

Abstract
Cyclooxygenase-2 (COX-2) catalyzes the oxygenation of arachidonic acid and the endocannabinoids, 2-arachidonoylglycerol and arachidonoylethanolamide. Evaluation of a series of COX-2 inhibitors revealed that many weak, competitive inhibitors of arachidonic acid oxygenation are potent inhibitors of endocannabinoid oxygenation. (R)-Enantiomers of ibuprofen, naproxen, and flurbiprofen, which are considered to be inactive as COX-2 inhibitors, are potent “substrate-selective inhibitors” of endocannabinoid oxygenation. Crystal structures of the COX-2-(R)-naproxen and COX-2-(R)-flurbiprofen complexes verified this unexpected binding and defined the orientation of the (R)-enantiomers relative to (S)-enantiomers. (R)-Profens selectively inhibited endocannabinoid oxygenation by lipopolysaccharide-stimulated dorsal root ganglion cells. Substrate-selective inhibition provides novel tools for investigating the role of COX-2 in endocannabinoid oxygenation and a possible explanation for the ability of (R)-profens to maintain endocannabinoid tone in models of neuropathic pain.
The endocannabinoids, 2-arachidonoylglycerol (2-AG) and arachidonoylethanolamide (AEA), exert analgesic and anti-inflammatory effects through their actions at the cannabinoid receptors, CB1 and CB2 1–3. They also are substrates for the fatty acid oxygenases, lipoxygenases and cyclooxygenases (COXs), and certain cytochromes P-450, which convert them to bioactive oxygenated metabolites 4–8. 2-AG and AEA are oxygenated efficiently to prostaglandin glycerol esters (PG-Gs) and prostaglandin ethanolamides (PG-EAs), respectively, by COX-2 but not COX-1. PG-Gs and PG-EAs activate calcium mobilization in macrophages and tumor cells, enhance miniature excitatory and inhibitory postsynaptic currents in neurons, induce mechanical allodynia, and stimulate thermal hyperalgesia 9–13. 2-AG and AEA are rapidly hydrolyzed by monoacylglycerol lipase (MAGL) or fatty acid amide hydrolase (FAAH), respectively, to arachidonic acid (AA), which terminates endocannabinoid signaling but produces a fatty acid that is converted to leukotrienes and prostaglandins inter alia 14,15. Thus, 2-AG and AEA are at the nexus of a complex network of bioactive lipid production, inactivation, and signaling (Supplementary Results, Supplementary Fig. 1).
The importance of endocannabinoids as naturally occurring analgesic agents provides a potential mechanism for inhibition of neuropathic pain – i.e., through the development of agents that prevent endocannabinoid metabolism at sites of neuroinflammation 16. FAAH inhibitors appear to be promising candidates in this regard, and MAGL inhibitors are also potential leads, although their broader range of cannabimimetic effects in animal models may limit their utility 17,18. Since COX-2 is induced at sites of neuroinflammation, non-steroidal anti-inflammatory drugs (NSAIDs), whether non-selective or selective for COX-2, may contribute to endocannabinoid-sparing by preventing COX-2-selective oxygenation of 2-AG and AEA 19.
We recently reported that ibuprofen and mefenamic acid are potent, non-competitive inhibitors of 2-AG oxygenation by COX-2 but weak, competitive inhibitors of AA oxygenation 20. Kinetic studies indicate that this “substrate-selective inhibition” results from negative cooperativity between the two monomers of the COX-2 homodimer. Binding of a molecule of inhibitor in one subunit inhibits the oxygenation of 2-AG, but not AA, in the other subunit 20,21. Inhibition of AA oxygenation by ibuprofen or mefenamic acid requires binding of an additional molecule of inhibitor in the second subunit 20.
In the present study, we surveyed a series of weak, reversible inhibitors and slow, tight-binding inhibitors for substrate-selectivity. Only weak, reversible inhibitors demonstrated substrate-selectivity. We discovered that (R)-arylpropionic acids, (i.e., profens), which are not believed to inhibit COX enzymes, are efficient substrate-selective inhibitors of 2-AG oxygenation by COX-2. Crystal structures of (R)-naproxen and (R)-flurbiprofen complexed to COX-2 indicated that they bind in an analogous fashion to the (S)-enantiomers. Site-directed mutagenesis identified Arg-120 as the key residue for inhibition of COX-2 by (R)-flurbiprofen. Induction of COX-2 in primary murine dorsal root ganglion cells (DRGs) led to the oxygenation of AA, 2-AG, and AEA to their respective prostaglandin products. (R)-Profens selectively inhibited the COX-2 mediated oxygenation of 2-AG and AEA but not AA. This substrate-selective inhibition led to an elevation in the levels of the endocanabinoids, 2-AG and AEA, in the DRGs.
Results
Survey of COX Inhibitors for Substrate-Selectivity
To explore the generality of substrate-selective inhibition, we compared the effects of different classes of NSAIDs on COX-2-dependent AA and 2-AG oxygenation. An assay was employed that measured PG or PG-G formation by LC-MS-MS following a 15 min preincubation of enzyme and inhibitor to ensure maximal inhibition of the oxygenation of both substrates. Saturating concentrations of both AA and 2-AG (50 μM) were used. The results are summarized in Table 1. Weak, reversible inhibitors of AA oxygenation were strong inhibitors of 2-AG oxygenation whereas slow, tight-binding inhibitors were potent inhibitors of both 2-AG and AA oxidation by COX-2 with comparable IC50’s for both substrates (Table 1). The ability of slow, tight binding inhibitors to inhibit 2-AG and AA oxygenation at comparable concentrations arises from the ability of a single molecule of inhibitor to block the oxygenation of both substrates 20–23. Within the inhibitor classes, no differences in behavior were observed for compounds that are COX-2-selective inhibitors or non-selective inhibitors of both COX enzymes (e.g., celecoxib vs diclofenac).
