

Abstract
Background
X-linked lymphoproliferative syndrome (XLP) is a rare inherited immunodeficiency by an extreme vulnerability to Epstein-Barr virus (EBV) infection, frequently resulting in hemophagocytic lymphohistiocytosis (HLH). XLP are now divided into type 1 (XLP-1) and type 2 (XLP-2), which are caused by mutations of SH2D1A/SLAM-associated protein (SAP) and X-linked inhibitor of apoptosis protein (XIAP) genes, respectively. The diagnosis of XLP in individuals with EBV-associated HLH (EBV-HLH) is generally difficult because they show basically similar symptoms to sporadic EBV-HLH. Although EBV-infected cells in sporadic EBV-HLH are known to be mainly in CD8+ T cells, the cell-type of EBV-infected cells in EBV-HLH seen in XLP patients remains undetermined.
Methods
EBV-infected cells in two patients (XLP-1 and XLP-2) presenting EBV-HLH were evaluated by in EBER-1 in situ hybridization or quantitative PCR methods.
Results
Both XLP patients showed that the dominant population of EBV-infected cells was CD19+ B cells, whereas EBV-infected CD8+ T cells were very few.
Conclusions
In XLP-related EBV-HLH, EBV-infected cells appear to be predominantly B cells. B cell directed therapy such as rituximab may be a valuable option in the treatment of EBV-HLH in XLP patients.
Keywords: B cells, Epstein Barr virus, Hemophagocytic lymphohistiocytosis, X-linked lymphoproliferative syndrome
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is clinically characterized by prolonged fever, hepatosplenomegaly, hypertriglyceridemia, systemic hypercytokinemia and cytopenia [1]. HLH consists of primary (familial) and secondary (infection, lymphoma or autoimmune disease-associated) types. Approximately half of all infection-associated HLH cases involves the Epstein-Barr virus (EBV) [2]. Most cases of EBV-HLH are sporadic, but a few cases may present the first presentation of X-linked lymphoproliferative syndrome (XLP) [3]. XLP is a rare, inherited immunodeficiency that is characterized by an extreme vulnerability to EBV infection and shows variable clinical phenotypes, including severe or fatal EBV-HLH (60%), malignant B-cell lymphoma (30%), and progressive dysgammaglobulinemia (30%) [3]. The first genes that is responsible for XLP was identified as the SH2D1A/SLAM-associated protein (SAP) gene in 1998 [4-6], and mutations in the X-linked inhibitor of apoptosis protein (XIAP) gene can also lead to the clinical phenotype of XLP in 2006 [7]. XLP is now considered to comprise two distinct diseases, namely XLP-1 (SAP deficiency) and XLP-2 (XIAP deficiency).
In addition to B cells, EBV can infect other cell types, including epithelial cells, T cells and natural killer (NK) cells [8]. Studies have shown that activated T cells, particularly CD8+ T cells, are the primary cellular target of EBV infection in sporadic EBV-HLH [9,10], which reflects the pathogenic role of EBV-infected CD8+ T cells in sporadic EBV-HLH. Patients with sporadic EBV-HLH are usually treated with immunochemotherapy, including dexamethasone, cyclosporine A and etoposide, and this therapy can be curable [11]. In contrast, XLP-related EBV-HLH is usually refractory to immunochemotherapy [3]. It is possible that the poor response of XLP-related EBV-HLH to immunochemotherapy can be attributed to the type of EBV-infected cells in this disease, which may differ from the cell type that infected in sporadic EBV-HLH. We investigated the affected cell type in EBV infection of two XLP (XLP-1 and XLP-2) patients with EBV-HLH. Our results demonstrate that the predominant EBV target cells in XLP-related EBV-HLH are CD19+ B cells, which appears to be distinct from sporadic EBV-HLH cases.
