

Abstract
Mitochondria control essential cellular activities including generation of ATP via oxidative phosphorylation. Mitochondrial DNA (mtDNA) mutations in the regulatory D-loop region and somatic mtDNA mutations are common in primary human cancers. The biological impact of a given mutation may vary, depending on the nature of the mutation and the proportion of mutant mtDNAs carried by the cell. Identification of mtDNA mutations in precancerous lesions supports their early contribution to cell transformation and cancer progression. Introduction of mtDNA mutations in transformed cells has been associated with increased ROS production and tumor growth. Studies reveal that increased and altered mtDNA plays a role in the development of cancer but further work is required to establish the functional significance of specific mitochondrial mutations in cancer and disease progression. This review offers some insight into the extent of mtDNA mutations, their functional consequences in tumorigenesis, mitochondrial therapeutics, and future clinical application.
Introduction
Defects in mitochondrial function have long been suspected to contribute to the development and possible progression of cancer. More than half a century ago, Warburg (1) initiated research on mitochondrial alterations in cancer and proposed a mechanism to explain the differences in energy metabolism between normal and cancer cells. He suggested that mitochondrial alterations could provide unique therapeutic targets in various cancer types (1–3). Since Warburg’s proposal, several cancer-related mitochondrial alterations have been identified.
To carry out its functions, a mitochondrion carries its own genome, which consists of 13 polypeptides of the electron transport chain (ETC) and 22 tRNA and 2 rRNA genes for its own protein synthesis (ref. 4; Fig. 1). The remaining protein subunits involved in the ETC complexes, along with those required for maintenance of mitochondrial DNA (mtDNA), are nuclear encoded, synthesized in the cytosol, and are specifically targeted to the mitochondria. Typically a mammalian cell contains 103 to 104 copies of mtDNA and this DNA can replicate independently of nuclear DNA (5). Human mtDNA is a 16.6 kb circular double-standard DNA molecule and is devoid of protective histones, although mitochondrial transcription factor A and single-stranded DNA-binding protein are cooperatively involved in the maintenance of mtDNA (6). Unlike nuclear DNA, which is inherited from both parents and in which genes are rearranged by recombination, there is usually no change in mtDNA from parent to offspring. Mitochondrial genes have an exclusively maternal mode of inheritance in mammals, therefore mtDNA lineages are clonal (7).
Figure 1.

Organization of the human mitochondrial genome. The 16.5 kb genome encodes 37 genes including 22 for mitochondrial tRNA (20 standard amino acids, pink arrows) and 2 for the rRNA (black arrows). The rest of the 13 genes encode for subunits of the different respiratory complexes I to IV (11 genes) and ATP synthase (2 genes). Complex I (NADH dehydrogenase) is composed of 7 mitochondrial subunits (ND1, ND2, ND3, ND4, ND5, ND6,and ND4L) and 37 nuclear subunits. Complex-II (succinate dehydrogenase) is composed of 4 subunits (all being nuclear encoded) and is both a component of the ETC and an enzyme of the Krebs cycle. Complex III (cytochrome c reductase) is a complex of 11 subunits. Only subunit, cytochrome b (purple arrow), is encoded by mtDNA, the remainder 10 are nuclear encoded. Complex IV (cytochrome c oxidase) is a complex of 13 different subunits, 3 (red arrows) are encoded by mtDNA, and 10 are nuclear encoded. The ATP synthase family is composed of 14 subunits; 2 (blue arrows) are mtDNA encoded. The other 14 are nuclear encoded. The displacement loop (D-loop, orange arrows) is the main noncoding area of the mtDNA where replication occurs. The region contains promoters for the transcription of RNA from the 2 strands of mtDNA. The heavy H strand has higher guanine content, and is transcribed from the PH promoter. The light L strand is transcribed from the PL promoter. Replication of the heavy strand by DNA polymerase commences from the OH replication origin; this eventually exposes the OL origin allowing replication of the light strand.
The mitochondrial ETC is a major source of reactive oxygen species (ROS). Elevated levels of ROS are associated with various diseases including cancer, and are generally associated with a cascade of redox signaling that leads to DNA damage. MtDNA is continuously exposed to ROS produced by the mitochondria and is also predisposed to chemical damage (8, 9), for example, by environmental factors such as UV, cigarette smoke, and ionizing radiation. MtDNA has a 10-fold higher accumulation of mutations than nuclear DNA (5). Cancer cells can carry mtDNA mutations and altered copy numbers of mtDNA, which in turn affect expression and activity of the ETC. A number of antioxidant defense mechanisms exist in mitochondria to remove ROS. Cells are normally able to defend themselves against ROS damage through ROS scavengers and antioxidant enzymes such as superoxide dismutases (SOD), catalases, and glutathione peroxidases, which are often deficient in tumor cells. As a result, persistent oxidative stress on cells leads to promotion of cancer growth and metastasis through induction of DNA damage which leads to mutations. Because of the susceptibility of mtDNA to damage, mitochondria also contain their own DNA repair systems. One important mtDNA repair mechanism is base excision repair (BER), and the detection of various BER enzymes in mitochondria highlights the importance ofmaintaining mtDNA integrity for normal cellular functioning (10, 11).
In this review, we have sought to focus on mitochondria-specific mechanisms that may promote cell survival and cancer progression in the presence of mtDNA alterations. Abnormalities in the mitochondrial genome may also arise from nuclear genetic disorders, which are reviewed elsewhere (12). It has also been well established that mitochondria play an important role in the regulation of programmed cell death (apoptosis) via release of proapoptotic agents and/or disruption of cellular energy metabolism (13–17), and this will also not be discussed further.
Heteroplasmy and Homoplasmy
Cells harbor multiple copies of mitochondrial genes as opposed to only 2 copies of each nuclear gene. Homoplasmy is a state in which all the mitochondria of a cell have the same genome, which may either be the wild type or a mutated one. As each cell contains many mitochondria and each mitochondrion 2 to 10 copies of mtDNA, it is possible that wild-type and mutant mtDNA can coexist in a state called heteroplasmy. Therefore, the biological impact of a given mutation could vary, depending on the proportion of mutant mtDNAs carried by each cell. In earlier studies no evidence of heteroplasmy was detected, probably due to the lower sensitivities of earlier techniques. However, using massively parallel sequencing-by-synthesis approach, one recent study reported profound heteroplasmy in the mtDNA of normal human cell (18, 19). MtDNA constantly undergoes mutation, with expansion or loss of mutated mtDNA copies, which may lead to homoplasmy (20, 21). Whether mtDNA point mutations occur by simple clonal expansion is debated. Although a large number of somatic mtDNA deletions are capable of clonal expansion in individual cells (22), somatic point mutations have not been shown to be able to clonally expand or reach homoplasmy in vivo, though there are some reportsin cell culture (23) and germline cells (24). Indirect evidence supporting the possibility of clonal expansion of somatic point mutation comes from genetically engineered heteroplasmic mouse studies (25).
If a mutation confers cell growth/survival advantage or facilitates mtDNA replication, such a mutation is likely to survive through selection and may eventually become dominant and evolve to become a homoplasmic mutant, by clonal expansion. It has been indicated that a single cell with a mutant mitochondrial genome may acquire a selective growth advantage during tumor evolution, allowing it to become the predominant cell type in the tumor cell population (26). Also, it is possible that some (if not all) germline mtDNA mutations are actually somatic mutations that occurred early during prenatal development of the individual and drifted to homoplasmy. As such, heteroplasmic mutations may reflect an intermediate stage in this process (Fig. 2). For an mtDNA mutation to have significant effect on the physiology of the cell it must reach a threshold level of 60% or more (27, 28), depending on the type of mutation. Also, it is insufficient for the cell to accumulate different mutations, as different mutations are likely to compensate each other’s deficiencies (29–31); however, there are limitations of transcomplementation (32). These studies strongly support the hypothesis that mutants would need to accumulate via clonal expansion of a single initial mutant. An alternative explanation to homoplasmic mtDNA mutations has been presented by Coller and colleagues (20), where using a mathematical model they show that the presence of a homoplasmic mitochondrial mutation in a tumor can result from random segregation of mutant genomes in many cell generations that occur during tumor development. The model further predicts that homoplasmic mitochondrial variations may arise in normal tissue derived from stem cells that have undergone a high number of cell division. This would mean that there is no need to have a selective advantage for a mutation in the mitochondrial genome to become dominant. Whether this exclusive model is valid or not is still a topic of extensive debate. If there is a high background of random mutation that provides no selective advantage, it is also to be considered that there are other mutations that alter mitochondrial function and cell physiology in a manner that has significant effect on tumor development.
Figure 2.

Homoplasmic and heteroplasmic mtDNA mutations due to environmental and genotoxic damages, along with nuclear (N) genetic changes; cells may incorporate somatic mtDNA mutations and acquire a state of heteroplasmy with both wild-type and mutated mtDNA copies. Further progression of tumor cells may be aided with a homoplasmic bias with mutant mtDNA copies or an admixture of both wild-type and mutant mtDNA, a state of heteroplasmy.
MtDNA Alterations as Markers of Cancer
Mitochondrial cellular content and mutations are emerging as new molecular markers for cancer detection and monitoring. It has been shown that damaged mtDNA in cell lines led to the rapid evolution of homoplasmic mutations (33). Recent refinements in techniques including next-generation sequencing (34) for the detection of mtDNA content or copy number combined with rapid high-throughput methods of mutation detection, have incited interest in clinical studies of various tissues and bodily fluids (35). Initial examination of human bladder, head and neck, breast (19), and primary lung tumors revealed a high frequency of mtDNA mutations (36). One recent study also showed a correlation between increased mtDNA copy and future risk of lung cancer in heavy smokers (37). The mutated mtDNA was readily detectable in cancer-paired bodily fluids (including urine, saliva, and sputum) from each type of cancer and was 19 to 220 times more abundant than mutated nuclear p53 DNA (36). Recent studies from our laboratory detected clonal mtDNA mutations in histologically normal respiratory mucosa and surgical margins of some smoker lung cancers and recurrent head and neck cancer patients, respectively (38, 39), supporting the presence of extensive altered cellular fields with aberrant mtDNA surrounding much smaller clinical neoplasms.
It is critical to know the timing of mtDNA mutation in the course of disease development and progression if such alterations are to be used as markers. MtDNA mutations have been detected in clinical samples from early stage patients, indicating that they could be useful in early cancer detection. In a comprehensive analysis, 93 premalignant lesions from the upper aerodigestive tract were examined for mtDNA D-loop mutation (40). Twenty-two percent of the hyperplasia, 33% of the mild dysplasia, 36% of the moderate dysplasia, 50% of the severe dysplasia, and 62% of the CIS (carcinoma in situ) lesions carried mutation in the D-loop region. In 2 other studies, the whole mitochondrial genome and D-loop regions were sequenced in gastric intraepithelial neoplasia and sequence variants were detected in 100% and 62% of cases, respectively (41, 42). Using D310 mutations as a clonal marker, investigators have detected changes in fine-needle aspirates in breast cancer (43) and in urine sediments from patients with bladder and prostate cancer (44, 45). A study comprising the buccal cells from 42 healthy smokers and 30 nonsmokers reported higher frequency of mtDNA mutation in the mtDNA of the smokers, though no clear connection has been established that smokers with mtDNA mutations are more likely to develop cancer (46).
By virtue of their clonal nature and high copy number, mitochondrial mutations could provide a powerful molecular marker for noninvasive early detection of cancer. In an interesting study, a specific mtDNA mutation (m.15296A>G; cytochrome b) was clonally detected in bone marrow samples collected during different time points from a leukemia patient (47), suggesting a possible role for this marker for monitoring disease progression. In light of this, it remains important to identify disease-associated clonal mtDNA mutations in different cancers at all stages utilizing appropriate detection platforms. With the advent of high-throughput approaches such as the Mitochip (48), larger more prospective studies may be carried out to determine the value of these mutations in early detection approaches. As most cancer cells harbor homoplasmic mitochondrial mutations, their ease of detection in bodily fluids and minute cellular samples such as biopsies offer varied opportunities for clinical use. As noted earlier, definitive diagnosis of clonal expansion through mtDNA testing would be helpful in designating difficult biopsies as malignant. Testing for mtDNA mutation in blood, urine, saliva, stool, or sputum DNA couldbeusedwithimaging studiestodetectearly cancers. Clonal mtDNA mutations could also be followed to indicate recurrence or to monitor response to therapy, and could even be therapeutic targets. Finally, abrogation of mtDNA mutations in patients undergoing chemoprevention could be used to monitor reversal of early benign lesion. Validation of tumor-associated mtDNA mutations by comparing matched normal and tumor samples followed by their detection in corresponding clinically available samples including paired bodily fluids will facilitate their development in prevention, early detection, and monitoring strategies.
MtDNA Mutations in Human Cancers
To date, whole genome or specific regions of human mtDNA (Fig. 1) have been sequenced in different tumor types for mutation detection. The mutations detected specifically in the regulatory noncoding D-loop or other coding regions of the mtDNA are discussed in the following text.