Table 1.
IC50 values for the inhibition of COX-2 oxygenation of AA and 2-AG by NSAIDs
| Inhibitor | 50 μM AA | 50 μM 2-AG | |
|---|---|---|---|
| reversible | Ibuprofenb,c | 7 μM | 20 nM |
| Mefenamic Acidb,c | 180 μM | 210 nM | |
| DM-INDO | > 25 μM | 250 nM | |
| Lumiracoxib | no inhib. | 40 nM | |
| Naproxenb | 4.5 μM | 430 nM | |
| SC-58076 | > 4 μM | 40 nM | |
| slow, tight binders | Diclofenac | 60 nM | 50 nM |
| Flurbiprofen | 130 nM | 30 nM | |
| INDO | 180 nM | 30 nM | |
| Celecoxib | 80 nM | 95 nM | |
| Rofecoxib | 520 nM | 85 nM |
Enzyme and inhibitor were pre-incubated for 15 min prior to the addition of 50 uM substrate for 30 s. Reactions were quenched with organic solvent containing deuterated internal standards. Product formation was analyzed by LC-MS-MS using selected reaction monitoring and normalized to DMSO control.
Substrate oxygenation was measured using an oxygen electrode.
Values for ibuprofen and mefenamic acid taken from 20.
Inhibition by (R)-Profens
Profens exhibit marked enantiospecificity for inhibition of AA oxygenation by COX enzymes. The (S)-enantiomers of naproxen, ibuprofen and flurbiprofen inhibit AA oxygenation by COX-1 and COX-2, but the (R)-enantiomers are either poor inhibitors or exhibit no inhibitory activity. Because we have observed that weak inhibitors of AA oxygenation can be strong inhibitors of 2-AG oxygenation, we evaluated the ability of the (R)-enantiomers of naproxen, ibuprofen and flurbiprofen to inhibit 2-AG oxygenation by COX-2. As shown in Supplementary Fig. 2 and Table 2, the (R)-enantiomers inhibited 2-AG oxidation. The inhibitors were more potent at concentrations of 2-AG near its Km (5 μM) than at saturation (50 μM). Importantly, (R)-naproxen, (R)-ibuprofen, and (R)-flurbiprofen did not inhibit AA oxidation by COX-2 at either 5 or 50 μM substrate concentrations.
Table 2.
(R)-Profen inhibition of 2-AG oxygenation by COX-2a
| Inhibitor | 50 μM 2-AG | 5 μM 2-AG |
|---|---|---|
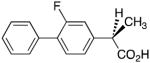
|
3.7 μM | 0.08 μM |
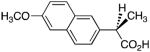
|
6.7 μM | 3.0 μM |
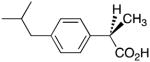
|
18 μM | 10 μM |
Enzyme and inhibitor were pre-incubated for 15 min prior to the addition of substrate for 30 s. Reactions were quenched with organic solvent containing deuterated internal standards. Product formation was analyzed by LC-MS-MS using selected reaction monitoring and normalized to DMSO control.
Structure-Function Analysis of (R)-Profen Inhibition
These findings illustrate that the (R)-enantiomers of arylpropionic acids bind to COX-2 and inhibit the oxygenation of 2-AG. This was surprising given prior results suggesting that steric clashes with active site residues prevent the binding of (R)-arylpropionic acids within the COX active site 24. Therefore, we attempted to crystallize complexes of each of the (R)-enantiomers bound to mCOX-2 to identify their binding sites. Diffraction-quality crystals were obtained with both (R)-naproxen and (R)-flurbiprofen using recently described methodology 25. The experimental electron density map for (R)-naproxen bound to COX-2 is shown in Fig. 1a and Supplementary Fig. 3. The inhibitor is located exclusively within the cyclooxygenase active site with its carboxylate moiety adjacent to Arg-120 and Tyr-355 at the mouth of the active site and the naphthyl ring projecting up into the center of the cyclooxygenase channel (Supplementary Fig. 3). This is typical of the orientation of arylpropionic acids in the COX active site as has been reported for (S)-ibuprofen complexed to COX-1, (S)-naproxen complexed to COX-2, and (S)-flurbiprofen complexed to COX-1 or COX-2 25–27.
Figure 1.

Crystal structure of (R)-naproxen within the mCOX-2 active site. (a) Stereoview of (R)-naproxen (green sticks) bound in the COX-2 active. The simulated annealing omit map (Fo-Fc) contoured at 3σ is displayed in the vicinity of (R)-naproxen. Protein residues are shown in cyan sticks. (b) Stereoview of the active site of the (R)-naproxen-COX-2 complex (green sticks) overlaid with the (S)-naproxen-COX-2 complex (grey sticks).
Previous reports based on site-directed mutagenesis and structure-activity studies have suggested that stable binding of (R)-arylpropionic acids within the active site of COX is prevented by unfavorable steric interactions between the α-methyl group and Tyr-355 at the base of the active site 24. Interestingly the α-methyl group of (R)-naproxen binds adjacent to Tyr-355 as illustrated in Fig. 1a. The (S)-naproxen structure was recently reported at 1.7 A, and an overlay of (R)-naproxen and (S)-naproxen bound in the active site of mCOX-2 is shown in Fig. 1b 25. The p-methoxy substituents and the two naphthyl rings are nearly superimposed and the two enantiomers appear to participate in many of the same interactions with surrounding protein residues. These include hydrogen-bonding with Arg-120 and Tyr-355 as well as hydrophobic interactions with Ala-527, Val-349, Gly-526, Trp-387, Tyr-385, and Leu-352. However, (R)-naproxen does participate in some interactions distinct from those of (S)-naproxen. The α-methyl group of (R)-naproxen participates in Van der Waals interactions with Ser-530 and Ser-353, but the α-methyl substituent of (S)-naproxen does not. The most significant difference in protein structure between the two complexes is the repositioning of Arg-120 and Tyr-355 to accommodate the α-methyl group in the binding of (R)-naproxen (RMSD: 0.47 and 0.45 A, respectively). This increases the hydrogen bond distance between Tyr-355 and the carboxylate of (R)-naproxen to 3.05 A compared to a distance of 2.44 A for (S)-naproxen, which may reduce the binding energy of the (R)-naproxen-COX-2 complex.