Patients, materials and methods
Patients
Three patients presented with clinical features of HLH, including persistent fever, hepatosplenomegaly, cytopenia, abnormal liver function, hyperferritinemia and elevated levels of soluble interleukin-2-receptor (Table 1). The clinical features of the patients fulfilled the diagnostic criteria for HLH [1], although hemophagocytosis in the bone marrow was not observed in patients 1 and 2. Patient 3 was previously reported as patient HLH3 [10]. The number of EBV-DNA copies in the peripheral blood was increased from the normal level of ≤ 1 × 102 copies/ml to 1.4 × 105, 5.7 × 103 and 1.4 ×106 copies/ml in patients 1, 2 and 3, respectively. Blood samples from the patients were obtained using standard ethical procedures with the approval of the Ethics Committee of the University of Toyama, and an analysis of the SH2D1A and XIAP genes was performed. Patient 1 showed a one-nucleotide insertion (239_240insA) in the SH2D1A gene that resulted in a frameshift and a premature stop codon (80KfsX22). Patient 2 carried a two-nucleotides deletion (1021_1022delAA) in the XIAP gene that resulted in a frameshift and a premature stop codon (N341YfsX7). Patient 3 had no mutations in the SH2D1A or XIAP gene.
Table 1.
Clinical and laboratory finding of the patients in this study
| Patient 1 | Patient 2 | Patient 3 | |
|---|---|---|---|
| Family history available | No | Yes | No |
| Age at the time of the study | 4 years | 21 months | 16 months |
| Age at onset | 3 years | 17 months | 16 month |
| Fever | Yes | Yes | Yes |
| Hepatomegaly | 4 cm | 5 cm | 2.5 cm |
| Splenomegaly | 2 cm | 3 cm | 1 cm |
| White blood cells (×109/L) | 11.6 | 6.36 | 3.03 |
| Neutrophils (×109/L) | 1.61 | 3.915 | 0.56 |
| Hemoglobin (g/dL) | 8.1 | 9.6 | 7.5 |
| Platelets (×109/L) | 95 | 56 | 30 |
| LDH (IU/L) | 449 | 1,693 | 1,698 |
| AST (IU/L) | 88 | 122 | 453 |
| ALT (IU/L) | 31 | 25 | 255 |
| Ferritin (μg/L) | 1,276 | 26,282 | 11,129 |
| sIL-2R (U/mL) | 3,162 | 2,880 | 14,334 |
| IgG (mg/dL) | 1,821 | 806 | 423 |
| IgA (mg/dL) | 302 | 124 | 32 |
| IgM (mg/dL) | 1,843 | 40 | 18 |
| Whole blood EBV-DNA (copies/mL) | 140,000 | 5,700 | 1,400,000 |
LDH, lactate dehydrogenase; AST, aspartate amino transferase; ALT, alanine amino transferase, sIL-2R, soluble interleukin-2 receptor; NA: not available.
Cell preparation
Peripheral blood mononuclear cells (PBMCs) were isolated from the the patients using Ficoll-Hypaque gradient centrifugation. Lymphocytes were prepared from the PBMCs by depleting the monocytes using anti-CD14 monoclonal antibody (mAb)-coated magnetic beads (Becton Dickinson, San Diego, CA) [10]. The CD19+ B cells, CD56+ NK cells, CD4+ T cells and CD8+ T cells were purified by positive selection from the lymphocytes using the respective mAb-coated magnetic beads. The purity of each isolated cell population was assessed by flow cytometriy analysis, and each sorted population was found to be higher than 85% pure.
In situ hybridization of EBVRNA
The presence of EBV was estimated by measuring the EBV-encoded small RNA 1 (EBER-1) mRNA using the in situ hybridization (ISH) method as described previously [10]. The sorted cells were cytocentrifuged onto silanized slides (Dako, Kyoto, Japan), and the presence of EBER-1 mRNA was determined by ISH using the alkaline phosphatase-conjugated EBER-1 antisense probe (5'-AGCAGAGTCTGGGAAGACAACCACAGACACCGTCCTCACC-3') or a sense probe.