D-loop alteration
The D-loop or displacement loop region is the main noncoding area of the mtDNA molecule, in which replication of mtDNA occurs. The region also contains promoters for transcription from the 2 strands of mtDNA. D-loop alterations might interfere with initiation of mtDNA replication, though there are no reports of precise consequence of D-loop mutations (49). An earlier study from our laboratory identified the C-tract, a poly-C mononucleotide repeat (CCCCCCC...TCCCCC) located between the nt303 and nt315 (regarded as D310) within the D-loop, as a mutational hot spot in primary tumors (50). The first stretch of cytosines is highly polymorphic ranging between 7-C to 9-C, with the most common sequence represented by 7-C. Deletion or insertion mutations in this region have been observed in approximately 22% of the tumors analyzed (50). To date, frequent somatic D-loop alterations (insertion/deletion) have been identified in all major cancer types (refs. 35, 41, 45, 49–58; Table 1). The number of cytosine in the D310 area seems to influence the incidence of mutations in the D310 sequence (49, 56). The majority of D310 mutations were observed when the number of cytosine was more than 7 (i.e., 8 or 9). Therefore, this variation in cytosine number could explain discrepancies among published results (26, 50, 52, 59–61).
Table 1.
D-loop mutation in different cancers
| Cancer type |
n/N, % of patients with mutation |
Method | Reference |
|---|---|---|---|
| Bladder cancer | 3/15, 20 | CS | 50 |
| 4/16, 25 | CS | 45 | |
| Breast cancer | 5/17, 29 | CS | 50 |
| 3/20, 15 | CS | 45 | |
| Colon cancer | 38.3% | CS | 49 |
| 7/25, 28 | CS | 50 | |
| 20/45, 44 | CS | 52 | |
| Gastric cancer | 15/31, 48 | CS | 58 |
| 5/8, 62 | CS | 50 | |
| 4/32, 12.5 | CS | 51 | |
| 15/24, 62 | CS | 41 | |
| HNSCC | 23/109, 21 | CS | 54 |
| 19/51, 37 | CS | 50 | |
| 6/13, 46 | CS | 36 | |
| HCC | 2/56, 3.6 | CS | 53 |
| 13/19, 68 | CS | 35 | |
| 28/71, 39.4 | CS | 55 | |
| 37/62, 59 | CS | 57 | |
| Lung cancer | 17/28, 61–CL | CS | 56 |
| 11/55, 20–PT | CS | 56 | |
| 16/100, 16 | CS | 50 |
Abbreviations: CS, conventional sequencing; CL, cell line; PT, primary tumors; HCC, hepatocellular carcinoma.
Most of the studies in Table 1 compared the mtDNA sequence in neoplastic cells to mtDNA in matched normal tissue or lymphocytes; however, the functional contribution of D-loop alterations in cancer is unclear. The different rates of D310 variations in different tumor types (45, 50) suggest the existence of alternative mechanisms for the generation of some D310 alterations, such as the rate of acquired mutations during tumor development and the number of mitochondria per cell. The fact that most D-loop alterations are confined to the polymorphic length range suggests that most D310 variants in tumors are unlikely to functionally impair mitochondria.
Somatic mtDNA mutations
Somatic mtDNA mutation was reported in colon cancer by Polyak and colleagues, though a whole-genome conventional sequencing approach (26). The whole mitochondrial genome was sequenced in 10 colon cancer cell lines. Seventy percent showed a total of 12 somatic mtDNA mutations which were homoplasmic in nature. All the mutations were also detected in the primary tumors from which the cell lines were derived. Interestingly, another complete sequencing of the mitochondrial genome revealed a high level of mtDNA mutations in normalcolonic crypt stem cells (21). The presence of mtDNA mutations in stem cells indicates that mutations actually develop prior to development of colon cancer, though it is yet to be established whether these mutations actually cause malignancy. Following the mtDNA analysis in colon cancer (26), numerous somatic mtDNA sequence variants using a whole genome approach were reported in various cancers including bladder (36), breast(62, 63), esophagus (62), head and neck (64–66), leukemia (47), lung (67), and thyroid cancers (refs. 68–70; Table 2). Majority of these human cancers (36, 62–65, 71) harbor some frequency of somatic mtDNA mutation which are homoplasmic, with low levels of heteroplasmy.
Table 2.
Somatic mtDNA mutation involving other mtDNA regions in different cancers
| Cancer type |
n/N, % of patients with mutation |
Method | Reference |
|---|---|---|---|
| Bladder cancer | 9/14, 64 | CS | 36 |
| Breast cancer | 15/36, 47 | CS | 62 |
| 4/20, 20 | Mitochip 2.0a | 63 | |
| Esophagus cancer |
12/31, 39 | CS | 62 |
| HNSCC | 41/83, 49 | Mitochip 2.0a | 66 |
| 4/12, 33.3 | CS | 64 | |
| 4/7, 57 | Mitochip 2.0a | 71 | |
| Leukemia | 9/24, 37.5 | CS | 47 |
| Lung cancer | 33/55, 60 | CS | 67 |
| Thyroid cancer | 26/45, 58 | CS | 69 |
| 34/66, 51.5 | CS | 70 | |
| 20/26, 76.9 | CS | 68 |
Abbreviation: CS, conventional sequencing.
Affymetrix Mitochip v2.0 platform (www.affymetrix.com) for mitochondrial whole genome sequencing.
A common deletion (4,977 base pair; located in ND2) studied by several groups in several cancer types, including breast (72, 73) and liver (57), indicated that the deletion accumulates more in the adjacent nontumor tissue than in the neoplastic cells. This may indicate that during carcinogenesis a strong selection pressure exists in favor of cells containing mitochondria with no deletions (57).
Importantly, most described mtDNA mutations in cancer are similar to naturally occurring polymorphic mtDNAs in the population and many of them have no apparent phenotype. These apparently neutral but homoplasmic somatic alterations point to simple clonal evolution of the affected cancer cell, presumably from another driving nuclear mutation or altered oncogenic pathway. As such, these changes are valuable clonal markers (see the following text) but are very unlikely to produce a real growth advantage for affected cells. How these simple variants arise and then become homoplasmic in individual cells is still an unknown process.
Several investigators have recently identified that a significant number of the mtDNA studies in Table 2 are based on “flawed sequencing results” (74). On the basis of a literature search and subsequent analysis of different phylogenetic trees, most errors seemed to result from inadequate database searches as well as sample mix-ups (74–79). These investigators identified several reported “mtDNA mutations,” to actually be neutral single nucleotide polymorphisms (SNP) belonging to different haplogroups (74–78). It has been suggested that a phylogenetic tool should be utilized for proper data analysis as well as for appropriate mtDNA study design (74). This will allow us to understand the distinction between neutral SNPs in the population and disease-associated mutations (74).
It should be noted that mutation detection depends on the sensitivity of the method used and the quality of the starting DNA. For example, a high incidence of mtDNA mutations were detected by using single cell analysis, whereas most of the mutations could not be detected when using DNA from pooled cells (80). Another consideration is that the majority of the reported mutations are synonymous. Subsequent functional studies of the specific and bona fide tumor-associated mtDNA mutations as carried out recently in uterocervical and bladdercancer (66, 81) would allow us to understand their role in cancer progression and impact on mitochondrial biology. Such studies provide a link between mitochondrial genomic mutations and induction of the malignant phenotype that has been a known feature of most tumors for centuries. These studies indicate that mitochondrial mutations may activate pathways involved in the development and maintenance of a malignant phenotype. Moreover, generation of increased cellular ROS results in a continuous genomic insult leading to mutations in the mitochondrial genome, which may alter the functioning of the oxidative phosphorylation chain.
Germline sequence variants were reported in different tumors and were suggested as an indicator of a high susceptibility genetic background which might facilitate concomitant somatic mutation in mtDNA and nDNA (82). In a recent study, mtDNA A10398G polymorphism and alcohol consumption were shown to be associated with breast cancer risk (83). However, the authors failed to categorize the participants into well-defined haplogroups (83) as done by Bai and colleagues (84). In light of the recent criticism of this study, the association between mtDNA polymorphism and breast cancer risk needs to be further refined. Thus, in mtDNA SNP-association studies, population categorization in respective haplogroups followed by careful evaluation of SNP distribution is warranted.
MtDNA copy number
In addition to mtDNA mutations and deletion, alteration in the mtDNA copy number has been studied in different tumor types. An increase in mtDNA copy number has been reported in Head and neck squamous cell carcinoma (HNSCC; ref. 85), papillary thyroid carcinoma (86, 87), and lung cancer (88). An increase in mtDNA copy number has also been associated with chronic lymphocytic leukemia (CLL), Burkett lymphoma, Epstein-Barr virus–transformed lymphoblastoid cell lines, non-Hodgkin’s lymphoma, and small lymphocytic lymphoma (89). An increase in mtDNA content has been suggested as a compensatory mechanism for mtDNA damage (85). On the contrary, a decrease in mtDNA copy number (mtDNA depletion) has been reported in number of tumor types including breast (86), kidney (90), liver (91), ovary (92), and gastric cancers (58), and was suggested to be associated with increased risk of disease development and progression (86, 90–92). The mtDNA copy number in cancers might depend on several factors (93), including the site of mutation in the mitochondrial genome. For example, mutations in the D-loop of the mitochondria, which control replication of mtDNA, may result in decreased copy number. Conversely, an increase in mtDNA copy number might occur as an adaptive response to mitochondrial dysfunction or due to mutations in nuclear genes involved in controlling mtDNA copy number indirectly. Finally, the likelihood of a more active mechanism controlling mtDNA copy number cannot be overlooked, as we still have not identified all factors participating in this process.
Chemotherapy and mtDNA Mutations
Mitochondrial defects have been implicated in the development and progression of cancer for several decades. Certain chemotherapeutic agents with DNA-damaging properties may cause mtDNA mutations, resulting in increased mitochondrial ROS generation. Chinese hamster ovary cells exposed to to cis-diamminedichloro-platinum(II) for 24 hours have shown 4- to 6-fold levels of DNA adducts in mtDNA compared with nuclear DNA (94). Studies in adenocarcinoma (95) showed that chemotherapeutic agents such as cisplatinum can accumulate in mitochondria and damage mtDNA in vitro. Also, studies in CLL showed that patients who received prior chemotherapy generated higher levels of cellular free radical than those who were untreated (96). Further studies from the same group also indicated that CLL patients who had received prior chemotherapy had higher frequency of heteroplasmic mtDNA mutations compared with the patients untreated with chemotherapy. The mtDNA mutations were associated with increased ROS generation (97). Studies by our group have indicated that exposure of cervical cancer cells to 2-methoxyestradiol (2-ME) resulted in an increase in mitochondrial membrane potential and apoptosis (98). These studies suggest that chemotherapy with certain DNA-damaging agents may cause mtDNA mutations, which initially appear heteroplasmic, and are associated with increased ROS generation. However, it is difficult to assess whether the mutations are a direct consequence of chemotherapy-induced DNA damage or whether the chemotherapy first increases the free radical generation, leading to oxidative damage to mtDNA, resulting in further increase in ROS generation due to a compromised respiratory chain. It is quite possible that both scenarios occur in vivo. As discussed earlier, both homo- and heteroplasmic mutations have been frequently observed in cancer cells, it is likely that drug-induced mtDNA mutations in cancer cells may initially appear in a heteroplasmic state and convert to homoplasmic state via in vivo selection process.
As the mitochondrion plays an essential role in ROS generation, increased ROS may also affect the sensitivity of cancer cells to chemotherapy (99). Clinical drug resistance is a multifactorial event, and mtDNA mutation is only one of many important contributing factors. An increase in ROS generation due to mtDNA mutation may also assist in the design of novel therapeutic strategies that preferentially kill cancer cells with ROS stress (100, 101). It is possible that respiratory inefficiency as a consequence of mtDNA alterations may contribute to the elevated glycolytic activity (Warburg effect) leading to constitutive oxidative stress frequently observed in malignant cells. Recent studies by Dang and colleagues (102) have described approaches to target the Warburg effect in cancer, where the authors have shown that mutant IDH1 may be used as potential therapeutic target as a metabolic genetic biomarker for drug development. A detailed description of such metabolic therapeutic approaches have been reviewed in ref. 103.
Another promising therapeutic approach to treat patients with mtDNA mutation in their tumor is based on allotopic gene expression (ref. 104; allotopic gene expression is based on expressing a mitochondrially encoded gene from nucleus transfected constructs as a fusion with an N-terminal mitochondrial target sequence). Allotopic gene expression is generally difficult due to the extreme hydrophobicity of these proteins, which prevents their import into mitochondria from the cytosol. An alternative to this approach is the use of inteins (self-splicing “protein introns”) as their insertion into such transgenes could greatly reduce the hydrophobicity of the encoded proteins, enabling import, with post import excision and restoration of the natural amino acid sequence. A detailed description of the use of inteins is reviewed in ref. 105. Recently, antioxidant therapy showed promise as anticancer therapy, and such agents were designated as “mitocans” (an acronym for mitochondria and cancer). Mitocans act via mitochondrial destabilization, with activation of mitochondrial mediators of apoptosis. These inducers of apoptosis elude the frequent mutations at mtDNA that occur in cancers which may be responsible in making tumors resistant to many established chemotherapeutic drugs (106, 107). Mitocans may be favorable in the fight against cancer because of low levels of side effects due to their greater target selectivity. Mitocans are still in the early phase of clinical testing, although preliminary data suggest a promising clinical outcome. Mitocans when used in combination with existing chemotherapeutic drugs offer opportunities for additive or synergistic therapeutic effects (108). Further studies are warranted to see whether they can induce the clonal depletion and exhaustion of aberrant mtDNA progenitor cell populations leading to a new therapeutic strategy.
