while not affecting the motility of cancer cells in vitro, indicates that the function of MUC4-NIDO domain is associated with the presence of extracellular matrix proteins (Figure 2 A and B). Therefore, during cancer cells metastasis, the MUC4-NIDO domain might have a role in rupturing the basement membrane integrity. However, no overall effect of MUC4 or MUC4-NIDO domain deleted proteins on the incidence of metastasis to sites like spleen, mesentery, peritoneum and diaphragm (Figure 3B, Table-1) might be due to the fact that unlike cancer cells that grow in situ and break the basement membrane for distant organ metastasis, the cancer cells in the orthotopic model don't face the epithelial basement membrane barrier at the primary tumor site. As a result of which, metastasis of the cancer cells to organs like, mesentery, peritoneum and diaphragm, which can mostly occur by peritoneal dissemination from the primary tumor site, might not get influenced due to absence of MUC4-NIDO domain and epithelial-basement membrane proteins interaction. Simultaneously, in the same experimental model, metastasis of MiaPaCa-MUC4 cells to the liver compared to none in MiaPaCa-MUC4-NIDOΔ group strongly supports our hypothesis, and this might be due to the fact that liver metastasis mostly occurs through hematogenous metastasis process, and even though the cancer cells may not face the epithelial basement membrane barrier in the orthotopic model of cancer metastasis, they will have to face the basement membrane of endothelial cells to complete a successful hematogenous metastasis. On the other way, MUC4 presence, aids the PC cells to easily intravasate and/or extravasate from the blood vessels, which is essential for distant organ metastasis (Figure 3B). This argument is further supported by the fact that, MUC4/MUC4-NIDO domain promotes extravasation of cancer cells in vitro (Figure 2C and D). Furthermore, several recent reports have demonstrated extravasation as a key step of cancer metastasis that is regulated by a variety of secreted or cell surface proteins (Ma et al. 2008; Karnoub et al. 2007).
Figure 6. Schematic representation for the proposed action of MUC4-NIDO domain in disrupting the basement membrane integrity.
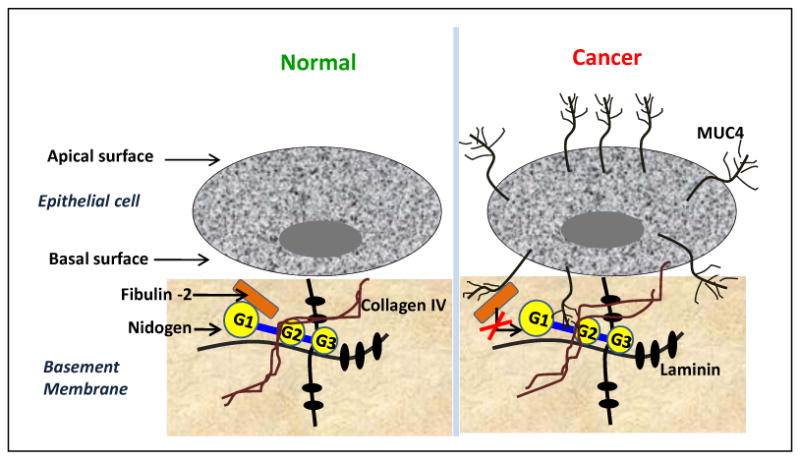
Normal pancreatic epithelial cells do not express MUC4; however, in pancreatic cancer, MUC4 is expressed all over the epithelial cells' surface. Through the NIDO domain, the pancreatic cancer-associated MUC4 present in close proximity to the basement membrane, interacts with fibulin-2, and possibly hinders the normal interaction between fibulin-2 and nidogen. Through this interaction MUC4 potentially disrupts the basement membrane integrity by yet undefined non-proteolytic process, as a result of which invasion and metastasis of PC cells is thus mediated by MUC4-NIDO and Fibulin 2 interaction.