Quantitative PCR for EBV DNA
Quantitative PCR was performed using AmpliTaq Gold and a real-time PCR 7300 system (Applied Biosystems, Foster City, CA) as described previously [12]. The PCR primers for detecting EBV DNA were selected from within the BALF5 gene, which encodes the viral DNA polymerase. The primers for amplifying the BALF5 gene sequences were as follows: forward, CGGAAGCCCTCTGGACTTC, and reverse, CCCTGTT TATCCGATGGAATG. The TaqMan probe was FAM-TATACACGCACGAGAAATGCGCC-BFQ. The PCR conditions were as follows: denaturation at 95°C for 2 minutes, annealing at 58°C for 15 seconds, and extension at 72°C for 15 seconds, and the products were subjected to 45 cycles of PCR amplification. The EBV DNA copy number was considered to be significant when more than 500 copies/μg of DNA were observed.
Flow cytometry analysis for the T cell receptor Vβ repertoire
Flow cytometry analysis of the T cell receptor (TCR) Vβ repertoire was performed as described previously [10]. In briefl, the PBMCs were incubated with the appropriate phycoerythrin-conjugated mAbs with specificity for TCR Vβ 1-23 (Immunotech, Marseille, France), fluorescein isothiocyanate-conjugated anti-CD8 (Becton Dickinson) and R-PE-Cy5-conjugated anti-CD4 (Dako) mAbs. The stained cells were analyzed using a flow cytometer. TCR Vβ expression is represented as the percentage of CD4+ or CD8+ cells for each receptor family.
Results
To determine the localization of EBV infection in the lymphocyte subpopulations of patient 1, CD4+ T cells, CD8+ T cells, CD19+ B cells and CD56+ NK cells were sorted using the immunomagnetic bead method and the presence of EBV was evaluated in each lymphocyte subpopulation by EBER-1 ISH (Figure 1A). EBER-1-positive cells were observed in 34.0% of the CD19+ B cells, whereas the remaining lymphocyte subpopulations contained fewer than 0.1% EBER-1-positive cells. Therefore, the EBV-infected cells in patient 1 were almost exclusively CD19+ B cells. In patient 3, EBER-1-positive cells constituted 75.5% of CD8+ T cells, however, they were not detected among CD4+ T cells and observed in a few of CD19+ B cells and CD56+ NK cells (2.8% and1 7.4%, respectively) (Figure 1B).
Figure 1.
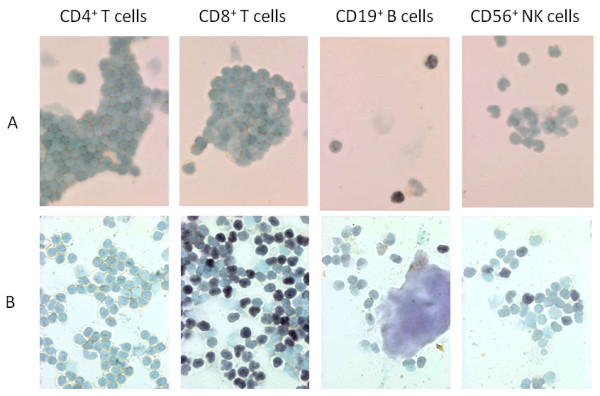
Cytospin preparations showing EBER-1 in situ hybridization of the lymphocyte subpopulations. Lymphocyte subpopulations from patients 1 and 3 were separated by magnetic bead sorting after immunostaining with anti-CD4, CD8, CD19 or CD56 mAbs. EBV infection in each subpopulation was determined using EBER-1 ISH. A, In patient 1, EBER-1-positive cells (shown by their dark nuclear staining) were detected in34.0% of the B cells but were not detected in CD4+ T cells, CD8+ T cells or CD56+ NK cells (< 0.1% each). B, In patient 3, EBER-1-positive cells were observed in 75.5% of CD8+ T cells, 2.8% of CD19+ B cells, and 17.4% of CD56+ NK cells, but not observed in CD4+ T cells [10].
The viral loads in the CD4+ T cells, CD8+ T cells, CD19+ B cells and CD56+ NK cells in patient 2 were determined by quantitative PCR. The number of EBV DNA genome copies in the CD19+ B cells was 1.8 × 104 copies/μg, and the copy number in the CD8+ T cells was 1.0 × 103 copies/μg. The EBV DNA genome could not be detected in either the CD4+ T cells or the CD56+ NK cells that were isolated from patient 2.