Like the naproxen enantiomers, the (R)- and (S)- enantiomers of flurbiprofen bind in a similar fashion within the COX-2 active site (Fig. 2a and Supplementary Figs. 4 and 5). (R)-Flurbiprofen interacts with Arg-120 and Tyr-355 at the base of the active site and with Ala-527, Val-349, Gly-526, Tyr-385, Leu-359, and Ser-530 in the hydrophobic channel. To probe the importance of individual residues in the inhibition of 2-AG oxygenation, we measured the inhibitory activity of (R)-flurbiprofen against a series of active site mutants. As shown in Fig. 2b, mutation of Arg-120 to Gln, which eliminates the ability of that residue to participate in ion-pairing interactions, abolishes (R)-flurbiprofen inhibition. In contrast, mutations of Tyr-355 to Phe, Glu-524 to Leu, or Ser-530 to Ala did not have significant effects on the IC50 values for (R)-flurbiprofen when compared to WT mCOX-2 (Fig. 2b). These data are consistent with the binding mode of (R)-flurbiprofen observed in the crystal structure and suggest that ion-pairing between the carboxylate and Arg-120 is a critical determinant of binding.
Figure 2.

Binding of (R)-flurbiprofen within the mCOX-2 active site. (a) Crystal structure of (R)-flurbiprofen (magenta) bound in the active site of mCOX-2 (cyan). The simulated annealing omit map (Fo-Fc) contoured at 3σ is shown surrounding (R)-flurbiprofen. (b) Comparison of the concentration-dependence of (R)-flurbiprofen inhibition of 2-AG oxygenation by wild-type or mutant mCOX-2s. Inhibition assays were performed as described under Methods. The data points (n=3) represent the mean ± SEM.
COX-2 Action in DRGs
(R)-Flurbiprofen exhibits analgesic activity in humans and inhibits neuropathic pain in rodents 28,29. It is inefficiently converted to (S)-flurbiprofen in vivo and does not display gastrointestinal toxicity, which is typically observed with compounds that inhibit COX-dependent prostaglandin synthesis 30,31. Interestingly, (R)-flurbiprofen has been reported to elevate AEA levels in the dorsal horn of rats surgically treated to induce nerve injury 29. The mode of action by which (R)-flurbiprofen causes analgesia and AEA elevation is uncertain, although it is a weak inhibitor of FAAH (IC50 = >1 mM in vitro) 29. Another possible explanation for the analgesic activity of (R)-flurbiprofen is that it inhibits the COX-2-selective metabolism of endocannabinoids. To evaluate this possibility and to test whether substrate-selective inhibition can be detected in intact cells stimulated to release physiological levels of 2-AG and AEA, cellular experiments were performed with DRGs. DRGs were harvested from E14 mouse embryos and plated onto collagen-coated dishes. After culturing for 3–5 days, they were treated overnight with granulocyte macrophage colony stimulating factor, followed by lipopolysaccharide, interferon γ, and 10 μM 15(S)-hydroxy-5,8,11,13-eicosatetraenoic acid for 6 hr. This resulted in a strong induction of COX-2, but not COX-1, in the DRGs and no increase in the levels of MAGL, ABHD6 or FAAH (Fig. 3a and Supplementary Fig. 6). The presence of COX-2 in the DRGs, located mainly in neuronal cell bodies, was verified by the uptake of a COX-2-selective fluorescent imaging agent (Fig. 3b). The design and synthesis of this compound, fluorocoxib A, was recently described along with studies validating its selective binding to COX-2 in cultured cells in vitro and inflammatory lesions and tumors in vivo 32.
Figure 3.

Analysis of DRGs. (a) Immunostaining and fluorescent imaging of neurons and glia from dissociated E14 DRGs. Stimulated DRGs were imaged using a fluorescent TuJ1 antibody to label neurons (green), DAPI to label nuclei (blue), and fluorocoxib A to label COX-2 (red). (b) Western blot analysis of basal versus stimulated DRGs comparing enzymes involved in endocannabinoid metabolism and prostaglandin synthesis. (c) LC-MS chromatographic peaks and SRM transitions for prostaglandins derived from AA, AEA, and 2-AG isolated from DRGs.
DRGs, activated as above for 3 hr, were treated with ionomycin for an additional 3 hr to stimulate substrate release. The substrates and products of COX-2 mediated oxygenation were extracted and identified by LC-MS-MS. Peaks were detected that coeluted with PG, PG-G, and PG-EA standards (Fig. 3c). In all cases, the major products were PGF2α and PGE2, their glyceryl esters, and ethanolamide derivatives. It is noteworthy that the stimulation of DRGs resulted in the generation of PG-EAs. This is the first time that these oxygenated metabolites of AEA have been detected in intact cells stimulated to release endogenous COX-2 substrates. The identity of the PG-EAs was verified by collision-induced dissociation and analysis of the fragment ions (Supplementary Fig. 7). Thus, DRGs release AA, 2-AG, and AEA and oxygenate them to PGF2α and PGE2 derivatives following stimulation with pro-inflammatory mediators.