Mitochondrial Production of ROS
Mitochondria play important roles in energy metabolism, generation of ROS, and apoptosis. The best characterized function of the mitochondria is oxidative phosphorylation in which nicotinamide adenine dinucleotide (NADH) and succinate generated in the citric acid cycle are oxidized, providing energy to power ATP synthase (complex V of ETC) for generation of ATP (Fig. 3). Thisis a multistep redox process that occurs on the mitochondrial inner membrane. In the process of ETC, a small percentage of electrons are prematurely leaked to oxygen, from complex I and/or complex III, resulting in the formation of toxic free radicals, commonly referred to as ROS. Considering that mtDNA lacks sizeable introns, most mutations occur in the coding regions, and are thus likely to be of biological consequence (109). Reduced expression of oxidative phosphorylation complexes (complexes II, III, and IV of the ETC, and ATP synthase) has been associated with various forms of cancer. This intricate energy conversion system is susceptible to malfunction by many causes, including mtDNA and/or nuclear gene mutations or oxidative damage of the enzymes and associated lipids, occurring either alone or in various combinations, leading to increased production of ROS (110–113).
Figure 3.

Schematic representation of mitochondria complexes. The ETC in the mitochondrion is the site of oxidative phosphorylation. The NADH and succinate generated in the citric acid cycle (TCA cycle) are oxidized, providing energy to power ATP synthase. The diagram shows the complexes involved in OXPHOS (oxidative phosphorylation). Complex I (also known as NADH coQ reductase or NADH dehydrogenase) accepts electrons from the citric acid cycle (TCA cycle) electron carrier NADH, and passes them to coQ (ubiquinone; labeled CoQ), which also receives electrons from complex II, (also known as succinate dehydrogenase or succinate-Q oxidoreductase). CoQ passes electrons to complex III, (also known as Q-cytochrome c oxidoreductase or cytochrome c reductase or cytochrome b complex), which passes them to cytochrome c (Cyt c). Cyt c passes electrons to complex IV (also known as cytochrome c oxidase), which uses the electrons and hydrogen ions to reduce molecular oxygen to water. The electrochemical proton gradient allows ATP synthase (ATPase or complex V) to use the flow of H+ through the enzyme back into the matrix to generate ATP from adenosine diphosphate (ADP). The last destination for an electron along this chain is an oxygen molecule. Normally the oxygen is reduced to produce water; however, few of the electrons passing through the chain escape and oxygen is instead reduced to the superoxide radical (ROS).
Complex I
NADH dehydrogenase (complex I) is located in the inner mitochondrial membrane and catalyzes the transfer of electrons from NADH to coenzyme Q (CoQ). It is composed of 46 subunits, 7 of which [NADH dehydrogenase (ND) 1–6 and NADH dehydrogenase 4L] are mitochondrially encoded (Fig. 3). The first report of a functionally significant mtDNA mutation in cancer was the deletion of 294 nucleotides in ND1 and preferential transcription of truncated ND1 in a patient with renal adenocarcinoma (114). Targeting mitochondria with nuclear-transcribed ND2 also results in increased ROS production (66). Transmitochondrial cybrids (cytoplasmic hybrids; see glossary) harboring the m.14487T>C mutation in ND6 had increased ROS production owing to complex I deficiency (115). Interestingly, increased ROS did not result in increased antioxidant defense, suggesting that complex I mutations may result in failure of the antioxidant defense system.
The role of mitochondrial mutations in complex I components in the progression of tumor metastasis was studied by using mouse tumor cell lines with high or low metastatic potential (116). Complex I activity decreased and metastatic potential was acquired in cybrids containing mtDNA from highly metastatic cells, whereas those containing mtDNA from cells with low metastatic potential had no change in complex I activity and lost metastatic potential. High metastatic potential was also conferred by mutant ND6. These observations suggest that defects in complex I and high metastatic potential are transferred simultaneously and that some pathogenic mutations in mtDNA may also induce complex I defects (116).
Accumulation of mtDNA mutations in benign oncocytoma is exclusively associated with loss of complex I and also complex III (ref. 117; the most potent sites for ROS generation in the TCA cycle). The fascinating feature of these oncocytomas is that nearly all mutations were somatic and homoplasmic in nature. The deficiency of complex I activity has also been reported by others (118, 119), further supporting the concept of clonal expansion of mtDNA mutations and suggesting that positive selection pressure is exerted on cells by mtDNA mutations that alter complex I activity.
Ubiquinol-cytochrome c reductase (complex III)
Overexpression of a 21–base pair deletion mutation of mitochondria encoded cytochrome b (complex III) in murine and human uroepithelial carcinoma/transformed cells resulted in increased ROS, lactate production, and oxygen consumption. An increased in vitro and in vivo tumor growth was also observed in these cells (81). Moreover, stable mutant clones showed increased invasive phenotypes in murine and human bladder cancer models and were able to kill normal splenocytes indicating an adaptation of the mutant cells to evade the immune system (81).
Cytochrome c oxidase (complex IV)
Defective oxidative phosphorylation, including defects in cytochrome c oxidase (complex IV) in cancers, which can increase ROS levels and mtDNA damage has also been reported (69, 120, 121). Low expression of mitochondrial COX II or relatively high expression of COX I and III (see glossary) have been reported in different cancer types and tumors (120–123).
ATP synthase (complex V)
Insights into the alteration of ATP6 (see glossary) has been reported in prostate cancer (122). Mouse tumors harboring ATP6 with an m.8993T>G mutation generated increased ROS and it was postulated that this may lead to an increase in DNA damage and tumor growth (122, 124). Petros and colleagues (124) also introduced ATP6m.8993T>G into the PC3 prostate cancer cell line through cybrid transfer and found that mutant cybrid tumors in nude mice were 7 times larger than those from wild-type cybrids, which had little perceptible growth. This mutation may also contribute to prognosis and bone metastasis in prostate cancer (125). ATP6-m.8993T>G, and also an m.9176T>C mutation, also decreased mitochondrial respiration and accelerated tumor growth by prevention of apoptosis in HeLa cell cybrids (126). These different mutations in ATP6 may lead to different functional abnormalities.
Table 3 describes the links to the various databases available for analyzing mtDNA alterations.
Table 3.
Important databases for analyzing mtDNA alterations
Antioxidant Defense Mechanisms
During the reduction of oxygen to water in mitochondria, approximately 1% to 2% of total oxygen consumption gives rise to ROS such as superoxide radicals (O2–) and hydrogen peroxide (H2O2). Oxidative stress may affect several cellular functions, like proliferation, genomic instability, alterations in cellular sensitivity to anticancer agents, invasion, and metastasis. The first line of defense that cells possess is the conversion of O2– to H2O2 and O2 by SODs; (Fig. 4). Overexpression of Mn-SOD leads to retardation of tumor growth (127) and resistance to O2– (128). Furthermore, modulation of signal transduction cascades, leading to connective tissue degradation, and finally cancer, has been linked with unbalanced expression of the Mn-SOD (128, 129). Generally, most cancer cells have been associated with low expression of Mn-SOD (130, 131), but some reports also indicate increased expression of this protein/enzyme (132–134). The discrepancy in the levels of SOD could be due to (a) the studies were conducted in different population groups; (b) the technique used to detect the enzyme levels varied between the studies; or (c) the studies were done in different cancer types and of different stages. Further, variable expression of Mn-SOD in different cancer types, polymorphisms affecting enzyme function, and a mutation in exon 3 have also been associated with unbalanced expression and decreased activity of the enzyme (135). Mn-SOD is protective up to a point, beyond which protection is lost and damage is increased (136). This is also supported by the observation thatabnormally high levels of Mn-SOD activity, whereas suppressing cell growth, increase the invasive potential of cancer cells, (137, 138) possibly owing to an imbalance between O2– production and H2O2 degradation.
Figure 4.

Antioxidant defense mechanism in mitochondria. Mitochondria is the primary site for generation of ATP, which is the energy needed for cellular machinery. In addition to energy, ROS are produced, which results in cellular damage. The most commonly known ROS are hydrogen peroxide (H2O2), superoxide (O2–), and hydroxyl ions (OH–). Superoxide generated in mitochondria is converted to H2O2 by the enzyme Mn-SOD. The H2O2 is further degraded to water by 2 defense mechanisms: (i) Glutathione peroxidase (GPx) catalyzes the reaction, whereby GSH reacts with H2O2 and converts it to glutathione disulfide (GSSG) and water (H2O). Glutathione reductase (GSR) then reduces the oxidized glutathione (GSSG) to GSH. (ii) As a second mode of defense the H2O2 is converted into H2O and molecular oxygen (O2) by the mitochondrial catalase. Any superoxide that escapes mitochondria is again attacked by the SOD (Cu/Zn) present in the cytosol, which is again converted to H2O2 and this peroxide is decomposed by GPx and Catalase present in the cytoplasm and peroxisomes, respectively. The last destination for an electron along this chain is an oxygen molecule. Normally the oxygen is reduced to produce water; however, few of the electrons passing through the chain leak resulting in the generation of O2–. The most common site for electron leak are complexes I and III. Superoxide is not particularly reactive by itself, but can inactivate specific enzymes or initiate the formation of OH– (depicted in red in dotted lines). Accumulation of OH– in mitochondria could lead to release of cyt C from mitochondria leading to apoptosis. The diagram also depicts the TCA cycle which takes place in the matrix of the mitochondria. To be noted: the diagram depicts NADH being generated by malate dehydrogenase, but in TCA cycle NADH is also generated by isocitrate dehydrogenase and α-ketoglutarate dehydrogenase.
H2O2 is not a radical because it lacks an unpaired electron, but it is capable of mediating oxidative damage. H2O2 is capable of diffusing across membranes, and in the presence of free transition metals is capable of reacting and forming OH– (hydroxyl radicals; Fenton Reaction). Removal of H2O2 is hence another and second critical step, mediated by glutathione peroxidase (GPx). GPx is a selenium-containing tetrameric glycoprotein, that is, a molecule with 4 selenocysteine amino acid residues, that reacts with glutathione to reduce H2O2 to H2O (Fig. 4). A high level of GPx has been reported in lung (139), gastrointestinal (140), and colorectal (141) cancers, suggesting that oxidative stress plays an important role in tumorigenesis. Contradictory to these findings, decreased concentrations of GPx were reported in CLL (142), prostate cancer (143), and bladder cancer (134). Upregulation of antioxidants induced by oxidative stress, such as GPx, may confer a selective growth advantage to tumor cells over their adjacent normal counterparts. Low or reduced GPx in tumor tissue could be an indication of poor enzymatic antioxidant defense system, which would make cells extremely vulnerable to ROS-induced damage. Further, the discrepancies noted earlier regarding levels of glutathione (GSH) and/or GPx could be due to heterogeneous tissue structure, storage condition of the samples or the method used in the analysis. Emerging studies also reveal regulation of GPx and its isoforms by methylation and deletions (143).
Another defense enzyme that can convert H2O2 to H2O is catalase. Catalase is present primarily in the peroxisome, and also in mitochondria from rat heart (144) and liver (145). Catalase has one of the highest turnover numbers of all enzymes; 1 molecule of catalase can convert approximately 6 million molecules of H2O2 to H2O and O2 per second. Both upregulation (133) and downregulation (146) of catalase, as well as decreased catalase activity (147), have been reported in lung cancer. One possibility for the discrepancy in the regulation of the enzyme could be the assay method used in the studies. Aberrant methylation in the catalase promoter may also downregulate catalase transcription (148). Attenuation of catalase in malignantly transformed cell lines is mainly responsible for the elevated ROS levels in these cells (149). Although downregulation of catalase has been observed in carcinomas, the potential consequences of the decrease largely remain unknown. One of the consequences of decreased catalase expression may be elevated transcription factor activity during tumor progression (reviewed in ref. 150). Reduction of catalase activity in liver tumors is one of the earliest biochemical changes observed in this cancer (151). Liver catalase activity is restored to normal levels by tumor removal (151), indicating that soluble factors from tumor cells may repress catalase expression.
These findings strongly suggest that ROS, especially H2O2, are not efficiently removed in most tumor tissues. Taken together, these studies indicate that (a) high level of SOD accompanied by decrease in catalase activity and (b) decreased levels of SOD along with decreased activity of GPx may lead to increased production of intracellular H2O2, or enhanced production of oxygen radicals leading to a favorable environment for DNA damage and promotion of cancer.
MtDNA Repair Machinery
To maintain proper genetic integrity and ultimately prevention of cancer, more elaborate mechanisms are in place to repair DNA damage in the nucleus compared with those present in the mitochondria. DNA repair is a crucial function necessary to maintain genomic stability. Thus, multiple DNA repair pathways exist, each associated with a specific class of lesion (152). The major source of endogenous DNA damage is ROS generated from oxidative phosphorylation. Exogenous sources of DNA damage include environmental agents such as UV light, ionizing radiation, chemicals, toxins, and pollutants. The manner in which oxidative DNA damage is handled varies in the mitochondria and nucleus (153).