Flow cytometry analysis of the TCR Vβ repertoire revealed a polyclonal pattern in patients 1 and 2 (Figure 2), which was in contrast to the skewed pattern that is most commonly seen in the CD8+ T cells of patients with sporadic EBV-HLH [10]. No clonal dominance in CD8+ T cells was demonstrated by mAb in patient 3, but TCR Vβ13.3 was predominantly found in the CD8+ T cells by complentarity-determining region 3 spectratyping [10].
Figure 2.
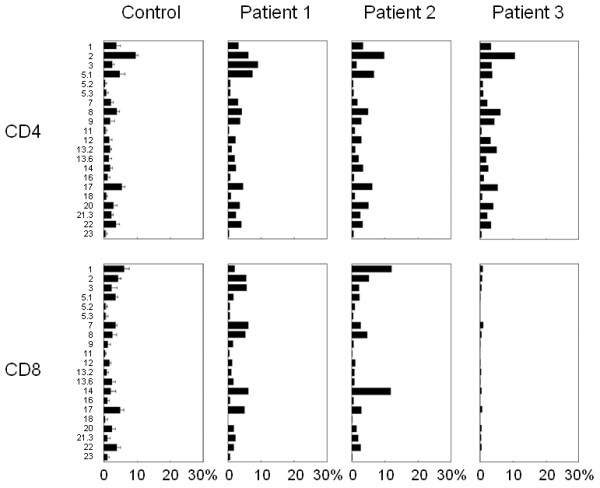
The results of the flow cytometric analysis of TCR Vβ. The expression profiles of the TCR Vβ subfamilies of patients 1, 2 and 3. The PBMCs were stained with mAbs for individual TCR Vβ, together with an anti-CD8 mAb. The percentage of the expression of each TCR Vβ within CD8+ T cells was analyzed by flow cytometry.
Discussion
XLP is a severe and rare immunodeficiency disease that is characterized by an extreme vulnerability to EBV infection and frequently results in HLH [3]. XLP was first described as X-linked progressive combined immunodeficiency in 1975 by Purtilo et al. [13]. To better understand and reflect the pathophysiology of this disease, the term "X-linked lymphoproliferative disease or syndrome" has now been used. The first gene to be linked to XLP in 1998 was SH2D1A which is located on Xq25 and encodes the SAP [4-6]. Importantly, in 2006, a mutation in the gene that encodes the XIAP was identified as a second XLP-linked gene [7]. Thus, XLP can be divided into XLP-1 (SAP deficiency) and XLP-2 (XIAP deficiency). Most XLP patients present with EBV-HLH. Pachlopnik Schmid et al. [14] reported that the incidence of HLH in XLP-1 and XLP-2 is 55 and 76%, respectively. Currently, hematopoietic stem cell transplantation (HSCT) is the only curative therapy for XLP. Therefore, an early definitive diagnosis and immediate treatment are extremely important for both life-saving intervention and an improved prognosis for XLP patients.
EBV infects the majority of the adult population worldwide and persists in B cells throughout the lifetime of normal individuals, usually without causing disease. EBV is the most common trigger for both the XLP-1 and XLP-2 phenotypes. Prior to being exposed to EBV, most patients with XLP can tolerate infections by other agents, although in vitro studies have demonstrated defects of T cell-mediated and humoral immunity. During an acute EBV infection, XLP patients develop normal or high levels of anti-viral capsid antigen IgM antibodies but usually lack heterophile antibodies. Initially, these patients fail to develop EBV-specific cytotoxic T cells, and this results in a massive and overwhelming polyclonal B cell proliferation involving lymphoid and other tissues [8]. SAP binds 2B4, which is a surface molecule involved in activation of NK cell-mediated cytotoxity. Therefore, SAP-deficient patient shows that NK cell function is impaired, allowing B cell proliferation [15]. SAP has proapoptotic function, and contributes to the maintenance of T cell homeostasis and to the elimination of potentially dangerous DNA-damaged cells. Thus, the loss of this function could be responsible for the uncontrol T cell proliferation in acute EBV infection [16].