Substrate-Selective Inhibition in DRGs
The levels of PGs, PG-Gs, and PG-EAs were quantified using stable isotope dilution methods with labeled internal standards. Increasing concentrations of (R)-flurbiprofen, (R)-ibuprofen, or (R)-naproxen added 1 hr before ionomycin inhibited the synthesis of endocannabinoid-derived eicosanoids, PG-Gs and PG-EAs, at concentrations that did not inhibit the synthesis of PGs. The concentration-dependences for inhibition of 2-AG oxygenation and AEA oxygenation were similar (Fig. 4). Thus, the substrate-selective inhibition of endocannabinoid oxygenation observed with purified COX-2 was also observed in intact DRGs stimulated with physiological agonists and endogenous substrates. The IC50’s for inhibition of 2-AG and AEA oxidation in DRG’s by the (R)-profens ((R)-flurbiprofen-2-AG, 5.8 ± 2.9 μM, (R)-flurbiprofen-AEA, 6.0 ± 2.7 μM; (R)-naproxen-2-AG, 8.9 ± 3.2 μM, (R)-naproxen-AEA, 11.8 ± 4.1 μM; (R)-ibuprofen-2-AG, 10.1 ± 4.7 μM, (R)-ibuprofen-AEA, 9.4 ± 4.3 μM) were closer to the IC50’s for inhibition of pure COX-2 at 50 μM 2-AG than at 5 μM 2-AG (Table 2). One might have anticipated a closer correspondence to values at low substrate concentrations in vitro but there were many differences between the in vitro and cellular assays including different incubation times (30 s vs 4 hr), the presence of serum in the cell culture medium, and the presence of multiple competing fatty acids and other lipids in the activated DRG’s. These may have had the effect of increasing the IC50’s of the (R)-profens in intact DRG’s compared to purified enzyme.
Figure 4.

Inhibition of eicosanoid synthesis in stimulated DRGs by (R)-flurbiprofen, (R)-naproxen, and (R)-ibuprofen. Product formation was monitored following the oxygenation of AA, 2-AG, and AEA by COX-2 to form PGs (−), PG-Gs (- -), and PG-EAs (• •) in DRGs. IC50’s were calculated using a non-linear regression. The data points represent percent inhibition with respect to control of two sets of three DRG culture plates from two independent DRG preparations for each (R)-profen. The data points (n=6) represent the mean ± SEM.
Concomitant with the inhibition of PG-G and PG-EA formation, (R)-flurbiprofen, (R)-ibuprofen, and (R)-naproxen treatment of stimulated DRGs increased the amounts of AEA and 2-AG measured in cell extracts but did not increase the amounts of AA (Fig. 5). Importantly, treatment of DRGs that were not stimulated with pro-inflammatory agonists did not increase the concentrations of 2-AG or AEA suggesting that they did not inhibit the catalytic activity of MAGL or ABHD6, which appear to be present in the cells (Fig. 3a). Interestingly, FAAH does not appear to be present in the DRGs (Fig. 3a). Indeed, incubation of increasing concentrations of the three (R)-profens with purified MAGL or FAAH in vitro caused no inhibition of their catalytic activities at concentrations up to 1 mM (Supplementary Figs. 8 and 9). Similar control experiments indicated that (R)-profens do not inhibit 15-lipoxygenase-1 oxygenation of AA or 2-AG at concentrations used in DRGs (Supplementary Figure 10). Finally, the enantiomeric composition of the (R)-profens recovered following a 4 hr incubation with DRGs was evaluated by chiral chromatography and shown to be >99% (R) (Supplementary Fig. 11). Thus, no racemization occurred during incubation with the cells so the substrate-selective inhibition of endocannabinoid oxygenation observed with the various profens was due to the (R)-enantiomers.
Figure 5.

Comparison of the effects of (R)-flurbiprofen, (R)-naproxen, and (R)-ibuprofen on substrate levels in basal versus stimulated DRGs. The data points represent the amount of AEA (blue), 2-AG (red), and AA (white) from two sets of three DRG culture plates from two independent DRG preparations for each (R)-profen. The fatty acid levels (n=6) are plotted as mean ± SEM and statistical significance was determined using a one-way ANOVA analysis. Statistically significant increases (P < .05) in both AEA and 2-AG are indicated by overhead bars.
Discussion
These results expand the range of compounds capable of substrate-selective inhibition of endocannabinoid oxygenation and indicate that it is limited to compounds characterized as rapid reversible inhibitors of COX-2. The results also illustrate that (R)-enantiomers of arylpropionic acid inhibitors, which are considered inactive as COX inhibitors because of their inability to inhibit AA oxygenation, actually bind to the enzyme and potently inhibit endocannabinoid oxygenation. Crystallographic studies indicate that the binding site of (R)-naproxen and (R)-flurbiprofen is exclusively within the COX-2 active site and functional studies indicate that ion-pairing to Arg-120 is critical for (R)-flurbiprofen binding. The potency of (R)-profen inhibition of endocannabinoid oxygenation is a dramatic illustration of the negative cooperativity between the two monomers of the COX-2 homodimer that results from binding an inhibitor molecule in a single monomer (Fig. 6) 20,21,23. Although COX-2 and COX-1 are structural homodimers, they behave as functional heterodimers with an allosteric site and a catalytic site 23. Kinetic analysis of substrate-selective inhibition of 2-AG oxygenation by (S)-ibuprofen suggests it is a non-competitive inhibitor that binds in the allosteric site 20. Thus, binding of (R)-profens likely occurs in the allosteric site and induces a conformational change that prevents endocannabinoid, but not AA oxygenation, in the catalytic site (Fig. 6).
Figure 6.