As mentioned earlier, continuous generation of DNA modifications by ROS occur at a much higher frequency in mitochondrial than nuclear DNA, making mtDNA more susceptible to mutation, which has been well documented in cancer. It is therefore not surprising that mitochondria have endogenous DNA repair mechanisms to remove the oxidatively damaged bases. The most versatile excision repair pathway, responsible for repairing most endogenous DNA lesions is BER (Fig. 5). To date, this is the only fully intact repair system to be reported in mitochondria (154). In the nucleus, 2 penultimate steps of BER are carried out by DNA polymerase-β, which exhibits 5′-deoxyribose-5-phosphate (5′-dRP) lyase and DNA polymerase activities. This is followed by joining of the repaired DNA strand by DNA ligase. The sole DNA polymerase known to be functional in the mitochondria is polymerase-γ (pol-γ) and it is assumed to function in mitochondrial BER (155). Human pol-γ is composed of a 140 kDa catalytic subunit and a 55 kDa accessory subunit. The catalytic subunit of pol-γ exhibits 5′-deoxyribose phosphatase lyase activity and can fill nucleotide gaps generated in the BER pathway (156). Mutations in either the polymerase or the exonuclease domain of pol-γ have been associated with increased frequency of mtDNA mutations (155). The molecular mechanism by which pol-γ ensures stability of the mitochondrial genome following mtDNA damage is still unknown. One aspect of poor repair of mtDNA has been attributed to the oxidation of pol-γ by H2O2 within mitochondria (157). Mouse studies reveal the absolute necessity of pol-γ for embryonic development and mtDNA maintenance (158). Homozygous knock-in mice expressing pol-γ lacking proof-reading activity (the catalytic subunit of pol-γ) developed an mtDNA mutator phenotype with a 3- to 5-fold increase in mtDNA pointmutations and deletions (159). This increase in somatic mtDNA mutations has been associated with many alterations, including ageing, but a higher incidence of cancer has not yet been reported (159). The pol-γ mutator mouse not only harbors mtDNA point mutations but also harbors linear mtDNAs, which cause replication pausing and chromosomal breakage leading to perturbed mtDNA replication and aging features (160). In contract to these studies Vermulst and colleagues (161) reported that a heterozygous pol-γ mutated mouse exhibited 500-fold higher mutation when compared with a normal mouse without any correlation with aging phenotype. In their subsequent studies (162) the same authors reported that in the homozygous pol-γ mutated mouse, there was a 7- to 11-fold increase in mtDNA mutation and accelerated aging compared with the wild-type or heterozygous mutator mouse. These studies suggest that there may be prevalent heterozygous carriers of pol-γ, who harbor a large number of undetected mitochondrial mutations and still attain normal aging.
Figure 5.
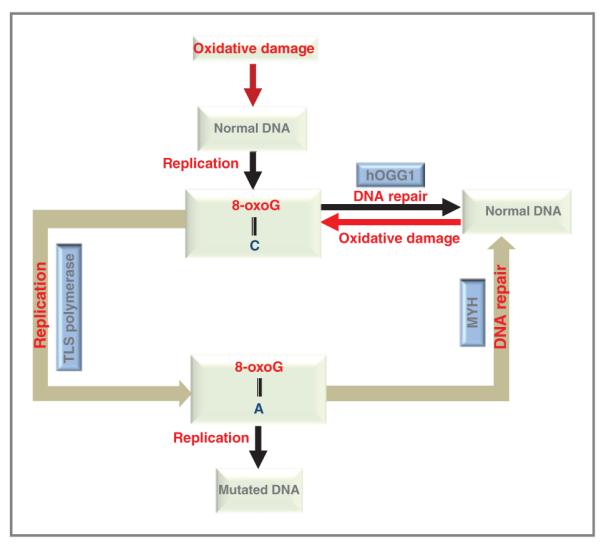
DNA repair pathway in mitochondria. A schematic representation of BER in mitochondria. In normal DNA guanine (G) pairs with cytosine (C); G:C. In case of oxidative DNA damage, guanine is directly oxidized to 8-oxoG, which pairs against cytosine (C). The oxidatively damaged base is repaired by DNA glycosylase, here the represented glycosylase is hOGG1 (8-oxoguanine DNA glycosylase which acts both as an N-glycosylase and an AP-lyase), which specifically removes the 8-oxoG opposite C and restores the normal DNA, that is, G:C. If unrepaired, the damaged base are replicated by translesion DNA synthesis (TLS) polymerases (181), which results in an 8-oxoG opposite adenine (A). In the second defense mechanism, repair enzymes like hMYH (Mut Y homologue) act in removing adenine opposite 8-oxoG thus eliminating G:C → T:A transversions. If the 8-oxoG:A is left unrepaired and undergoes DNA replication it results in mutated DNA, that is, G:C to T:A.
DNA glycosylases are also involved in the repair of mtDNA by BER. The first DNA glycosylase discovered was uracil DNA glycosylase (UDG) which excises uracil from DNA. Uracil may also arise due to spontaneous cytosine deamination or misincorporation of dUMP opposite adenine during DNA replication, leading to a G:C to A: T transition mutation. There are 2 isoforms of UDG-– nuclear and mitochondrial, which are generated by transcription from different promoters and alternate splicing. Deficiency of mitochondria-specific UDG in yeast was shown to be mutagenic to mtDNA (163). Recent studies also indicate the existence of UDG in mammalian mitochondria (164, 165). Upregulation of mitochondrial UDG was reported in response to oxidative stress highlighting the importance of the enzyme in mitochondria (166).
Exposure to oxidative damage often leads to conversion of guanine to 8-oxo-7,8-dihydroguanine (8-oxoG), resulting in the incorporation of dATP opposite 8-oxoG leading to G:C to T:A transversion (167). In humans, 8-oxoG is removed by hOgg1, a BER DNA glycosylase/Ap lyase that specifically incises 8-oxoG opposite cytosine (168). The hOGG1 gene encodes 2 major isoforms, α-hOGG1 and β-hOGG1, which are products of alternative splicing. α-hOGG1 is nuclear whereas β-hOGG1 is mitochondrial (169). We and others have shown that mtDNA repair and cellular survival is enhanced by targeting recombinant hOGG1 to the mitochondria in HeLa cells (which normally expresses low levels of hOGG1;ref. 98, 168, 170, 171). Targeting hOGG1 to mitochondria also protects cells from menadione-induced oxidative stress, apoptosis evoked by xanthine oxidase, fatty acid–induced apoptosis and nitric oxide–induced mtDNA damage (98). These studies have confirmed that hOGG1, when expressed in mitochondria, enhances mtDNA repair and protects cells from damage-induced death. Our previous studies in lung cancer also showed that the expression level of hOGG1 can determine cellular survival on exposure to oxidative damaging agents like H2O2 (172). The protective effect of hOGG1 against 1,3 bis(2 chloroethyl) 1 nitrosourea in lung cancer has also been documented (173). Furthermore, we found that targeting hOGG1 to mitochondria of cervical cancer cells protects the cells from injury inflicted by 2-methoxy estradiol (98). Studies in breast (174, 175) and pancreatic (176) cancers reveal defective repair of 8-oxoG, pointing to the importance of this lesion in different cancer types. Taken together these studies highlight the importance of mitochondrial hOGG1 in cellular protection against DNA damaging agents.
Contrary to these, Zhang and colleagues (177) found that overexpression of mitochondrial hOgg1 in hepatoma cells increased mtDNA damage, membrane potential, and ROS production, and decreased survival of cells exposed to the chemotherapeutic agent cisplatin. Human mammary adenocarcinoma cells when targeted with increased expression of, N-methylpurine DNA glycosylase (MPG–another DNA glycosylase, involved in BER) in mitochondria, increased the lethal action of methyl methane sulfonate (MMS), at the same time increased cellular mortality was also observed in the absence of MMS (178). Recent studies also indicate sensitization of cells to a chemotherapeutic alkylating agent by overexpression of MPG (179). In all of these cases where an increase in DNA damage is observedwith overexpression of BER enzymes, it is hypothesized that it causes an imbalance in BER, leading to accumulation of AP sites and or single/double-strand breaks, which remains unrepaired by mitochondria (probably because the ratio of incision by dRPase exceeds that of nick sealing by DNA ligase). These studies indicate the importance of proper functioning of the BER pathway in mitochondria.
Conclusion
Frequent mtDNA alterations have been documented in different stages of cancer progression, strongly suggesting a functional correlation between mtDNA alterations and tumorigenesis. The analysis of mtDNA mutations for cancer detection has some advantages as compared with the detection of alterations in nuclear DNA. Mitochondrial mutations are essentially homoplasmic, and there is evidence that tumor cells have higher mtDNA content than normal cells. A functional contribution for at least some of these key mutations, pertaining to the ETC complexes have been documented in prostate, cervical, head and neck and bladder cancer cells. Under normal physiologic conditions, ROS generated by oxidative phosphorylation is scavenged by enzymatic or nonenzymatic antioxidants. An unfortunate loop appears to form in mitochondria subverted by cancer cells. Increases in ROS lead to mutations, and the mutations caused by ROS and its defective repair capacity clearly contribute to the development and progression of cancer. In this regard, the importance of repair pathways in mitochondria, and their role in mtDNA integrity and cancer progression cannot be ignored. Moreover, proficient communication between the nucleus and mitochondria is necessary for regulation of proteins encoded by the nucleus and then translocated into the mitochondria.
Many unanswered questions remain. The relationship between mtDNA alteration and clinical disease still remains to be fully elucidated. Also, the interplay between nuclear and mitochondrial genes needs much more careful investigation. Genetic and epigenetic changes in the nucleus and their association with mtDNA polymorphisms are still largely unknown. Identifying nuclear genes controlling mtDNA copy number followed by functional changes associated with mtDNA copy number or mutations need to be investigated. These and other areas of investigation may provide a more comprehensive understanding of the role of mitochondria in tumorigenesis (180). Mitochondria-specific drug targeting in case of defective BER in cancer cells, may allow enhanced efficacy of other therapies. Moreover, increased mtDNA and clonal mtDNA mutations in preneoplastic and neoplastic cells deserve further attention as molecular markers for prevention and early detection of cancer.
Acknowledgments
Grant Support This work was supported by Early Detection Research Network (UO1 CA084986) and Spore and Head and Neck Cancer (P50DE019032); FAMRI-funded Young Clinical Scientist Award (072017_YCSA); and US-Egypt Joint Science and Technology fund (58-3148-169).
Glossary
- Oxidative stress
An excess of ROS, caused by an imbalance between the rate of reduction and oxidation of oxygen, leading to free radical generation and damage to cellular components such as DNA and lipids.
- Turnover number (also termed kcat)
Defined as the maximum number of molecules of substrate that an enzyme can convert to product per catalytic site per unit of time.
- Apurinic apyrimidinic (AP) site
A location in DNA that has neither a purine nor pyrimidine base, usually due to DNA damage. If left unrepaired AP sites can lead to mutation during DNA replication as a random nucleotide will be inserted into the strand synthesized opposite them.
- Base excision repair (BER)
A cellular mechanism that repairs damaged DNA. BER involves flipping the mutated base out of the DNA helix and repairing the base alone. There are 2 main enzymes used, DNA glycosylases and AP endonucleases.
- Cybrid
A eukaryotic cell line produced by the fusion of a whole cell with a cytoplast. Cytoplasts are enucleated cells that contain mitochondria. Cybrids are generated by fusing cytoplasts with a cell line that lacks mtDNA.
- Fenton reaction
Ferrous Iron(II) is oxidized by hydrogen peroxide to ferric iron(III), a hydroxyl radical and a hydroxyl anion. Ferric(III) is then reduced back to ferrous(II), and a peroxide radical and a proton is generated by the same hydrogen peroxide
- Sub-G1 DNA content
A reduction in DNA content below 2n, often a result of DNA fragmentation.
- Translesion DNA synthesis (TLS)
A mechanism during DNA replication in which the standard DNA polymerase is temporarily exchanged for a specialized polymerase that can synthesize DNA across base damage on the template strand.
- Menadione
A polycyclic aromatic ketone, DNA damaging agent.
- Mitochip array
A high-throughput whole mitochondrial genome sequencing array system developed by Affymetrix (www.Affymetrix.com) capable of detection of somatic and germline mtDNA sequence variation.
- Buccal cells
Cells isolated from buccal mucosa of the oral cavity.
- Haplotype
Configurations of alleles within regions on the chromosome that tend to be inherited together. Little genetic variability is observed within this region among individuals in a population.
- Synonymous
A DNA mutation that does not result in a change in the amino acid.
- Nonsynonymous
A DNA mutation that results in a change in the amino acid.
- MtDNA content
Represents the copy number of mtDNA in cells or tissues. It can be determined quantitatively by real-time PCR by using specific primers amplifying mtDNA encoded (COX-I/COX-II) and nDNA encoded genes (β-actin as control).The alteration in mtDNA content may represent a compensatory mechanism for a change in respiratory function.
- COX I, II, and III
The mtDNA-encoded subunits of complex IV.
- ATP 6
An mtDNA-encoded gene for ATPase 6 of the ATP synthase, part of complex V.
Footnotes
Note: Current address for S. Dasgupta: Department of Human and Molecular Genetics, VCU Institute of Molecular Medicine, Virginia Commonwealth University, School of Medicine, 1220 East Broad Street, Richmond, VA 23298.