B cells are the usual cellular targets of EBV in a primary EBV infection such as infectious mononucleosis and in the sero-positive normal host [8]. After the interaction of the viral surface glycoproteins with the CD21 receptor, EBV entry into B cells is mediated by HLA class II and other co-receptors. However, in cases of sporadic EBV-HLH, EBV infects primarily T cells and NK cells [9,10,17]. The mechanism of T cell infection by EBV in HLH is still unclear, but one hypothesis is that, in specific situations, CD8+ T cells express CD21, which can mediate EBV infection. Although T cells do not express the glycoprotein, they contain mRNA for CD21 [18]. In sporadic EBV-HLH cases, EBV infection into B cells is delayed but occurs during every case of cured EBV-HLH [17]. To the best of our knowledge, this is the first report of EBV infection status in two different types of XLP patients with EBV-HLH. The present study shows that the primary EBV-infected cells in XLP-related EBV-HLH are CD19+ B cells and not T cells or NK cells, which are a primary target of EBV infection in sporadic EBV-HLH.
For decades, clinicians and investigators have been puzzled by the differential diagnosis between XLP and sporadic EBV-HLH when they encountered a young boy presenting with EBV- HLH. We believe the different EBV target cells can provide additional information to help discriminate between XLP and sporadic EBV-HLH. An evaluation of specific cell type that is infected by EBV should be considered when target therapy is applied. Most patients with sporadic EBV-HLH can achieve remission by immunochemotherapy; however, patients with XLP are usually refractory to this therapy. Recently, B cell-directed therapy using an anti-CD20 mAb (rituximab) was performed in patients with XLP-1 [19]. Two XLP patients who presented with acute EBV infection were successfully treated with rituximab and were free from EBV-HLH and lymphoma for a prolonged period. In addition, rituximab combined with methylprednisolone and intravenous immunoglobulin were administered to an XLP-1 patient with EBV-HLH, and the patient achieved a remission [20]. Patient 1 was also associated with EBV-associated encephalitis and lymphoproliferative disorder. The patient's lymphoproliferative disorder was treated with rituximab, but he died of the disease. Patient 2 was successfully treated with dexamethasone and immunoglobulin. Our data suggest that B cell target therapy can be a viable therapeutic option for an initial stage of EBV-HLH in both XLP-1and XLP-2 patients.
Abbreviations
EBER: EBV-encoded small RNA; EBV: Epstein-Barr virus; HLH: Hemophagocytic lymphohistiocytosis; HSCT: Hematopoietic stem cell transplantation; ISH: In situ Hybridization; mAb: Monoclonal antibody; NK: Natural killer; PBMC: Peripheral blood mononuclear cells; SAP: SLAM-associated protein; TCR: T cell receptor; XIAP: X-linked inhibitor of apoptosis; XLP: X-linked lymphoproliferative syndrome.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
XY and HK wrote the manuscript. XY, TW, KI and NN performed the experimental studies. TM, MF, HK and FK managed the patients' care. SF, AY, XDZ and TM revised the manuscript. XY, TW and KI contributed equally to this study. All the authors read and approved the final manuscript.
Contributor Information
Xi Yang, Email: double-cow@163.com.
Taizo Wada, Email: taizo@staff.kanazawa-u.ac.jp.
Ken-Ichi Imadome, Email: imadome@nch.go.jp.
Naonori Nishida, Email: naonori_n1231@yahoo.co.jp.
Takeo Mukai, Email: tm11407@kchnet.or.jp.
Mitsuhiro Fujiwara, Email: mf7315@kchnet.or.jp.
Haruka Kawashima, Email: spring_harucafe@yahoo.co.jp.
Fumiyo Kato, Email: fkatohpd@dnh.twmu.ac.jp.
Shigeyoshi Fujiwara, Email: shige@nch.go.jp.
Akihiro Yachie, Email: yachie@med.kanazawa-u.ac.jp.