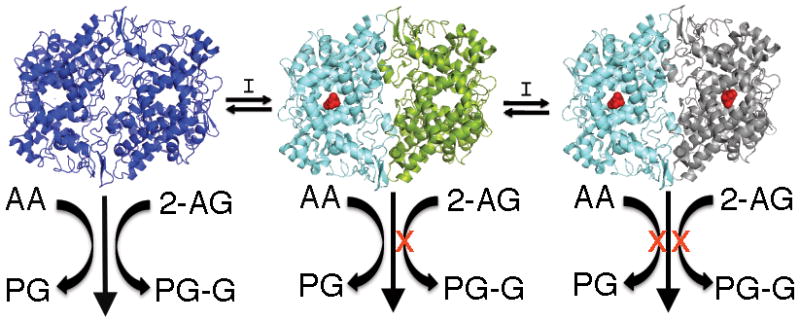
The mechanism of COX-2 substrate-selective inhibition of endocannabinoid oxygenation by rapid, reversible inhibitors. Inhibitor binding in one subunit of the homodimer induces a conformational change in the second subunit that blocks 2-AG and AEA oxygenation but not AA oxygenation. In order to inhibit oxygenation of AA, another molecule of inhibitor must bind in the second subunit. For slow, tight-binding inhibitors, the conformational changes induced by binding a single inhibitor molecule are sufficient to inhibit the oxygenation of all substrates.
COX-2 oxygenation of 2-AG and AEA in intact cells produces PG-G and PG-EA derivatives that exhibit a range of biological activities 9,13,33–35. The receptors responsible for these effects have not been identified but they appear to be distinct from classic PG receptors 9,36. Thus, COX-2-dependent endocannabinoid oxygenation may represent a novel mechanism for generating lipid signaling molecules dependent on different sets of agonists and phospholipases than are responsible for COX-2- (or COX-1-) dependent PG formation. Testing this hypothesis in cellular systems or animal models has been difficult because of the lack of specific reagents that can differentiate COX-2-dependent 2-AG and AEA oxygenation from AA oxygenation. The high degree of substrate-selective inhibition exhibited by (R)-profens suggests they may be valuable probes for dissecting the specific contributions to cellular physiology or pathophysiology of endocannabinoid oxygenation by COX-2 from those of AA oxygenation.
The metabolism of endocannabinoids and its relationship to signaling involves a complex set of enzymes and receptors 2,37. Following their biosyntheses from phospholipid precursors, AEA binds to the CB1 receptor and TRPV1 whereas 2-AG binds to the CB1 and CB2 receptors to stimulate cellular responses. The levels of AEA and 2-AG are primarily controlled through hydrolysis by FAAH and MAGL, respectively, although other enzymes will also hydrolyze 2-AG (e.g., ABHD6, ABHD12 and carboxyl esterase 1) 38,39. AEA and 2-AG are also oxygenated by COX-2, lipoxygenases, and cytochromes P-450 and it is conceivable that, under certain conditions, sufficient oxygenation could occur to further lower endocannabinoid levels. COX-2 is a particularly attractive candidate to modulate endocannabinoid levels because it is highly induced by a range of agents including pro-inflammatory stimuli. Fig. 3 illustrates that COX-2 is induced in DRGs stimulated with pro-inflammatory agents; in contrast, the levels of MAGL, FAAH, and ABHD6 are not increased. Stimulation with pro-inflammatory agents resulted in significant COX-2 mediated oxygenation of AA, 2-AG, and AEA, but no oxygenation was observed in the absence of pretreatment with pro-inflammatory stimuli. Incubation of DRGs with (R)-profens selectively inhibited oxygenation of 2-AG and AEA compared to AA (Fig. 4). (R)-Profens also increased the levels of 2-AG and AEA but not the levels of AA (Fig. 5). Interestingly, (R)-profens did not increase the levels of 2-AG and AEA in DRGs that were not pretreated with pro-inflammatory stimuli (Fig. 5). This is consistent with COX-2 reducing endocannabinoid levels by oxygenation to PG-Gs and PG-EAs and with (R)-profens preventing endocannabinoid depletion by selectively inhibiting their oxygenation.
These findings uncover a potential mechanism for the analgesic activity of (R)-flurbiprofen. The ability of (R)-flurbiprofen to selectively inhibit AEA and 2-AG oxygenation in DRGs correlates to its ability to elevate AEA levels at sites of neuroinflammation in the spinal cord 29. Although FAAH and MAGL are likely responsible for the basal turnover of endocannabinoids in non-inflammed tissue, diurnal fluctuations lead to increases in COX-2 in regions of the brain and induction of inflammation in the peripheral or central nervous system by nerve injury results in elevated levels of COX-2 in the inflamed tissue 19,40. COX-2 induction may contribute to the depletion of AEA and 2-AG and blockage of this depletion by substrate-selective inhibition of COX-2 by (R)-flurbiprofen could spare endocannabinoid levels and induce analgesia. Consistent with this mechanism, the analgesic effect of (R)-flurbiprofen is prevented by CB1 receptor antagonists despite the fact that (R)-flurbiprofen does not activate the CB1 receptor 29. This highlights the importance of maintenance of endocannabinoid tone in the analgesic action of (R)-flurbiprofen.
Methods
Materials
Wild-type, R120Q, E524L, S530A and Y355F mCOX-2 protein were expressed in insect cells and purified as described previously 41. Human MAGL was purchased from Cayman Chemical (Ann Arbor, MI). Humanized rat FAAH was a generous gift of R. Stevens and B. Cravatt (Scripps Research Institute). Human 15-lipoxygenase-1 was a generous gift of A. Brash (Vanderbilt University School of Medicine).