A. Chatterjee and S. Dasgupta contributed equally to this work.
References
- 1.Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–70. [PubMed] [Google Scholar]
- 2.Warburg O. On the origin of cancer cells. Science. 1956;123:309–14. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 3.Warburg O. Origin of cancer cells. Oncologia. 1956;9:75–83. [PubMed] [Google Scholar]
- 4.Attardi G, Schatz G. Biogenesis of mitochondria. Annu Rev Cell Biol. 1988;4:289–333. doi: 10.1146/annurev.cb.04.110188.001445. [DOI] [PubMed] [Google Scholar]
- 5.Lightowlers RN, Chinnery PF, Turnbull DM, Howell N. Mammalian mitochondrial genetics: heredity, heteroplasmy and disease. Trends Genet. 1997;13:450–5. doi: 10.1016/s0168-9525(97)01266-3. [DOI] [PubMed] [Google Scholar]
- 6.Takamatsu C, Umeda S, Ohsato T, Ohno T, Abe Y, Fukuoh A, et al. Regulation of mitochondrial D-loops by transcription factor A and single-stranded DNA-binding protein. EMBO Rep. 2002;3:451–6. doi: 10.1093/embo-reports/kvf099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Giles RE, Blanc H, Cann HM, Wallace DC. Maternal inheritance of human mitochondrial DNA. Proc Natl Acad Sci U S A. 1980;77:6715–9. doi: 10.1073/pnas.77.11.6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cadet J, Berger M, Douki T, Ravanat JL. Oxidative damage to DNA: formation, measurement, and biological significance. Rev Physiol Biochem Pharmacol. 1997;131:1–87. doi: 10.1007/3-540-61992-5_5. [DOI] [PubMed] [Google Scholar]
- 9.Dizdaroglu M. Chemical determination of free radical-induced damage to DNA. Free Radic Biol Med. 1991;10:225–42. doi: 10.1016/0891-5849(91)90080-m. [DOI] [PubMed] [Google Scholar]
- 10.Bohr VA, Stevnsner T, de Souza-Pinto NC. Mitochondrial DNA repair of oxidative damage in mammalian cells. Gene. 2002;286:127–34. doi: 10.1016/s0378-1119(01)00813-7. [DOI] [PubMed] [Google Scholar]
- 11.Shokolenko IN, Alexeyev MF, Robertson FM, LeDoux SP, Wilson GL. The expression of Exonuclease III from E. coli in mitochondria of breast cancer cells diminishes mitochondrial DNA repair capacity and cell survival after oxidative stress. DNA Repair (Amst) 2003;2:471–82. doi: 10.1016/s1568-7864(03)00019-3. [DOI] [PubMed] [Google Scholar]
- 12.Shoubridge EA. Nuclear genetic defects of oxidative phosphorylation. Hum Mol Genet. 2001;10:2277–84. doi: 10.1093/hmg/10.20.2277. [DOI] [PubMed] [Google Scholar]
- 13.Pietsch EC, Sykes SM, McMahon SB, Murphy ME. The p53 family and programmed cell death. Oncogene. 2008;27:6507–21. doi: 10.1038/onc.2008.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rong Y, Distelhorst CW. Bcl-2 protein family members: versatile regulators of calcium signaling in cell survival and apoptosis. Annu Rev Physiol. 2008;70:73–91. doi: 10.1146/annurev.physiol.70.021507.105852. [DOI] [PubMed] [Google Scholar]
- 15.Harris MH, Thompson CB. The role of the Bcl-2 family in the regulation of outer mitochondrial membrane permeability. Cell Death Differ. 2000;7:1182–91. doi: 10.1038/sj.cdd.4400781. [DOI] [PubMed] [Google Scholar]
- 16.Joza N, Susin SA, Daugas E, Stanford WL, Cho SK, Li CY, et al. Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature. 2001;410:549–54. doi: 10.1038/35069004. [DOI] [PubMed] [Google Scholar]
- 17.Shi Y. A structural view of mitochondria-mediated apoptosis. Nat Struct Biol. 2001;8:394–401. doi: 10.1038/87548. [DOI] [PubMed] [Google Scholar]
- 18.He Y, Wu J, Dressman DC, Iacobuzio-Donahue C, Markowitz SD, Velculescu VE, et al. Heteroplasmic mitochondrial DNA mutations in normal and tumour cells. Nature. 2010;464:610–4. doi: 10.1038/nature08802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fendt L, Niederstatter H, Huber G, Zelger B, Dunser M, Seifarth C, et al. Accumulation of mutations over the entire mitochondrial genome of breast cancer cells obtained by tissue microdissection. Breast Cancer Res Treat. 2010 doi: 10.1007/s10549-010-1092-8. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 20.Coller HA, Khrapko K, Bodyak ND, Nekhaeva E, Herrero-Jimenez P, Thilly WG. High frequency of homoplasmic mitochondrial DNA mutations in human tumors can be explained without selection. Nat Genet. 2001;28:147–50. doi: 10.1038/88859. [DOI] [PubMed] [Google Scholar]
- 21.Taylor RW, Barron MJ, Borthwick GM, Gospel A, Chinnery PF, Samuels DC, et al. Mitochondrial DNA mutations in human colonic crypt stem cells. J Clin Invest. 2003;112:1351–60. doi: 10.1172/JCI19435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khrapko K, Bodyak N, Thilly WG, van Orsouw NJ, Zhang X, Coller HA, et al. Cell-by-cell scanning of whole mitochondrial genomes in aged human heart reveals a significant fraction of myocytes with clonally expanded deletions. Nucleic Acids Res. 1999;27:2434–41. doi: 10.1093/nar/27.11.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dunbar DR, Moonie PA, Jacobs HT, Holt IJ. Different cellular back-grounds confer a marked advantage to either mutant or wild-type mitochondrial genomes. Proc Natl Acad Sci U S A. 1995;92:6562–6. doi: 10.1073/pnas.92.14.6562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jenuth JP, Peterson AC, Fu K, Shoubridge EA. Random genetic drift in the female germline explains the rapid segregation of mammalian mitochondrial DNA. Nat Genet. 1996;14:146–51. doi: 10.1038/ng1096-146. [DOI] [PubMed] [Google Scholar]
- 25.Jenuth JP, Peterson AC, Shoubridge EA. Tissue-specific selection for different mtDNA genotypes in heteroplasmic mice. Nat Genet. 1997;16:93–5. doi: 10.1038/ng0597-93. [DOI] [PubMed] [Google Scholar]
- 26.Polyak K, Li Y, Zhu H, Lengauer C, Willson JK, Markowitz SD, et al. Somatic mutations of the mitochondrial genome in human colorectal tumours. Nat Genet. 1998;20:291–3. doi: 10.1038/3108. [DOI] [PubMed] [Google Scholar]
- 27.Attardi G, Yoneda M, Chomyn A. Complementation and segregation behavior of disease-causing mitochondrial DNA mutations in cellular model systems. Biochim Biophys Acta. 1995;1271:241–8. doi: 10.1016/0925-4439(95)00034-2. [DOI] [PubMed] [Google Scholar]
- 28.Hayashi J, Ohta S, Kikuchi A, Takemitsu M, Goto Y, Nonaka I. Introduction of disease-related mitochondrial DNA deletions into HeLa cells lacking mitochondrial DNA results in mitochondrial dysfunction. Proc Natl Acad Sci U S A. 1991;88:10614–8. doi: 10.1073/pnas.88.23.10614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hayashi J, Takemitsu M, Goto Y, Nonaka I. Human mitochondria and mitochondrial genome function as a single dynamic cellular unit. J Cell Biol. 1994;125:43–50. doi: 10.1083/jcb.125.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takai D, Isobe K, Hayashi J. Transcomplementation between different types of respiration-deficient mitochondria with different pathogenic mutant mitochondrial DNAs. J Biol Chem. 1999;274:11199–202. doi: 10.1074/jbc.274.16.11199. [DOI] [PubMed] [Google Scholar]
- 31.Yoneda M, Miyatake T, Attardi G. Complementation of mutant and wild-type human mitochondrial DNAs coexisting since the mutation event and lack of complementation of DNAs introduced separately into a cell within distinct organelles. Mol Cell Biol. 1994;14:2699–712. doi: 10.1128/mcb.14.4.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Enriquez JA, Cabezas-Herrera J, Bayona-Bafaluy MP, Attardi G. Very rare complementation between mitochondria carrying different mitochondrial DNA mutations points to intrinsic genetic autonomy of the organelles in cultured human cells. J Biol Chem. 2000;275:11207–15. doi: 10.1074/jbc.275.15.11207. [DOI] [PubMed] [Google Scholar]
- 33.Mambo E, Gao X, Cohen Y, Guo Z, Talalay P, Sidransky D. Electrophile and oxidant damage of mitochondrial DNA leading to rapid evolution of homoplasmic mutations. Proc Natl Acad Sci U S A. 2003;100:1838–43. doi: 10.1073/pnas.0437910100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Castle JC, Biery M, Bouzek H, Xie T, Chen R, Misura K, et al. DNA copy number, including telomeres and mitochondria, assayed using next-generation sequencing. BMC Genomics. 2010;11:244. doi: 10.1186/1471-2164-11-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nomoto S, Yamashita K, Koshikawa K, Nakao A, Sidransky D. Mitochondrial D-loop mutations as clonal markers in multicentric hepatocellular carcinoma and plasma. Clin Cancer Res. 2002;8:481–7. [PubMed] [Google Scholar]
- 36.Fliss MS, Usadel H, Caballero OL, Wu L, Buta MR, Eleff SM, et al. Facile detection of mitochondrial DNA mutations in tumors and bodily fluids. Science. 2000;287:2017–9. doi: 10.1126/science.287.5460.2017. [DOI] [PubMed] [Google Scholar]
- 37.Hosgood HD, III, Liu CS, Rothman N, Weinstein SJ, Bonner MR, Shen M, et al. Mitochondrial DNA copy number and lung cancer risk in a prospective cohort study. Carcinogenesis. 2010;31:847–9. doi: 10.1093/carcin/bgq045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dasgupta S, Koch R, Westra WH, Califano JA, Ha PK, Sidransky D, et al. Mitochondrial DNA mutation in normal margins and tumors of recurrent head and neck squamous cell carcinoma patients. Cancer Prev Res. 2010;3:1205–11. doi: 10.1158/1940-6207.CAPR-10-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dasgupta S, Yung RC, Westra WH, Rini DA, Brandes J, Sidransky D. Following mitochondrial footprints through a long mucosal path to lung cancer. PLoS One. 2009;4:e6533. doi: 10.1371/journal.pone.0006533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ha PK, Tong BC, Westra WH, Sanchez-Cespedes M, Parrella P, Zahurak M, et al. Mitochondrial C-tract alteration in premalignantlesions of the head and neck: a marker for progression and clonal proliferation. Clin Cancer Res. 2002;8:2260–5. [PubMed] [Google Scholar]
- 41.Rigoli L, Di Bella C, Verginelli F, Falchetti M, Bersiga A, Rocco A, et al. Histological heterogeneity and somatic mtDNA mutations in gastric intraepithelial neoplasia. Mod Pathol. 2008;21:733–41. doi: 10.1038/modpathol.2008.58. [DOI] [PubMed] [Google Scholar]
- 42.Sui G, Zhou S, Wang J, Canto M, Lee EE, Eshleman JR, et al. Mitochondrial DNA mutations in preneoplastic lesions of the gastrointestinal tract: a biomarker for the early detection of cancer. Mol Cancer. 2006;5:73. doi: 10.1186/1476-4598-5-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parrella P, Xiao Y, Fliss M, Sanchez-Cespedes M, Mazzarelli P, Rinaldi M, et al. Detection of mitochondrial DNA mutations in primary breast cancer and fine-needle aspirates. Cancer Res. 2001;61:7623–6. [PubMed] [Google Scholar]
- 44.Jeronimo C, Nomoto S, Caballero OL, Usadel H, Henrique R, Varzim G, et al. Mitochondrial mutations in early stage prostate cancer and bodily fluids. Oncogene. 2001;20:5195–8. doi: 10.1038/sj.onc.1204646. [DOI] [PubMed] [Google Scholar]
- 45.Parrella P, Seripa D, Matera MG, Rabitti C, Rinaldi M, Mazzarelli P, et al. Mutations of the D310 mitochondrial mononucleotide repeat in primary tumors and cytological specimens. Cancer Lett. 2003;190:73–7. doi: 10.1016/s0304-3835(02)00578-5. [DOI] [PubMed] [Google Scholar]
- 46.Tan D, Goerlitz DS, Dumitrescu RG, Han D, Seillier-Moiseiwitsch F, Spernak SM, et al. Associations between cigarette smoking and mitochondrial DNA abnormalities in buccal cells. Carcinogenesis. 2008;29:1170–7. doi: 10.1093/carcin/bgn034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.He L, Luo L, Proctor SJ, Middleton PG, Blakely EL, Taylor RW, et al. Somatic mitochondrial DNA mutations in adult-onset leukaemia. Leukemia. 2003;17:2487–91. doi: 10.1038/sj.leu.2403146. [DOI] [PubMed] [Google Scholar]