Xiaodong Zhao, Email: zhaoxd530@yahoo.com.cn.
Toshio Miyawaki, Email: toshio65@med.u-toyama.ac.jp.
Hirokazu Kanegane, Email: kanegane@med.u-toyama.ac.jp.
Acknowledgements
The authors would like to thank Ms. Chikako Sakai and Mr. Hitoshi Moriuchi for their excellent technical assistances. This study was supported by a grant-in-aid for scientific research from the Ministry of Education, Culture, Sports, Science and Technology, Japan (TM, HK), and a grant from the Ministry of Health, Labour, and Welfare, Japan (TM).
References
- Henter JI, Horne A, Aricó M, Egeler RM, Fillipovich AH, Imashuku S, Ladisch S, McClain K, Webb D, Winiarski J. et al. HLH-2004: Diagnostic and therapeutic guideline for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124–131. doi: 10.1002/pbc.21039. [DOI] [PubMed] [Google Scholar]
- Ishii E, Ohga S, Imashuku S, Yasukawa M, Tsuda H, Miura I, Yamamoto K, Horiuchi H, Takada K, Ohshima K. et al. Nationwide survey of hemaphagocytic lymphohistiocytosis in Japan. Int J Hematol. 2007;86:58–65. doi: 10.1532/IJH97.07012. [DOI] [PubMed] [Google Scholar]
- Seemayer TA, Gross TG, Egeler RM, Pirruccello SJ, Davis JR, Kelly CM, Okano M, Lanyi A, Sumegi J. X-linked lymphoproliferative disease: twenty- five years after the discovery. Pediatr Res. 1995;38:471–478. doi: 10.1203/00006450-199510000-00001. [DOI] [PubMed] [Google Scholar]
- Coffey AJ, Brooksbank RA, Brandau O, Oohashi T, Howell GR, Bye JM, Cahn AP, Durham J, Heath P, Wray P. et al. Host response to EBV infection in X-linked lymphoproliferative disease results from mutations in an SH2-domain encoding gene. Nat Genet. 1998;20:129–135. doi: 10.1038/2424. [DOI] [PubMed] [Google Scholar]
- Sayos J, Wu C, Morra M, Wang N, Zhang X, Allen D, Van-Schaik S, Notarangelo L, Geha R, Roncarolo MG. et al. The X-linked lymphoproliferative-disease gene product SAP regulates signals induced through the co-receptor SLAM. Nature. 1998;395:462–469. doi: 10.1038/26683. [DOI] [PubMed] [Google Scholar]
- Nichols KE, Harkin DP, Levitz S, Krainer M, Kolquist KA, Genovese C, Bernard A, Ferguson M, Zuo L, Snyder E. et al. Inactivating mutations in an SH2 domain-encoding gene in X-linked lymphoproliferative syndrome. Proc Natl Acad Sci USA. 1998;95:13765–13770. doi: 10.1073/pnas.95.23.13765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigaud S, Fondanèche MC, Lambert N, Pasquier B, Mateo V, Soulas P, Galicier L, Le-Deist F, Rieux-Laucat F, Revy P. et al. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature. 2006;444:110–114. doi: 10.1038/nature05257. [DOI] [PubMed] [Google Scholar]
- Tosato G, Taga K, Angiolillo AL, Sgadari C. Epstein-Barr virus as an agent of haematological disease. Baillière Clin Haematol. 1995;8:165–199. doi: 10.1016/S0950-3536(05)80237-9. [DOI] [PubMed] [Google Scholar]
- Kawaguchi H, Miyashita T, Herbst H, Niedobitek G, Asada M, Tsuchida M, Hanada R, Kinoshita A, Sakurai M, Kobayashi N. et al. Epstein-Barr virus-infected T lymphocytes in Epstein-Barr virus-associated hemophagocytic syndrome. J Clin Invest. 1993;92:1444–1450. doi: 10.1172/JCI116721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toga A, Wada T, Sakakibara Y, Mase S, Araki R, Tone T, Toma T, Kurokawa T, Yanagisawa R, Tamura K. et al. Clinical significance of cloned expansion and CD5 down-regulation in Epstein-Barr Virus (EBV)-infected CD8+ T lymphocytes in EBV-associated hemophagocytic lymphohistiocytosis. J Infect Dis. 2010;201:1923–1932. doi: 10.1086/652752. [DOI] [PubMed] [Google Scholar]