Inhibition of COX-2 mediated metabolism of AA and 2-AG
Various concentrations of inhibitor (or DMSO) were incubated with mCOX-2 (200 nM) for 5 – 15 min in 100 mM Tris-HCl with 0.5 mM phenol, pH 8.0. For mutant mCOX-2 experiments, the enzyme concentration was adjusted such that the turnover was approximately equal to wild-type enzyme. The pre-incubation time was determined based on previous reports regarding the time required to achieve maximal inhibition, and was performed at room temperature except for the final three minutes, which was at 37 °C 20,42. Following the pre-incubation of enzyme and inhibitor, AA or 2-AG was added for 30 s at 37 °C. The reaction was quenched with ice-cold ethyl acetate containing 0.5 % acetic acid (v/v) and 1 μM PGE2-d4 and PGE2-G-d5. The solution was then vigorously mixed and cooled on ice. The organic layer was separated and evaporated to near-dryness under nitrogen. For analysis, the samples were reconstituted in 1:1 MeOH:H2O and chromatographed using a Luna C18(2) column (50 × 2 mm, 3 μm) (Phenomenex, Torrance, CA) with an isocratic elution method consisting of 66% 5 mM ammonium acetate pH = 3.3 (solvent A) and 34% ACN containing 6% solvent A (solvent B) at a flow rate of 0.375 mL/min. MS/MS was conducted on a Quantum triple quadrupole mass spectrometer operated in positive ion mode utilizing a selected reaction monitoring method with the following transitions - m/z 370→317 for PGE2/D2, m/z 374 → 321 for PGE2-d4, m/z 444 → 391 for PGE2/D2-G and 449 → 396 for PGE2/D2-G-d5. Peak areas for analytes were normalized to the appropriate internal standard to determine the amount of product formation, and the amount of inhibition was determined by normalization to a DMSO control.
Crystallization, data collection, structure determination and refinement
Protein crystallization was performed as described 25. Data sets were collected on an ADSC Quantum-315 CCD using the synchrotron radiation X-ray source tuned at a wavelength of 0.97929 A and an operating temperature of 100 K at beamline 24ID-E of the Advance Photon Source at Argonne National Lab, Chicago, USA. Diffraction data were processed with HKL2000 43. Initial phases were determined by molecular replacement using a search model (PDB 3NT1) with MOLREP 44. Solutions having two molecules in the asymmetric unit for (R)-naproxen and four molecules in the asymmetric unit for (R)-flurbiprofen were obtained. The models were improved with iterative rounds of model building in Coot and refinement in PHENIX 45,46. Data collection and refinement statistics are reported in Supplemental Table 1. In the Ramachandran plot, 93.7% of all residues are in the most favored region for the (R)-naproxen-mCOX-2 structure and 94.0% of all residues appear in the most favored region for the (R)-flurbiprofen-mCOX-2 structure. The Estimated Coordinate Errors are 0.25 A for (R)-naproxen-mCOX-2 and 0.29 A for (R)-flurbiprofen-mCOX-2. Molecular graphics (Figs. 3 and 4) were illustrated with PyMOL 47. The coordinates are deposited at RCSB Protein Data Bank. ID codes are 3Q7D for (R)-naproxen-mCOX-2 and 3RR3 for (R)-flurbiprofen-mCOX-2.
DRG preparation
DRG culture and staining for neurons and glia was performed as described 48 using a protocol approved by the Vanderbilt Institutional Animal Care and Use Committee. Staining for COX-2 was performed as described 32. Treatment with inflammatory stimuli was performed as described in the text. Extraction and analysis of PGs, PG-Gs, and PG-EAs was performed as described 49.
Supplementary Material
Acknowledgments
This work was supported by research (CA89450, GM15431, NS064278) and training grants (DA022873, DA031572) from the National Institutes of Health. It is based upon research conducted at the Advanced Photon Source on the Northeastern Collaborative Access Team beamlines, which are supported by award RR-15301 from the National Center for Research Resources at the National Institutes of Health. Use of the Advanced Photon Source is supported by the U.S. Department of Energy, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. We are grateful to J. Harp for assistance with crystallography, K. Masuda, M. Brown, R. Stevens, and B. Cravatt for a sample of FAAH, A. Brash for a sample of 15-lipoxygenase and J. Uddin for a sample of fluorocoxib A.
Footnotes
Author Contributions
J.J.P., K.C.D. and L.J.M. originated the project. K.C.D. performed all in vitro WT COX-2 inhibition experiments. D.J.H. performed all in vitro mutant COX-2 inhibition experiments. K.C.D., J.M., and S.B. determined the COX-2-(R)-naproxen crystal structure and K.C.D. and S.B. determined the (R)-flurbiprofen crystal structure. Primary DRGs were harvested and cultured by J.L.S. and D.J.H. in the laboratory of B.D.C. D.J.H. designed and executed COX-2 inhibition experiments in DRGs as well as the imaging studies. D.J.H. and J.L.S. performed western blot analysis of DRGs. J.A.O. reviewed the data and offered critical commentary, L.J.M. oversaw the research and wrote the manuscript, which was reviewed and edited by all authors.
Competing Financial Interests
The authors declare no competing financial interests.