- 48.Maitra A, Cohen Y, Gillespie SE, Mambo E, Fukushima N, Hoque MO, et al. The Human MitoChip: a high-throughput sequencing microarray for mitochondrial mutation detection. Genome Res. 2004;14:812–9. doi: 10.1101/gr.2228504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lievre A, Chapusot C, Bouvier AM, Zinzindohoue F, Piard F, Roignot P, et al. Clinical value of mitochondrial mutations in colorectal cancer. J Clin Oncol. 2005;23:3517–25. doi: 10.1200/JCO.2005.07.044. [DOI] [PubMed] [Google Scholar]
- 50.Sanchez-Cespedes M, Parrella P, Nomoto S, Cohen D, Xiao Y, Esteller M, et al. Identification of a mononucleotide repeat as a major target for mitochondrial DNA alterations in human tumors. Cancer Res. 2001;61:7015–9. [PubMed] [Google Scholar]
- 51.Burgart LJ, Zheng J, Shu Q, Strickler JG, Shibata D. Somatic mitochondrial mutation in gastric cancer. Am J Pathol. 1995;147:1105–11. [PMC free article] [PubMed] [Google Scholar]
- 52.Habano W, Sugai T, Yoshida T, Nakamura S. Mitochondrial gene mutation, but not large-scale deletion, is a feature of colorectal carcinomas with mitochondrial microsatellite instability. Int J Cancer. 1999;83:625–9. doi: 10.1002/(sici)1097-0215(19991126)83:5<625::aid-ijc10>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 53.Hung WY, Lin JC, Lee LM, Wu CW, Tseng LM, Yin PH, et al. Tandem duplication/triplication correlated with poly-cytosine stretch variation in human mitochondrial DNA D-loop region. Mutagenesis. 2008;23:137–42. doi: 10.1093/mutage/gen002. [DOI] [PubMed] [Google Scholar]
- 54.Lievre A, Blons H, Houllier AM, Laccourreye O, Brasnu D, Beaune P, et al. Clinicopathological significance of mitochondrial D-Loop mutations in head and neck carcinoma. Br J Cancer. 2006;94:692–7. doi: 10.1038/sj.bjc.6602993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nishikawa M, Nishiguchi S, Shiomi S, Tamori A, Koh N, Takeda T, et al. Somatic mutation of mitochondrial DNA in cancerous and noncancerous liver tissue in individuals with hepatocellular carcinoma. Cancer Res. 2001;61:1843–5. [PubMed] [Google Scholar]
- 56.Suzuki M, Toyooka S, Miyajima K, Iizasa T, Fujisawa T, Bekele NB, et al. Alterations in the mitochondrial displacement loop in lung cancers. Clin Cancer Res. 2003;9:5636–41. [PubMed] [Google Scholar]
- 57.Wheelhouse NM, Lai PB, Wigmore SJ, Ross JA, Harrison DJ. Mitochondrial D-loop mutations and deletion profiles of cancerous and noncancerous liver tissue in hepatitis B virus-infected liver. Br J Cancer. 2005;92:1268–72. doi: 10.1038/sj.bjc.6602496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu CW, Yin PH, Hung WY, Li AF, Li SH, Chi CW, et al. Mitochondrial DNA mutations and mitochondrial DNA depletion in gastric cancer. Genes Chromosomes Cancer. 2005;44:19–28. doi: 10.1002/gcc.20213. [DOI] [PubMed] [Google Scholar]
- 59.Alonso A, Martin P, Albarran C, Aquilera B, Garcia O, Guzman A, et al. Detection of somatic mutations in the mitochondrial DNA control region of colorectal and gastric tumors by heteroduplex and singlestrand conformation analysis. Electrophoresis. 1997;18:682–5. doi: 10.1002/elps.1150180504. [DOI] [PubMed] [Google Scholar]
- 60.Habano W, Nakamura S, Sugai T. Microsatellite instability in the mitochondrial DNA of colorectal carcinomas: evidence for mismatch repair systems in mitochondrial genome. Oncogene. 1998;17:1931–7. doi: 10.1038/sj.onc.1202112. [DOI] [PubMed] [Google Scholar]
- 61.Hibi K, Nakayama H, Yamazaki T, Takase T, Taguchi M, Kasai Y, et al. Detection of mitochondrial DNA alterations in primary tumors and corresponding serum of colorectal cancer patients. Int J Cancer. 2001;94:429–31. doi: 10.1002/ijc.1480. [DOI] [PubMed] [Google Scholar]
- 62.Gochhait S, Bhatt A, Sharma S, Singh YP, Gupta P, Bamezai RN. Concomitant presence of mutations in mitochondrial genome and p53 in cancer development–a study in north Indian sporadic breast and esophageal cancer patients. Int J Cancer. 2008;123:2580–6. doi: 10.1002/ijc.23817. [DOI] [PubMed] [Google Scholar]
- 63.Jakupciak JP, Maggrah A, Maragh S, Maki J, Reguly B, Maki K, et al. Facile whole mitochondrial genome resequencing from nipple aspirate fluid using MitoChip v2.0. BMC Cancer. 2008;8:95. doi: 10.1186/1471-2407-8-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mithani SK, Taube JM, Zhou S, Smith IM, Koch WM, Westra WH, et al. Mitochondrial mutations are a late event in the progression of head and neck squamous cell cancer. Clin Cancer Res. 2007;13:4331–5. doi: 10.1158/1078-0432.CCR-06-2613. [DOI] [PubMed] [Google Scholar]
- 65.Prior SL, Griffiths AP, Baxter JM, Baxter PW, Hodder SC, Silvester KC, et al. Mitochondrial DNA mutations in oral squamous cell carcinoma. Carcinogenesis. 2006;27:945–50. doi: 10.1093/carcin/bgi326. [DOI] [PubMed] [Google Scholar]
- 66.Zhou S, Kachhap S, Sun W, Wu G, Chuang A, Poeta L, et al. Frequency and phenotypic implications of mitochondrial DNA mutations in human squamous cell cancers of the head and neck. Proc Natl Acad Sci U S A. 2007;104:7540–5. doi: 10.1073/pnas.0610818104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jin X, Zhang J, Gao Y, Ding K, Wang N, Zhou D, et al. Relationship between mitochondrial DNA mutations and clinical characteristics in human lung cancer. Mitochondrion. 2007;7:347–53. doi: 10.1016/j.mito.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 68.Abu-Amero KK, Alzahrani AS, Zou M, Shi Y. Association of mitochondrial DNA transversion mutations with familial medullary thyroid carcinoma/multiple endocrine neoplasia type 2 syndrome. Oncogene. 2006;25:677–84. doi: 10.1038/sj.onc.1209094. [DOI] [PubMed] [Google Scholar]
- 69.Gasparre G, Porcelli AM, Bonora E, Pennisi LF, Toller M, Iommarini L, et al. Disruptive mitochondrial DNA mutations in complex I subunits are markers of oncocytic phenotype in thyroid tumors. Proc Natl Acad Sci U S A. 2007;104:9001–6. doi: 10.1073/pnas.0703056104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Maximo V, Soares P, Lima J, Cameselle-Teijeiro J, Sobrinho-Simoes M. Mitochondrial DNA somatic mutations (point mutations and large deletions) and mitochondrial DNA variants in human thyroid pathology: a study with emphasis on Hurthle cell tumors. Am J Pathol. 2002;160:1857–65. doi: 10.1016/S0002-9440(10)61132-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhou S, Kassauei K, Cutler DJ, Kennedy GC, Sidransky D, Maitra A, et al. An oligonucleotide microarray for high-throughput sequencing of the mitochondrial genome. J Mol Diagn. 2006;8:476–82. doi: 10.2353/jmoldx.2006.060008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tseng LM, Yin PH, Chi CW, Hsu CY, Wu CW, Lee LM, et al. Mitochondrial DNA mutations and mitochondrial DNA depletion in breast cancer. Genes Chromosomes Cancer. 2006;45:629–38. doi: 10.1002/gcc.20326. [DOI] [PubMed] [Google Scholar]
- 73.Bianchi MS, Bianchi NO, Bailliet G. Mitochondrial DNA mutations in normal and tumor tissues from breast cancer patients. Cytogenet Cell Genet. 1995;71:99–103. doi: 10.1159/000134072. [DOI] [PubMed] [Google Scholar]
- 74.Salas A, Yao YG, Macaulay V, Vega A, Carracedo A, Bandelt HJ. A critical reassessment of the role of mitochondria in tumorigenesis. PLoS Med. 2005;2:e296. doi: 10.1371/journal.pmed.0020296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bandelt HJ, Salas A. Contamination and sample mix-up can best explain some patterns of mtDNA instabilities in buccal cells and oral squamous cell carcinoma. BMC Cancer. 2009;9:113. doi: 10.1186/1471-2407-9-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bandelt HJ, Salas A, Bravi CM. Whatisa ‘novel’ mtDNA mutation–and does ‘novelty’ really matter? J Hum Genet. 2006;51:1073–82. doi: 10.1007/s10038-006-0066-5. [DOI] [PubMed] [Google Scholar]
- 77.Bandelt HJ, Salas A, Taylor RW, Yao YG. Exaggerated status of “novel” and “pathogenic” mtDNA sequence variants due to inadequate database searches. Hum Mutat. 2009;30:191–6. doi: 10.1002/humu.20846. [DOI] [PubMed] [Google Scholar]
- 78.Bandelt HJ, Yao YG, Salas A. The search of ‘novel’ mtDNA mutations in hypertrophic cardiomyopathy: MITOMAPping as a risk factor. Int J Cardiol. 2008;126:439–42. doi: 10.1016/j.ijcard.2007.02.049. [DOI] [PubMed] [Google Scholar]
- 79.Palanichamy MG, Zhang YP. Potential pitfalls in MitoChip detected tumor-specific somatic mutations: a call for caution when interpreting patient data. BMC Cancer. 2010;10:597. doi: 10.1186/1471-2407-10-597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yao YG, Ogasawara Y, Kajigaya S, Molldrem JJ, Falcao RP, Pintao MC, et al. Mitochondrial DNA sequence variation in single cells from leukemia patients. Blood. 2007;109:756–62. doi: 10.1182/blood-2006-01-011007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dasgupta S, Hoque MO, Upadhyay S, Sidransky D. Mitochondrial cytochrome B gene mutation promotes tumor growth in bladder cancer. Cancer Res. 2008;68:700–6. doi: 10.1158/0008-5472.CAN-07-5532. [DOI] [PubMed] [Google Scholar]
- 82.DeHaan C, Habibi-Nazhad B, Yan E, Salloum N, Parliament M, Allalunis-Turner J. Mutation in mitochondrial complex I ND6 subunit is associated with defective response to hypoxia in human glioma cells. Mol Cancer. 2004;3:19. doi: 10.1186/1476-4598-3-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pezzotti A, Kraft P, Hankinson SE, Hunter DJ, Buring J, Cox DG. The mitochondrial A10398G polymorphism, interaction with alcohol consumption, and breast cancer risk. PLoS One. 2009;4:e5356. doi: 10.1371/journal.pone.0005356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bai RK, Leal SM, Covarrubias D, Liu A, Wong LJ. Mitochondrial genetic background modifies breast cancer risk. Cancer Res. 2007;67:4687–94. doi: 10.1158/0008-5472.CAN-06-3554. [DOI] [PubMed] [Google Scholar]
- 85.Kim MM, Clinger JD, Masayesva BG, Ha PK, Zahurak ML, Westra WH, et al. Mitochondrial DNA quantity increases with histopathologic grade in premalignant and malignant head and neck lesions. Clin Cancer Res. 2004;10:8512–5. doi: 10.1158/1078-0432.CCR-04-0734. [DOI] [PubMed] [Google Scholar]
- 86.Mambo E, Chatterjee A, Xing M, Tallini G, Haugen BR, Yeung SC, et al. Tumor-specific changes in mtDNA content in human cancer. Int J Cancer. 2005;116:920–4. doi: 10.1002/ijc.21110. [DOI] [PubMed] [Google Scholar]
- 87.Rogounovitch TI, Saenko VA, Shimizu-Yoshida Y, Abrosimov AY, Lushnikov EF, Roumiantsev PO, et al. Large deletions in mitochondrial DNA in radiation-associated human thyroid tumors. Cancer Res. 2002;62:7031–41. [PubMed] [Google Scholar]
- 88.Bonner MR, Shen M, Liu CS, Divita M, He X, Lan Q. Mitochondrial DNA content and lung cancer risk in Xuan Wei, China. Lung Cancer. 2009;63:331–4. doi: 10.1016/j.lungcan.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lan Q, Lim U, Liu CS, Weinstein SJ, Chanock S, Bonner MR, et al. A prospective study of mitochondrial DNA copy number and risk of non-Hodgkin lymphoma. Blood. 2008;112:4247–9. doi: 10.1182/blood-2008-05-157974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xing J, Chen M, Wood CG, Lin J, Spitz MR, Ma J, et al. Mitochondrial DNA content: its genetic heritability and association with renal cell carcinoma. J Natl Cancer Inst. 2008;100:1104–12. doi: 10.1093/jnci/djn213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lee HC, Li SH, Lin JC, Wu CC, Yeh DC, Wei YH. Somatic mutations in the D-loop and decrease in the copy number of mitochondrial DNA in human hepatocellular carcinoma. Mutat Res. 2004;547:71–8. doi: 10.1016/j.mrfmmm.2003.12.011. [DOI] [PubMed] [Google Scholar]