Tumor cells interact with various components of the basement membrane, which is a key event during the process of invasion and metastasis. Earlier studies have shown that fibulin-2 contributes to the basement membrane of epithelial and endothelial cells (Yi et al. 2007). Importantly, fibulin-2 has been shown to be a major component of murine liver blood vessels (Piscaglia et al. 2009). Therefore, elimination of normal fibulin-2 function may create a favorable environment (particularly in liver) for the extravasation of metastatic PC cells (Figure 6). In the present study, the binding experiment suggests that the MUC4 NIDO domain interacts with fibulin-2 in vitro, and this is further confirmed by in vivo pancreatic cancer tissue colocalization studies (Figure 5B, 5A). We do realize that the present study doesn't show the binding affinity of the MUC4-NIDO domain for fibulin-2 compared to nidogen-NIDO domain. However, we believe that in pancreatic cancer cells overexpression of MUC4 and its localization on the whole cell surface may present a greater number of MUC4-NIDO domains around the fibulin-2 proteins, and thus, competitively inhibit nidogen-fibulin 2 interactions. At the same time, further studies are essential to map these interaction sites since the NIDO domain of both MUC4 and nidogen may interact on the same motif present in fibulin2. Interestingly, previous studies have shown that fibulin-2 loss promotes breast cancer cells metastasis and nidogen is under-expressed in gastric and colon cancer tissues (Ulazzi et al. 2011). Therefore, MUC-4 mediated inhibition of nidogen and fibulin-2 interaction in pancreatic cancer mimics loss of fibulin-2 and nidogen function in breast and gastric/colon cancer respectively.
Another significant finding of this study is that deletion of the NIDO-domain did not affect the MUC4-mediated cell's growth rate in vitro and in vivo (Supplementary figure 2, Figure 3B). This is expected, because our previous studies have shown that MUC4 promotes cells proliferation mostly through the activation of MAPK pathway, and this may be through its association with the HER2 (Chaturvedi et al. 2007a; Chaturvedi et al. 2008b), a process in which the NIDO domain may not have any role. However, we do realize that further experimental evidences are required to validate these predictions. Furthermore, the current data suggests that the observed difference in liver metastasis between MiaPaCa-MUC4 and MiaPaCa-MUC4-NIDOΔ groups is not due to the secondary effects of tumor growth or angiogenesis; rather it is the difference in their intrinsic invasive property.
To our knowledge, no study has thus far demonstrated the in vivo effect of MUC4 expression on tumor metastasis by using transfectants that express the MUC4 protein without a specific domain. From our studies we have shown that the MUC4-NIDO domain-mediated liver metastasis may be partly due to its interaction with the endogenous fibulin-2 protein. This study has provided first evidence for the domain-mediated role of MUC4 in Pancreatic cancer metastasis. Further investigation is needed to delineate the other possible mechanisms behind the MUC4-NIDO domain-mediated metastasis. Taken together, the data in this study support the concept that MUC4 is a relevant therapeutic target in pancreatic cancer.
Materials and methods
Generation of MUC4-NIDO domain deletion construct
The construction of the MUC4-NIDO domain deletion construct was performed in three steps. A MUC4 construct that we had generated before (Moniaux et al. 2007) was used as a template for PCR amplification of two DNA fragments at the extremities of the NIDO domain. The upstream fragment has EcoRV and HindIII at the 5′ and 3′end of the PCR product respectively. And the downstream fragment has HindIII and NotI at the 5′ and 3′ end of the PCR product respectively. The two fragments were digested with HindIII and ligated with each other. This NIDO-deleted fragment was digested with EcoRV and HindIII and ligated in the EcoRV and NotI digested MUC4-plasmid to obtain a MUC4-NIDO deletion construct. Each junction was sequenced to check the open reading frame.
Expression of MUC4-NIDO Domain Deletion Construct
MiaPaCa pancreatic cancer cell line was stably transfected with plasmid DNA using the lipofectamine method (Invitrogen). Single colonies were obtained by zeocin selection and expanded for screening. Cells were analyzed for MUC4 protein expression by immunobloting using human MUC4 peptide anti-mouse monoclonal antibody (8G7) (Singh et al. 2004).
FACS analysis
Cells were released with non-enzymatic cell dissociation solution (Sigma, MO, USA) fixed with freshly prepared 2% paraformaldehyde for 0.5 h, blocked with 5% normal goat serum for 0.3 h, and incubated with antibody against MUC4 for 1 h at room temperature. After the incubation with FITC-conjugated secondary antibody for 0.5 h, the expression of cell-surface MUC4 was analyzed by flow cytometry.