Additional Information
Supplementary information is available online at http://www.nature.com/naturechemicalbiology/. Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
References
- 1.Di Marzo V, De Petrocellis L, Bisogno T. The biosynthesis, fate and pharmacological properties of endocannabinoids. Handb Exp Pharmacol. 2005;168:147–185. doi: 10.1007/3-540-26573-2_5. [DOI] [PubMed] [Google Scholar]
- 2.Piomelli D. The molecular logic of endocannabinoid signalling. Nat Rev Neurosci. 2003;4:873–884. doi: 10.1038/nrn1247. [DOI] [PubMed] [Google Scholar]
- 3.Kogan NM, Mechoulam R. The chemistry of endocannabinoids. J Endocrinol Invest. 2006;29:3–14. [PubMed] [Google Scholar]
- 4.Ueda N, et al. Lipoxygenase-catalyzed oxygenation of arachidonylethanolamide, a cannabinoid receptor agonist. Biochim Biophys Acta. 1995;1254:127–134. doi: 10.1016/0005-2760(94)00170-4. [DOI] [PubMed] [Google Scholar]
- 5.Yu M, Ives D, Ramesha CS. Synthesis of prostaglandin E2 ethanolamide from anandamide by cyclooxygenase-2. J Biol Chem. 1997;272:21181–21186. doi: 10.1074/jbc.272.34.21181. [DOI] [PubMed] [Google Scholar]
- 6.Kozak KR, Rowlinson SW, Marnett LJ. Oxygenation of the endocannabinoid, 2-arachidonylglycerol, to glyceryl prostaglandins by cyclooxygenase-2. J Biol Chem. 2000;275:33744–33749. doi: 10.1074/jbc.M007088200. [DOI] [PubMed] [Google Scholar]
- 7.Snider NT, Walker VJ, Hollenberg PF. Oxidation of the endogenous cannabinoid arachidonoyl ethanolamide by the cytochrome P450 monooxygenases: physiological and pharmacological implications. Pharmacol Rev. 2010;62:136–154. doi: 10.1124/pr.109.001081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen JK, et al. Identification of novel endogenous cytochrome P450 arachidonate metabolites with high affinity for cannabinoid receptors. J Biol Chem. 2008;283:24514–24524. doi: 10.1074/jbc.M709873200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nirodi CS, Crews BC, Kozak KR, Morrow JD, Marnett LJ. The glyceryl ester of prostaglandin E2 mobilizes calcium and activates signal transduction in RAW264.7 cells. Proc Natl Acad Sci USA. 2004;101:1840–1845. doi: 10.1073/pnas.0303950101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sang N, Zhang J, Chen C. PGE2 glycerol ester, a COX-2 oxidative metabolite of 2-arachidonoyl glycerol, modulates inhibitory synaptic transmission in mouse hippocampal neurons. J Physiol. 2006;572:735–745. doi: 10.1113/jphysiol.2006.105569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sang N, Zhang J, Chen C. COX-2 oxidative metabolite of endocannabinoid 2-AG enhances excitatory glutamatergic synaptic transmission and induces neurotoxicity. J Neurochem. 2007;102:1966–1977. doi: 10.1111/j.1471-4159.2007.04668.x. [DOI] [PubMed] [Google Scholar]
- 12.Hu SS, Bradshaw HB, Chen JS, Tan B, Walker JM. Prostaglandin E2 glycerol ester, an endogenous COX-2 metabolite of 2-arachidonoylglycerol, induces hyperalgesia and modulates NFkappaB activity. Br J Pharmacol. 2008;153:1538–1549. doi: 10.1038/bjp.2008.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Richie-Jannetta R, et al. Structural determinants for calcium mobilization by prostaglandin E2 and prostaglandin F2α glyceryl esters in RAW 264.7 cells and H1819 cells. Prost Other Lip Med. 2010;92:19–24. doi: 10.1016/j.prostaglandins.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cravatt BF, et al. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- 15.Dinh TP, Kathuria S, Piomelli D. RNA interference suggests a primary role for monoacylglycerol lipase in the degradation of the endocannabinoid 2-arachidonoylglycerol. Mol Pharmacol. 2004;66:1260–1264. doi: 10.1124/mol.104.002071. [DOI] [PubMed] [Google Scholar]
- 16.Piomelli D, Giuffrida A, Calignano A, Rodriguez de Fonseca F. The endocannabinoid system as a target for therapeutic drugs. Trends Pharmacol Sci. 2000;21:218–224. doi: 10.1016/s0165-6147(00)01482-6. [DOI] [PubMed] [Google Scholar]
- 17.Cravatt BF, Lichtman AH. Fatty acid amide hydrolase: an emerging therapeutic target in the endocannabinoid system. Curr Opin Chem Biol. 2003;7:469–475. doi: 10.1016/s1367-5931(03)00079-6. [DOI] [PubMed] [Google Scholar]
- 18.Long JZ, et al. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat Chem Biol. 2009;5:37–44. doi: 10.1038/nchembio.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guay J, Bateman K, Gordon R, Mancini J, Riendeau D. Carrageenan-induced paw edema in rat elicits a predominant prostaglandin E2 (PGE2) response in the central nervous system associated with the induction of microsomal PGE2 synthase-1. J Biol Chem. 2004;279:24866–24872. doi: 10.1074/jbc.M403106200. [DOI] [PubMed] [Google Scholar]
- 20.Prusakiewicz JJ, Duggan KC, Rouzer CA, Marnett LJ. Differential sensitivity and mechanism of inhibition of COX-2 oxygenation of arachidonic acid and 2-arachidonoylglycerol by ibuprofen and mefenamic acid. Biochemistry. 2009;48:7353–7355. doi: 10.1021/bi900999z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rimon G, et al. Coxibs interfere with the action of aspirin by binding tightly to one monomer of cyclooxygenase-1. Proc Natl Acad Sci USA. 2010;107:28–33. doi: 10.1073/pnas.0909765106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kulmacz RJ, Lands WEM. Stoichiometry and kinetics of the interaction of prostaglandin H synthase with anti inflammatory agents. J Biol Chem. 1985;260:12572–12578. [PubMed] [Google Scholar]
- 23.Dong L, et al. Human cyclooxygenase-2 is a sequence homodimer that functions as a conformational heterodimer. J Biol Chem. 2011;286:19035–19046. doi: 10.1074/jbc.M111.231969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhattacharyya DK, Lecomte M, Rieke CJ, Garavito RM, Smith WL. Involvement of arginine 120, glutamate 524, and tyrosine 355 in the binding of arachidonate and 2-phenylpropionic acid inhibitors to the cyclooxygenase active site of ovine prostaglandin endoperoxide H synthase-1. J Biol Chem. 1996;271:2179–2184. doi: 10.1074/jbc.271.4.2179. [DOI] [PubMed] [Google Scholar]