- 92.Wang Y, Liu VW, Tsang PC, Chiu PM, Cheung AN, Khoo US, et al. Microsatellite instability in mitochondrial genome of common female cancers. Int J Gynecol Cancer. 2006;16(Suppl 1):259, 66. doi: 10.1111/j.1525-1438.2006.00412.x. [DOI] [PubMed] [Google Scholar]
- 93.Montier LL Clay, Deng JJ, Bai Y. Number matters: control of mammalian mitochondrial DNA copy number. J Genet Genomics. 2009;36:125–31. doi: 10.1016/S1673-8527(08)60099-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Olivero OA, Semino C, Kassim A, Lopez-Larraza DM, Poirier MC. Preferential binding of cisplatin to mitochondrial DNA of Chinese hamster ovary cells. Mutat Res. 1995;346:221–30. doi: 10.1016/0165-7992(95)90039-x. [DOI] [PubMed] [Google Scholar]
- 95.Talarico T, Cullinane CM, Gray PJ, Webster LK, Deacon GB, Phillips DR. Nuclear and mitochondrial distribution of organoamidoplatinum (II) lesions in cisplatin-sensitive and -resistant adenocarcinoma cells. Anticancer Drug Des. 2001;16:135–41. [PubMed] [Google Scholar]
- 96.Zhou Y, Hileman EO, Plunkett W, Keating MJ, Huang P. Free radical stress in chronic lymphocytic leukemia cells and its role in cellular sensitivity to ROS-generating anticancer agents. Blood. 2003;101:4098–104. doi: 10.1182/blood-2002-08-2512. [DOI] [PubMed] [Google Scholar]
- 97.Carew JS, Zhou Y, Albitar M, Carew JD, Keating MJ, Huang P. Mitochondrial DNA mutations in primary leukemia cells after chemotherapy: clinical significance and therapeutic implications. Leukemia. 2003;17:1437–47. doi: 10.1038/sj.leu.2403043. [DOI] [PubMed] [Google Scholar]
- 98.Chatterjee A, Chang X, Nagpal JK, Chang S, Upadhyay S, Califano J, et al. Targeting human 8-oxoguanine DNA glycosylase to mitochondria protects cells from 2-methoxyestradiol-induced-mitochondria-dependent apoptosis. Oncogene. 2008;27:3710–20. doi: 10.1038/onc.2008.3. [DOI] [PubMed] [Google Scholar]
- 99.Wang X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001;15:2922–33. [PubMed] [Google Scholar]
- 100.Golab J, Nowis D, Skrzycki M, Czeczot H, Baranczyk-Kuzma A, Wilczynski GM, et al. Antitumor effects of photodynamic therapy are potentiated by 2-methoxyestradiol. A superoxide dismutase inhibitor. J Biol Chem. 2003;278:407–14. doi: 10.1074/jbc.M209125200. [DOI] [PubMed] [Google Scholar]
- 101.Wood L, Leese MR, Leblond B, Woo LW, Ganeshapillai D, Purohit A, et al. Inhibition of superoxide dismutase by 2-methoxyoestradiol analogues and oestrogen derivatives: structure-activity relationships. Anticancer Drug Des. 2001;16:209–15. [PubMed] [Google Scholar]
- 102.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–44. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yen KE, Bittinger MA, Su SM, Fantin VR. Cancer-associated IDH mutations: biomarker and therapeutic opportunities. Oncogene. 2010;29:6409–17. doi: 10.1038/onc.2010.444. [DOI] [PubMed] [Google Scholar]
- 104.Shokolenko IN, Alexeyev MF, LeDoux SP, Wilson GL. The approaches for manipulating mitochondrial proteome. Environ Mol Mutagen. 2010;51:451–61. doi: 10.1002/em.20570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.de Grey AD. Mitochondrial gene therapy: an arena for the biomedical use of inteins. Trends Biotechnol. 2000;18:394–9. doi: 10.1016/s0167-7799(00)01476-1. [DOI] [PubMed] [Google Scholar]
- 106.Neuzil J, Dong LF, Ramanathapuram L, Hahn T, Chladova M, Wang XF, et al. Vitamin E analogues as a novel group of mitocans: anti-cancer agents that act by targeting mitochondria. Mol Aspects Med. 2007;28:607–45. doi: 10.1016/j.mam.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 107.Ralph SJ, Neuzil J. Mitochondria as targets for cancer therapy. Mol Nutr Food Res. 2009;53:9–28. doi: 10.1002/mnfr.200800044. [DOI] [PubMed] [Google Scholar]
- 108.Morisaki T, Katano M. Mitochondria-targeting therapeutic strategies for overcoming chemoresistance and progression of cancer. Curr Med Chem. 2003;10:2517–21. doi: 10.2174/0929867033456431. [DOI] [PubMed] [Google Scholar]
- 109.Carew JS, Huang P. Mitochondrial defects in cancer. Mol Cancer. 2002;1:9. doi: 10.1186/1476-4598-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bonora E, Porcelli AM, Gasparre G, Biondi A, Ghelli A, Carelli V, et al. Defective oxidative phosphorylation in thyroid oncocytic carcinoma is associated with pathogenic mitochondrial DNA mutations affecting complexes I and III. Cancer Res. 2006;66:6087–96. doi: 10.1158/0008-5472.CAN-06-0171. [DOI] [PubMed] [Google Scholar]
- 111.Simonnet H, Alazard N, Pfeiffer K, Gallou C, Beroud C, Demont J, et al. Low mitochondrial respiratory chain content correlates with tumor aggressiveness in renal cell carcinoma. Carcinogenesis. 2002;23:759–68. doi: 10.1093/carcin/23.5.759. [DOI] [PubMed] [Google Scholar]
- 112.Lee HC, Wei YH. Mitochondrial DNA instability and metabolic shift in human cancers. Int J Mol Sci. 2009;10:674–701. doi: 10.3390/ijms10020674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Pagniez-Mammeri H, Lombes A, Brivet M, Ogier-de Baulny H, Landrieu P, Legrand A, et al. Rapid screening for nuclear genes mutations in isolated respiratory chain complex I defects. Mol Genet Metab. 2009;96:196–200. doi: 10.1016/j.ymgme.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 114.Horton TM, Petros JA, Heddi A, Shoffner J, Kaufman AE, Graham SD, Jr, et al. Novel mitochondrial DNA deletion found in a renal cell carcinoma. Genes Chromosomes Cancer. 1996;15:95–101. doi: 10.1002/(SICI)1098-2264(199602)15:2<95::AID-GCC3>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 115.Gonzalo R, Garcia-Arumi E, Llige D, Marti R, Solano A, Montoya J, et al. Free radicals-mediated damage in transmitochondrial cells harboring the T14487C mutation in the ND6 gene of mtDNA. FEBS Lett. 2005;579:6909–13. doi: 10.1016/j.febslet.2005.11.034. [DOI] [PubMed] [Google Scholar]
- 116.Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, Imanishi H, et al. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science. 2008;320:661–4. doi: 10.1126/science.1156906. [DOI] [PubMed] [Google Scholar]
- 117.Gasparre G, Hervouet E, de Laplanche E, Demont J, Pennisi LF, Colombel M, et al. Clonal expansion of mutated mitochondrial DNA is associated with tumor formation and complex I deficiency in the benign renal oncocytoma. Hum Mol Genet. 2008;17:986–95. doi: 10.1093/hmg/ddm371. [DOI] [PubMed] [Google Scholar]
- 118.Mayr JA, Meierhofer D, Zimmermann F, Feichtinger R, Kogler C, Ratschek M, et al. Loss of complex I due to mitochondrial DNA mutations in renal oncocytoma. Clin Cancer Res. 2008;14:2270–5. doi: 10.1158/1078-0432.CCR-07-4131. [DOI] [PubMed] [Google Scholar]
- 119.Simonnet H, Demont J, Pfeiffer K, Guenaneche L, Bouvier R, Brandt U, et al. Mitochondrial complex I is deficient in renal oncocytomas. Carcinogenesis. 2003;24:1461–6. doi: 10.1093/carcin/bgg109. [DOI] [PubMed] [Google Scholar]
- 120.Cavelier L, Jazin EE, Eriksson I, Prince J, Bave U, Oreland L, et al. Decreased cytochrome-c oxidase activity and lack of age-related accumulation of mitochondrial DNA deletions in the brains of schizophrenics. Genomics. 1995;29:217–24. doi: 10.1006/geno.1995.1234. [DOI] [PubMed] [Google Scholar]
- 121.Sun AS, Cederbaum AI. Oxidoreductase activities in normal rat liver, tumor-bearing rat liver, and hepatoma HC-252. Cancer Res. 1980;40:4677–81. [PubMed] [Google Scholar]
- 122.Abril J, de Heredia ML, Gonzalez L, Cleries R, Nadal M, Condom E, et al. Altered expression of 12S/MT-RNR1, MT-CO2/COX2, andMT-ATP6 mitochondrial genes in prostate cancer. Prostate. 2008;68:1086–96. doi: 10.1002/pros.20771. [DOI] [PubMed] [Google Scholar]
- 123.Heerdt BG, Halsey HK, Lipkin M, Augenlicht LH. Expression of mitochondrial cytochrome c oxidase in human colonic cell differentiation, transformation, and risk for colonic cancer. Cancer Res. 1990;50:1596–600. [PubMed] [Google Scholar]
- 124.Petros JA, Baumann AK, Ruiz-Pesini E, Amin MB, Sun CQ, Hall J, et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc Natl Acad Sci U S A. 2005;102:719–24. doi: 10.1073/pnas.0408894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Arnold RS, Sun CQ, Richards JC, Grigoriev G, Coleman IM, Nelson PS, et al. Mitochondrial DNA mutation stimulates prostate cancer growth in bone stromal environment. Prostate. 2009;69:1–11. doi: 10.1002/pros.20854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Shidara Y, Yamagata K, Kanamori T, Nakano K, Kwong JQ, Manfredi G, et al. Positive contribution of pathogenic mutations in the mitochondrial genome to the promotion of cancer by prevention from apoptosis. Cancer Res. 2005;65:1655–63. doi: 10.1158/0008-5472.CAN-04-2012. [DOI] [PubMed] [Google Scholar]
- 127.Li S, Yan T, Yang JQ, Oberley TD, Oberley LW. The role of cellular glutathione peroxidase redox regulation in the suppression of tumor cell growth by manganese superoxide dismutase. Cancer Res. 2000;60:3927–39. [PubMed] [Google Scholar]
- 128.Wenk J, Brenneisen P, Wlaschek M, Poswig A, Briviba K, Oberley TD, et al. Stable overexpression of manganese superoxide dismutase in mitochondria identifies hydrogen peroxide as a major oxidant in the AP-1-mediated induction of matrix-degrading metalloprotease-1. J Biol Chem. 1999;274:25869–76. doi: 10.1074/jbc.274.36.25869. [DOI] [PubMed] [Google Scholar]
- 129.Behrend L, Henderson G, Zwacka RM. Reactive oxygen species in oncogenic transformation. Biochem Soc Trans. 2003;31:1441–4. doi: 10.1042/bst0311441. [DOI] [PubMed] [Google Scholar]
- 130.Baker AM, Oberley LW, Cohen MB. Expression of antioxidant enzymes in human prostatic adenocarcinoma. Prostate. 1997;32:229–33. doi: 10.1002/(sici)1097-0045(19970901)32:4<229::aid-pros1>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 131.Bostwick DG, Alexander EE, Singh R, Shan A, Qian J, Santella RM, et al. Antioxidant enzyme expression and reactive oxygen species damage in prostatic intraepithelial neoplasia and cancer. Cancer. 2000;89:123–34. [PubMed] [Google Scholar]
- 132.Hempel N, Ye H, Abessi B, Mian B, Melendez JA. Altered redox status accompanies progression to metastatic human bladder cancer. Free Radic Biol Med. 2008 doi: 10.1016/j.freeradbiomed.2008.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yoo DG, Song YJ, Cho EJ, Lee SK, Park JB, Yu JH, et al. Alteration of APE1/ref-1 expression in non-small cell lung cancer: the implications of impaired extracellular superoxide dismutase and catalase anti-oxidant systems. Lung Cancer. 2008;60:277–84. doi: 10.1016/j.lungcan.2007.10.015. [DOI] [PubMed] [Google Scholar]
- 134.Badjatia N, Satyam A, Singh P, Seth A, Sharma A. Altered antioxidant status and lipid peroxidation in Indian patients with urothelial bladder carcinoma. Urol Oncol. 2010;28:360–7. doi: 10.1016/j.urolonc.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 135.Kinnula VL, Crapo JD. Superoxide dismutases in malignant cells and human tumors. Free Radic Biol Med. 2004;36:718–44. doi: 10.1016/j.freeradbiomed.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 136.Nelson SK, Bose SK, McCord JM. The toxicity of high-dose superoxide dismutase suggests that superoxide can both initiate and terminate lipid peroxidation in the reperfused heart. Free Radic Biol Med. 1994;16:195–200. doi: 10.1016/0891-5849(94)90143-0. [DOI] [PubMed] [Google Scholar]
- 137.Mantovani G, Maccio A, Madeddu C, Mura L, Gramignano G, Lusso MR, et al. Quantitative evaluation of oxidative stress, chronic inflammatory indices and leptin in cancer patients: correlation with stage and performance status. Int J Cancer. 2002;98:84–91. doi: 10.1002/ijc.10143. [DOI] [PubMed] [Google Scholar]