- Imashuku S, Hibi S, Ohara T, Iwai A, Sako M, Kato M, Arakawa H, Sotomatsu M, Kataoka S, Asami K. et al. Effective control of Epstein-Barr virus-related hemophagocytic lymphohistiocytosis with immunochemotherapy. Blood. 1999;93:1869–1874. [PubMed] [Google Scholar]
- Kimura H, Hoshino Y, Kanegane H, Tsuge I, Okamura T, Kawa K, Morishima T. Clinical and virologic characteristics of chronic active Epstein-Barr virus infection. Blood. 2001;98:280–286. doi: 10.1182/blood.V98.2.280. [DOI] [PubMed] [Google Scholar]
- Purtilo DT, Cassel CK, Yang JP, Harper R. X-linked recessive progressive combined variable immunodeficiency (Duncan's disease) Lancet. 1975;1:935–940. doi: 10.1016/s0140-6736(75)92004-8. [DOI] [PubMed] [Google Scholar]
- Pachlopnik Schmid J, Canioni D, Moshous D, Moshous D, Touzot F, Mahlaoui N, Hauck F, Kanegane H, Lopez-Granados E, Mejstrikova E. et al. Clinical similarities and differences of patients with X-linked lymphoproliferative syndrome type 1 (XLP-1/SAP deficiency) versus type 2 (XLP-2/XIAP deficiency) Blood. 2011;117:1522–1529. doi: 10.1182/blood-2010-07-298372. [DOI] [PubMed] [Google Scholar]
- Parolini S, Bottino C, Falco M, Augugliaro R, Giliani S, Franceschini R, Ochs HD, Wolf H, Bonnefoy JY, Biassoni R. et al. X-linked lymphoproliferative disease: 2B4 molecules displaying inhibitory rather than activating function are responsible for the inhibit of natural killer cells to kill Epstein-Barr virus-infected cells. J Exp Med. 2000;192:337–346. doi: 10.1084/jem.192.3.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy N, Matskova L, Kis LL, Hellmah U, Klein G, Klein E. The proapoptotic function of SAP provides a clue to the clinical picture of X-linked lymphoproliferative disease. Proc Natl Acad Sci USA. 2009;106:11966–11971. doi: 10.1073/pnas.0905691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara Y, Yachie A. Cell type specific infection of Epstein-Barr virus (EBV) in EBV-associated hemophagocytic lymphohistiocytosis and chronic active EBV infection. Crit Rev Oncol Hematol. 2002;44:283–294. doi: 10.1016/S1040-8428(02)00119-1. [DOI] [PubMed] [Google Scholar]
- Braun M, Melchers I, Peter HH, Illges H. Human B and T lymphocytes have similar amounts of CD21 mRNA, but differ in surface expression of the CD21 glycoprotein. Int Immunol. 1998;10:1197–1202. doi: 10.1093/intimm/10.8.1197. [DOI] [PubMed] [Google Scholar]
- Milone MC, Tsai DE, Hodinka RL, Silverman LB, Malbran A, Wasik MA, Nichols KE. Treatment of primary Epstein-Barr virus infection in patients with X-linked lymphoproliferative disease using B-cell-directed therapy. Blood. 2005;105:994–996. doi: 10.1182/blood-2004-07-2965. [DOI] [PubMed] [Google Scholar]
- Bond J, Shahdadpuri R, Mc Mahon C, O'Marcaigh A, Cotter M, Smith O. Successful treatment to acute Epstein-Barr virus infection associated with X-linked lymphoproliferative disorder with rituximab. Pediatr Blood Cancer. 2007;49:761–762. doi: 10.1002/pbc.21081. [DOI] [PubMed] [Google Scholar]