Motility and Invasion Assay
For motility assays, 1×106 MiaPaCa-MUC4 and MiaPaCa-MUC4-NIDOΔ cells suspended separately in serum-free medium were plated in the top chamber of polyethylene teraphthalate membranes (six-well insert, pore size 8mm) (Becton Dickinson). The lower chamber of the well was added with the 2.0mL of 10% serum-containing medium and the cells were allowed to migrate for 22 h under chemotactic drive. After incubation, the cells that did not migrate through the pores in the membrane were removed by scraping the membrane with a cotton swab. The migrated cells on the lower side of the membrane were stained with Diff-Quick cell stain kit (Dade-Behring) and photographed in 10 random fields of view at 10× magnification. Cell numbers were counted and expressed as the average number of cells/field of view. For invasion assay, cells (5.0 × 105) were seeded on Matrigel-coated membrane inserts (BD Biosciences, Bedford, MA). The bottom chamber contained 2.0 mL of serum-supplemented medium as a chemo attractant. After incubation for 22h at 37°C, the cells that had invaded through the matrigel-coated membrane were fixed and stained using a Diff-Quick reagent kit. After air drying the membrane, the cells were counted at a magnification of 10× in 10 random fields of view under a microscope. Three independent experiments were done in each case. The data was represented as the average of the three independent experiments with the standard deviation of the average indicated.
Trans-endothelial invasion
To test the effect of MUC4-NIDO domain deletion on the extravasation process of pancreatic cancer cells, with a few modifications, we followed Cold Spring Harbor. Protocol (Ma and Wang 2008). Human umbilical vein endothelial cells (HUVECs) were cultured in transwell inserts with 8 micron pore filters (BD Falcon) for 3 days to allow tight formation of cell monolayers. To determine the integrity of the HUVECs monolayer, high-molecular-weight FITC-dextran (4 ul; Sigma) was added to the insert and further process was carried out as mentioned in the original protocol (mentioned above). MiaPaCa-derived cells, labeled with DIO (Molecular Probes, Inc, Eugene, OR, USA), were applied to the HUVECs for 16 h at 37°C. The cells at the upper side of the transwell membrane were removed with cotton swabs and fluorescent cells that migrated to the bottom side of the transwell membrane were counted by using fluorescence microscope.
For all the above experiments, non-transfected parental MiaPaCa cells were also taken as an additional control. The results for these cells were almost same as corresponding empty vector trasfected MiaPaCa cells (data not shown). Therefore, for further studies, only transfected control cells were used.
In-vivo tumor growth and metastasis
To investigate the functional consequences of the MUC4-NIDO domain deletion on the tumorigenicity and metastatic property of pancreatic cancer cells, orthotopic implantation was carried out. Ten to twelve week old immunodeficient female mice were purchased from the Animal Production Area of the National Cancer Institute-Frederick Cancer Research and Development Center (Frederick, MD). The mice were treated in accordance with the Institutional Animal Care and Use Committee (IACUC) guidelines. The MiaPaCa derived cells were harvested from sub-confluent cultures by a brief exposure to 0.25% trypsin and 0.02% EDTA. After neutralizing the effect of trypsin with 10% fetal bovine serum, the cells were washed once in PBS. Cells were resuspended in PBS at a concentration of 2×106 cells/50μl. Single-cell suspensions of >90% viability was used for the injections. Animals (eight animals per group) were anesthetized with intra-peritoneal injection of ketamine and xylazine mixture (4:1). The abdomen was cleaned with iodine solution and 1cm incision was made to expose the pancreas. Two million cells suspended in 50 μl of PBS were injected into the head of the pancreas by using a 30-guage needle. The abdominal wound was closed in two layers with catgut and wound clips. Animals were monitored twice weekly. To determine the tumor growth and metastasis, mice were sacrificed by CO2 asphyxiation and autopsied on the 65th day after the implantation of the tumor cells. After inspection of macroscopical tumor growth, the pancreas was resected and the weight was assessed in milligrams (mg). Regional and distant lymph nodes, lung, liver as well as other organs suspected for harboring metastasis were routinely formalin fixed, embedded, sectioned, and stained with hematoxylin and eosin using standard techniques for microscopic examination. In primary tumors, expression of MUC4 and MUC4-without the NIDO domain protein was checked by Immunoblot analysis (Supplementary figure 3).