- 138.Ray G, Batra S, Shukla NK, Deo S, Raina V, Ashok S, et al. Lipid peroxidation, free radical production and antioxidant status in breast cancer. Breast Cancer Res Treat. 2000;59:163–70. doi: 10.1023/a:1006357330486. [DOI] [PubMed] [Google Scholar]
- 139.Saydam N, Kirb A, Demir O, Hazan E, Oto O, Saydam O, et al. Determination of glutathione, glutathione reductase, glutathione peroxidase and glutathione S-transferase levels in human lung cancer tissues. Cancer Lett. 1997;119:13–9. doi: 10.1016/s0304-3835(97)00245-0. [DOI] [PubMed] [Google Scholar]
- 140.Scibior D, Skrzycki M, Podsiad M, Czeczot H. Glutathione level and glutathione-dependent enzyme activities in blood serum of patients with gastrointestinal tract tumors. Clin Biochem. 2008;41:852–8. doi: 10.1016/j.clinbiochem.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 141.Saygili EI, Akcay T, Konukoglu D, Papilla C. Glutathione and glutathione-related enzymes in colorectal cancer patients. J Toxicol Environ Health A. 2003;66:411–5. doi: 10.1080/15287390306448. [DOI] [PubMed] [Google Scholar]
- 142.Bakan N, Taysi S, Yilmaz O, Bakan E, Kuskay S, Uzun N, et al. Glutathione peroxidase, glutathione reductase, Cu-Zn superoxide dismutase activities, glutathione, nitric oxide, and malondialdehyde concentrations in serum of patients with chronic lymphocytic leukemia. Clin Chim Acta. 2003;338:143–9. doi: 10.1016/j.cccn.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 143.Yu YP, Yu G, Tseng G, Cieply K, Nelson J, Defrances M, et al. Glutathione peroxidase 3, deleted or methylated in prostate cancer, suppresses prostate cancer growth and metastasis. Cancer Res. 2007;67:8043–50. doi: 10.1158/0008-5472.CAN-07-0648. [DOI] [PubMed] [Google Scholar]
- 144.Radi R, Turrens JF, Chang LY, Bush KM, Crapo JD, Freeman BA. Detection of catalase in rat heart mitochondria. J Biol Chem. 1991;266:22028–34. [PubMed] [Google Scholar]
- 145.Salvi M, Battaglia V, Brunati AM, Rocca N La, Tibaldi E, Pietrangeli P, et al. Catalase takes part in rat liver mitochondria oxidative stress defense. J Biol Chem. 2007;282:24407–15. doi: 10.1074/jbc.M701589200. [DOI] [PubMed] [Google Scholar]
- 146.Kaynar H, Meral M, Turhan H, Keles M, Celik G, Akcay F. Glutathione peroxidase, glutathione-S-transferase, catalase, xanthine oxidase, Cu-Zn superoxide dismutase activities, total glutathione, nitric oxide, and malondialdehyde levels in erythrocytes of patients with small cell and non-small cell lung cancer. Cancer Lett. 2005;227:133–9. doi: 10.1016/j.canlet.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 147.Ho J Chung-man, Zheng S, Comhair SA, Farver C, Erzurum SC. Differential expression of manganese superoxide dismutase and catalase in lung cancer. Cancer Res. 2001;61:8578–85. [PubMed] [Google Scholar]
- 148.Sun Y, Colburn NH, Oberley LW. Depression of catalase gene expression after immortalization and transformation of mouse liver cells. Carcinogenesis. 1993;14:1505–10. doi: 10.1093/carcin/14.8.1505. [DOI] [PubMed] [Google Scholar]
- 149.Gupta A, Butts B, Kwei KA, Dvorakova K, Stratton SP, Briehl MM, et al. Attenuation of catalase activity in the malignant phenotype plays a functional role in an in vitro model for tumor progression. Cancer Lett. 2001;173:115–25. doi: 10.1016/s0304-3835(01)00656-5. [DOI] [PubMed] [Google Scholar]
- 150.Gupta A, Rosenberger SF, Bowden GT. Increased ROS levels contribute to elevated transcription factor and MAP kinase activities in malignantly progressed mouse keratinocyte cell lines. Carcinogenesis. 1999;20:2063–73. doi: 10.1093/carcin/20.11.2063. [DOI] [PubMed] [Google Scholar]
- 151.Yamaguchi Y, Sato K, Endo H. Depression of catalase gene expression in the liver of tumor bearing nude mice. Biochem Biophys Res Commun. 1992;189:1084–9. doi: 10.1016/0006-291x(92)92315-o. [DOI] [PubMed] [Google Scholar]
- 152.Larsen NB, Rasmussen M, Rasmussen LJ. Nuclear and mitochondrial DNA repair: similar pathways? Mitochondrion. 2005;5:89–108. doi: 10.1016/j.mito.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 153.Oka S, Ohno M, Tsuchimoto D, Sakumi K, Furuichi M, Nakabeppu Y. Two distinct pathways of cell death triggered by oxidative damage to nuclear and mitochondrial DNAs. EMBO J. 2008;27:421–32. doi: 10.1038/sj.emboj.7601975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Maynard S, Schurman SH, Harboe C, de Souza-Pinto NC, Bohr VA. Base excision repair of oxidative DNA damage and association with cancer and aging. Carcinogenesis. 2009;30:2–10. doi: 10.1093/carcin/bgn250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Copeland WC, Ponamarev MV, Nguyen D, Kunkel TA, Longley MJ. Mutations in DNA polymerase gamma cause error prone DNA synthesis in human mitochondrial disorders. Acta Biochim Pol. 2003;50:155–67. [PubMed] [Google Scholar]
- 156.Longley MJ, Prasad R, Srivastava DK, Wilson SH, Copeland WC. Identification of 5′-deoxyribose phosphate lyase activity in human DNA polymerase gamma and its role in mitochondrial base excision repair in vitro. Proc Natl Acad Sci U S A. 1998;95:12244–8. doi: 10.1073/pnas.95.21.12244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Graziewicz MA, Day BJ, Copeland WC. The mitochondrial DNA polymerase as a target of oxidative damage. Nucleic Acids Res. 2002;30:2817–24. doi: 10.1093/nar/gkf392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hance N, Ekstrand MI, Trifunovic A. Mitochondrial DNA polymerase gamma is essential for mammalian embryogenesis. Hum Mol Genet. 2005;14:1775–83. doi: 10.1093/hmg/ddi184. [DOI] [PubMed] [Google Scholar]
- 159.Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–23. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 160.Bailey LJ, Cluett TJ, Reyes A, Prolla TA, Poulton J, Leeuwenburgh C, et al. Mice expressing an error-prone DNA polymerase in mitochondria display elevated replication pausing and chromosomal breakage at fragile sites of mitochondrial DNA. Nucleic Acids Res. 2009;37:2327–35. doi: 10.1093/nar/gkp091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Vermulst M, Bielas JH, Kujoth GC, Ladiges WC, Rabinovitch PS, Prolla TA, et al. Mitochondrial point mutations do not limit the natural lifespan of mice. Nat Genet. 2007;39:540–3. doi: 10.1038/ng1988. [DOI] [PubMed] [Google Scholar]
- 162.Vermulst M, Wanagat J, Kujoth GC, Bielas JH, Rabinovitch PS, Prolla TA, et al. DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat Genet. 2008;40:392–4. doi: 10.1038/ng.95. [DOI] [PubMed] [Google Scholar]
- 163.Chatterjee A, Singh KK. Uracil-DNA glycosylase-deficient yeast exhibit a mitochondrial mutator phenotype. Nucleic Acids Res. 2001;29:4935–40. doi: 10.1093/nar/29.24.4935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Nilsen H, Otterlei M, Haug T, Solum K, Nagelhus TA, Skorpen F, et al. Nuclear and mitochondrial uracil-DNA glycosylases are generated by alternative splicing and transcription from different positions in the UNG gene. Nucleic Acids Res. 1997;25:750–5. doi: 10.1093/nar/25.4.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Akbari M, Visnes T, Krokan HE, Otterlei M. Mitochondrial base excision repair of uracil and AP sites takes place by single-nucleotide insertion and long-patch DNA synthesis. DNA Repair (Amst) 2008;7:605–16. doi: 10.1016/j.dnarep.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 166.Akbari M, Otterlei M, Pena-Diaz J, Krokan HE. Different organization of base excision repair of uracil in DNA in nuclei and mitochondria and selective upregulation of mitochondrial uracil-DNA glycosylase after oxidative stress. Neuroscience. 2007;145:1201–12. doi: 10.1016/j.neuroscience.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 167.Cheng KC, Cahill DS, Kasai H, Nishimura S, Loeb LA. 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G–-T and A–-C substitutions. J Biol Chem. 1992;267:166–72. [PubMed] [Google Scholar]
- 168.Chatterjee A, Mambo E, Zhang Y, Deweese T, Sidransky D. Targeting of mutant hogg1 in mammalian mitochondria and nucleus: effect on cellular survival upon oxidative stress. BMC Cancer. 2006;6:235. doi: 10.1186/1471-2407-6-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Nishioka K, Ohtsubo T, Oda H, Fujiwara T, Kang D, Sugimachi K, et al. Expression and differential intracellular localization of two major forms of human 8-oxoguanine DNA glycosylase encoded by alternatively spliced OGG1 mRNAs. Mol Biol Cell. 1999;10:1637–52. doi: 10.1091/mbc.10.5.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Dobson AW, Xu Y, Kelley MR, LeDoux SP, Wilson GL. Enhanced mitochondrial DNA repair and cellular survival after oxidative stress by targeting the human 8-oxoguanine glycosylase repair enzyme to mitochondria. J Biol Chem. 2000;275:37518–23. doi: 10.1074/jbc.M000831200. [DOI] [PubMed] [Google Scholar]
- 171.Rachek LI, Grishko VI, Musiyenko SI, Kelley MR, LeD oux SP, Wilson GL. Conditional targeting of the DNA repair enzyme hOGG1 into mitochondria. J Biol Chem. 2002;277:44932–7. doi: 10.1074/jbc.M208770200. [DOI] [PubMed] [Google Scholar]
- 172.Mambo E, Chatterjee A, de Souza-Pinto NC, Mayard S, Hogue BA, Hoque MO, et al. Oxidized guanine lesions and hOgg1 activity in lung cancer. Oncogene. 2005;24:4496–508. doi: 10.1038/sj.onc.1208669. [DOI] [PubMed] [Google Scholar]
- 173.He YH, Xu Y, Kobune M, Wu M, Kelley MR, Martin WJ., 2nd Escherichia coli FPG and human OGG1 reduce DNA damage and cytotoxicity by BCNU in human lung cells. Am J Physiol Lung Cell Mol Physiol. 2002;282:L50–5. doi: 10.1152/ajplung.00316.2001. [DOI] [PubMed] [Google Scholar]
- 174.Mambo E, Nyaga SG, Bohr VA, Evans MK. Defective repair of 8-hydroxyguanine in mitochondria of MCF-7 and MDA-MB-468 human breast cancer cell lines. Cancer Res. 2002;62:1349–55. [PubMed] [Google Scholar]
- 175.Nyaga SG, Lohani A, Jaruga P, Trzeciak AR, Dizdaroglu M, Evans MK. Reduced repair of 8-hydroxyguanine in the human breast cancer cell line, HCC1937. BMC Cancer. 2006;6:297. doi: 10.1186/1471-2407-6-297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Nyaga SG, Lohani A, Evans MK. Deficient repair of 8-hydroxyguanine in the BxPC-3 pancreatic cancer cell line. Biochem Biophys Res Commun. 2008;376:336–40. doi: 10.1016/j.bbrc.2008.08.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Zhang H, Mizumachi T, Carcel-Trullols J, Li L, Naito A, Spencer HJ, et al. Targeting human 8-oxoguanine DNA glycosylase (hOGG1) to mitochondria enhances cisplatin cytotoxicity in hepatoma cells. Carcinogenesis. 2007;28:1629–37. doi: 10.1093/carcin/bgm072. [DOI] [PubMed] [Google Scholar]
- 178.Fishel ML, Seo YR, Smith ML, Kelley MR. Imbalancing the DNA base excision repair pathway in the mitochondria; targeting and overexpressing N-methylpurine DNA glycosylase in mitochondria leads to enhanced cell killing. Cancer Res. 2003;63:608–15. [PubMed] [Google Scholar]
- 179.Trivedi RN, Wang XH, Jelezcova E, Goellner EM, Tang JB, Sobol RW. Human methyl purine DNA glycosylase and DNA polymerase beta expression collectively predict sensitivity to temozolomide. Mol Pharmacol. 2008;74:505–16. doi: 10.1124/mol.108.045112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Chinnery PF, Schon EA. Mitochondria. J Neurol Neurosurg Psychiatry. 2003;74:1188–99. doi: 10.1136/jnnp.74.9.1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Waters LS, Minesinger BK, Wiltrout ME, D’Souza S, Woodruff RV, Walker GC. Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol Mol Biol Rev. 2009;73:134–54. doi: 10.1128/MMBR.00034-08. [DOI] [PMC free article] [PubMed] [Google Scholar]