- 25.Duggan KC, et al. Molecular basis for cyclooxygenase inhibition by the non-steroidal anti-inflammatory drug, naproxen. J Biol Chem. 2010;285:34950–34959. doi: 10.1074/jbc.M110.162982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Selinsky BS, Gupta K, Sharkey CT, Loll PJ. Structural analysis of NSAID binding by prostaglandin H2 synthase: time-dependent and time-independent inhibitors elicit identical enzyme conformations. Biochemistry. 2001;40:5172–5180. doi: 10.1021/bi010045s. [DOI] [PubMed] [Google Scholar]
- 27.Kurumbail RG, et al. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature. 1996;384:644–648. doi: 10.1038/384644a0. [DOI] [PubMed] [Google Scholar]
- 28.Loetsch J, Geisslinger G, Mohammadian P, Brune K, Kobal G. Effects of flurbiprofen enantiomers on pain-related chemo-somatosensory evoked potentials in human subjects. Brit J Clin Pharmacol. 1995;40:339–346. doi: 10.1111/j.1365-2125.1995.tb04556.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bishay P, Schmidt H, Marian C, Haussler A, Wijnvoord N, Ziebell S, Metzner J, Koch M, Myrczek T, Bechmann I. R-Flurbiprofen Reduces Neuropathic Pain in Rodents by Restoring Endogenous Cannabinoids. PLoS One. 2010;5:e10628. doi: 10.1371/journal.pone.0010628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jamali F, Berry BW, Tehrani MR, Russell AS. Stereoselective pharmacokinetics of flurbiprofen in humans and rats. J Pharm Sci. 1988;77:666–669. doi: 10.1002/jps.2600770805. [DOI] [PubMed] [Google Scholar]
- 31.Brune K, Geisslinger G, Menzel-Soglowek S. Pure enantiomers of 2-arylpropionic acids: tools in pain research and improved drugs in rheumatology. J Clin Pharmacol. 1992;32:944–952. doi: 10.1002/j.1552-4604.1992.tb04643.x. [DOI] [PubMed] [Google Scholar]
- 32.Uddin MJ, et al. Selective visualization of cyclooxygenase-2 in inflammation and cancer by targeted fluorescent imaging agents. Cancer Res. 2010;70:3618–3627. doi: 10.1158/0008-5472.CAN-09-2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rouzer CA, Marnett LJ. Glycerylprostaglandin synthesis by resident peritoneal macrophages in response to a zymosan stimulus. J Biol Chem. 2005;280:26690–26700. doi: 10.1074/jbc.M501021200. [DOI] [PubMed] [Google Scholar]
- 34.Yang H, Chen C. Cyclooxygenase-2 in synaptic signaling. Curr Pharm Des. 2008;14:1443–1451. doi: 10.2174/138161208784480144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Woodward DF, et al. The pharmacology and therapeutic relevance of endocannabinoid derived cyclo-oxygenase (COX)-2 products. Pharmacol Ther. 2008;120:71–80. doi: 10.1016/j.pharmthera.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 36.Liang Y, Woodward DF, Guzman VM. Identification and pharmacological characterization of the prostaglandin FP receptor and FP receptor variant complexes. Brit J Pharmacol. 2008;154:1079–1093. doi: 10.1038/bjp.2008.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Di Marzo V. The endocannabinoid system: its general strategy of action, tools for its pharmacological manipulation and potential therapeutic exploitation. Pharmacol Res. 2009;60:77–84. doi: 10.1016/j.phrs.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 38.Blankman JL, Simon GM, Cravatt BF. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem Biol. 2007;14:1347–1356. doi: 10.1016/j.chembiol.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xie S, et al. Inactivation of Lipid Glyceryl Ester Metabolism in Human THP1 Monocytes/Macrophages by Activated Organophosphorus Insecticides: Role of Carboxylesterases 1 and 2. Chem Res Toxicol. 2010;23:1890–1904. doi: 10.1021/tx1002194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Glaser ST, Kaczocha M. Cyclooxygenase-2 mediates anandamide metabolism in the mouse brain. J Pharmacol Exp Ther. 2010;335:380–388. doi: 10.1124/jpet.110.168831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rowlinson SW, Crews BC, Lanzo CA, Marnett LJ. The binding of arachidonic acid in the cyclooxygenase active site of mouse prostaglandin endoperoxide synthase-2 (COX-2): a putative L-shaped binding conformation utilizing the top channel region. J Biol Chem. 1999;274:23305–23310. doi: 10.1074/jbc.274.33.23305. [DOI] [PubMed] [Google Scholar]
- 42.Kalgutkar AS, Kozak KR, Crews BC, Hochgesang J, GP, Marnett LJ. Covalent modification of cyclooxygenase-2 (COX-2) by 2-(acetoxyphenyl)alkyl sulfides, a new class of selective COX-2 inactivators. J Med Chem. 1998;41:4800–4818. doi: 10.1021/jm980303s. [DOI] [PubMed] [Google Scholar]
- 43.Otwinowski Z, Minor W. Methods Enzymol. 1997;276A:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 44.Vagin A, Teplyakov A. An approach to multi-copy search in molecular replacement. Acta Crystallogr D Biol Crystallogr. 2000;56:1622–1624. doi: 10.1107/s0907444900013780. [DOI] [PubMed] [Google Scholar]
- 45.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystall D: Biol Crystall. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 46.Adams PD, et al. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 47.DeLano WL. DeLano Scientific LLC. 2009. [Google Scholar]
- 48.Wu HH, et al. Glial precursors clear sensory neuron corpses during development via Jedi-1, an engulfment receptor. Nat Neurosci. 2009;12:1534–1541. doi: 10.1038/nn.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kingsley PJ, Marnett LJ. LC-MS-MS analysis of neutral eicosanoids. Methods Enzymol. 2007;433:91–112. doi: 10.1016/S0076-6879(07)33005-X. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.