Between the two groups of mice, incidence of metastasis was compared using Fisher's exact test and Student t-test was done to find the difference in tumor weights obtained from both groups.
Attachment assay
Before carrying out this experiment, 96-well cell culture plates were coated with 20 ug/mL of fibulin-2 or 25 μg/mL polylysine (Sigma) overnight at 4°C, followed by blocking with 1% (w/v) BSA in DMEM at 37°C for 1h. Wells coated with serum albumin and poly-L-lysine served as negative and positive controls, respectively. The wells were decanted and rinsed two times with PBS and harvested by using a non-enzymatic dissociation buffer, followed by resuspension at a density of 2.5×105 cells/ml. A total of 100 ul of the cell suspension was seeded in triplicates onto the fibulin-2-coated 96 well plates and incubated for 1 h at 37°C. After incubation, the cell suspension was discarded and the wells were gently washed with PBS. The adhered cells were labeled with DIO dye for 1 h at 37°C. The fluorescence of the samples was measured by using the fluorescence plate reader at an excitation wavelength of 484 nm and emission wavelength of 501nm. The fluorescence intensity obtained in the negative control (BSA) was subtracted from the values observed with fibulin-2 coated plates. A relative fluorescence (RFU) was calculated for all treatments with respect to the intensity value obtained with the positive control (poly-L-lysine-coated plates). The significance of each binding assay was evaluated using the t test, assuming unequal variances. P-values lower than 0.05 were considered statistically significant.
Supplementary Material
Acknowledgments
The authors on this work are supported by the grants from National Institutes of Health (CA78590, CA111294, CA127297, CA133774 and CA131944). We thank Ms. Kristi L. Berger for editing the manuscript. The authors acknowledge the invaluable technical support from Mr. Erik Moore, Kavita Mallya and Navneet Momi. We also thank Janice A. Tayor and James R. Talaska of the confocal laser scanning microscope core facility at the UNMC, for their support.
Footnotes
Conflict of interest: The authors declare no conflict of interest.
Reference List
- Aumailley M, Battaglia C, Mayer U, Reinhardt D, Nischt R, Timpl R, Fox JW. Kidney Int. 1993;43:7–12. doi: 10.1038/ki.1993.3. [DOI] [PubMed] [Google Scholar]
- Bacac M, Stamenkovic I. Annu Rev Pathol. 2008;3:221–247. doi: 10.1146/annurev.pathmechdis.3.121806.151523. [DOI] [PubMed] [Google Scholar]
- Casey RC, Skubitz AP. Clin Exp Metastasis. 2000;18:67–75. doi: 10.1023/a:1026519016213. [DOI] [PubMed] [Google Scholar]
- Chaturvedi P, Singh AP, Batra SK. FASEB J. 2008a;22:966–981. doi: 10.1096/fj.07-9673rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi P, Singh AP, Chakraborty S, Chauhan SC, Bafna S, Meza JL, Singh PK, Hollingsworth MA, Mehta PP, Batra SK. Cancer Res. 2008b;68:2065–2070. doi: 10.1158/0008-5472.CAN-07-6041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi P, Singh AP, Moniaux N, Senapati S, Chakraborty S, Meza JL, Batra SK. Mol Cancer Res. 2007a;5:309–320. doi: 10.1158/1541-7786.MCR-06-0353. [DOI] [PubMed] [Google Scholar]
- Chaturvedi P, Singh AP, Moniaux N, Senapati S, Chakraborty S, Meza JL, Batra SK. Mol Cancer Res. 2007b;5:309–320. doi: 10.1158/1541-7786.MCR-06-0353. [DOI] [PubMed] [Google Scholar]
- DiMagno EP, Reber HA, Tempero MA. Gastroenterology. 1999;117:1464–1484. doi: 10.1016/s0016-5085(99)70298-2. [DOI] [PubMed] [Google Scholar]
- Duraisamy S, Ramasamy S, Kharbanda S, Kufe D. Gene. 2006;373:28–34. doi: 10.1016/j.gene.2005.12.021. [DOI] [PubMed] [Google Scholar]
- Fox JW, Mayer U, Nischt R, Aumailley M, Reinhardt D, Wiedemann H, Mann K, Timpl R, Krieg T, Engel J, et al. EMBO J. 1991;10:3137–3146. doi: 10.1002/j.1460-2075.1991.tb04875.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotary K, Li XY, Allen E, Stevens SL, Weiss SJ. Genes Dev. 2006;20:2673–2686. doi: 10.1101/gad.1451806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Xu J, Ward E. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, Richardson AL, Polyak K, Tubo R, Weinberg RA. Nature. 2007;449:557–563. doi: 10.1038/nature06188. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Tatum L, Altman NH, Carothers Carraway CA, Carraway KL. Int J Cancer. 2000;87:480–486. doi: 10.1002/1097-0215(20000815)87:4<480::aid-ijc4>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Langley RR, Fidler IJ. Endocr Rev. 2007;28:297–321. doi: 10.1210/er.2006-0027. [DOI] [PubMed] [Google Scholar]
- Li D, Xie K, Wolff R, Abbruzzese JL. Lancet. 2004;363:1049–1057. doi: 10.1016/S0140-6736(04)15841-8. [DOI] [PubMed] [Google Scholar]
- Ma C, Rong Y, Radiloff DR, Datto MB, Centeno B, Bao S, Cheng AW, Lin F, Jiang S, Yeatman TJ, Wang XF. Genes Dev. 2008;22:308–321. doi: 10.1101/gad.1632008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, Wang XF. Cold Spring Harb Protoc. 2008;2008 db. [Google Scholar]
- Moniaux N, Chaturvedi P, Varshney GC, Meza JL, Rodriguez-Sierra JF, Aubert JP, Batra SK. Br J Cancer. 2007;97:345–357. doi: 10.1038/sj.bjc.6603868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen DX, Bos PD, Massague J. Nat Rev Cancer. 2009;9:274–284. doi: 10.1038/nrc2622. [DOI] [PubMed] [Google Scholar]
- Piscaglia F, Dudas J, Knittel T, Di RP, Kobold D, Saile B, Zocco MA, Timpl R, Ramadori G. Cell Tissue Res. 2009;337:449–462. doi: 10.1007/s00441-009-0823-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponnusamy MP, Lakshmanan I, Jain M, Das S, Chakraborty S, Dey P, Batra SK. Oncogene. 2010;29:5741–5754. doi: 10.1038/onc.2010.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ries A, Gohring W, Fox JW, Timpl R, Sasaki T. Eur J Biochem. 2001;268:5119–5128. doi: 10.1046/j.0014-2956.2001.02437.x. [DOI] [PubMed] [Google Scholar]
- Rowe RG, Weiss SJ. Trends Cell Biol. 2008;18:560–574. doi: 10.1016/j.tcb.2008.08.007. [DOI] [PubMed] [Google Scholar]
- Senapati S, Chaturvedi P, Sharma P, Venkatraman G, Meza JL, El-Rifai W, Roy HK, Batra SK. Br J Cancer. 2008;99:949–956. doi: 10.1038/sj.bjc.6604632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senapati S, Das S, Batra SK. Trends Biochem Sci. 2010;35:236–245. doi: 10.1016/j.tibs.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh AP, Chaturvedi P, Batra SK. Cancer Res. 2007a;67:433–436. doi: 10.1158/0008-5472.CAN-06-3114. [DOI] [PubMed] [Google Scholar]
- Singh AP, Chauhan SC, Andrianifahanana M, Moniaux N, Meza JL, Copin MC, van S I, Hollingsworth MA, Aubert JP, Batra SK. Oncogene. 2007b;26:30–41. doi: 10.1038/sj.onc.1209764. [DOI] [PubMed] [Google Scholar]
- Singh AP, Moniaux N, Chauhan SC, Meza JL, Batra SK. Cancer Res. 2004;64:622–630. doi: 10.1158/0008-5472.can-03-2636. [DOI] [PubMed] [Google Scholar]
- Ulazzi L, Sabbioni S, Miotto E, Veronese A, Angusti A, Gafà R, Manfredini S, Farinati F, Sasaki T, Lanza G, Negrini M. Molecular Cancer. 2011;6 doi: 10.1186/1476-4598-6-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi CH, Smith DJ, West WW, Hollingsworth MA. Am J Pathol. 2007;170:1535–1545. doi: 10.2353/ajpath.2007.060478. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.