

Abstract
An improved HILIC (hydrophilic interaction liquid chromatography) method has been developed to separate members of a closely related family of chemoprotective phytochemicals called glucosinolates. This method exploits the emergence of a second generation of HILIC chemistry, using a silica-based permanently zwitterionic stationary phase. These columns are more robust, durable, and glucosinolates separations are more reproducible than with the original polyhydroxyethyl aspartamide columns. Furthermore, the HILIC system that we report herein permits much greater alteration of the mobile phase composition for customized separation of glucosinolates from plant extracts, across a wide spectrum of polarity.
Keywords: glucoraphanin, broccoli, glucoiberin, chemoprotection, phytochemical, HPLC, sulforaphane glucosinolate
1. Introduction
Glucosinolates are plant secondary products, or phytochemicals, that are found in cruciferous vegetables and other related edible plants [1–5]. They are the precursors of cancer protective isothiocyanates. A variety of methods have been developed for separating glucosinolates, based upon the wide range of side chains (more than 120 of them have been identified) attached to a thioglucoside hydroxysulfate moiety (Fig 1). Early HPLC-based identification utilized the methodology of Wathelet and others whereby glucosinolates are enzymatically desulfated and the resulting desulfo-glucosinolates are separated on a C18 column [6]. This method has long been used for detection of rapeseed-specific glucosinolates [7], but it suffers from the fact that glucosinolates separated in this manner are no longer biologically active; nor can they be converted to isothiocyanates or other biologically active compounds. Thus, the bioassay of low levels of glucosinolate or the scale-up of separation methodology to yield commercially useful reagents is not possible. Unequivocal identification of glucosinolates with current chromatographic techniques is best accomplished by a combination of isocratic paired-ion (IPC) or gradient HPLC, with photodiode array (UV) detection and mass spectroscopic confirmation. A variety of such methods have been published [8–11], thus enabling investigators to identify and quantify known glucosinolates and to discover new glucosinolates in plant extracts.
Figure 1.
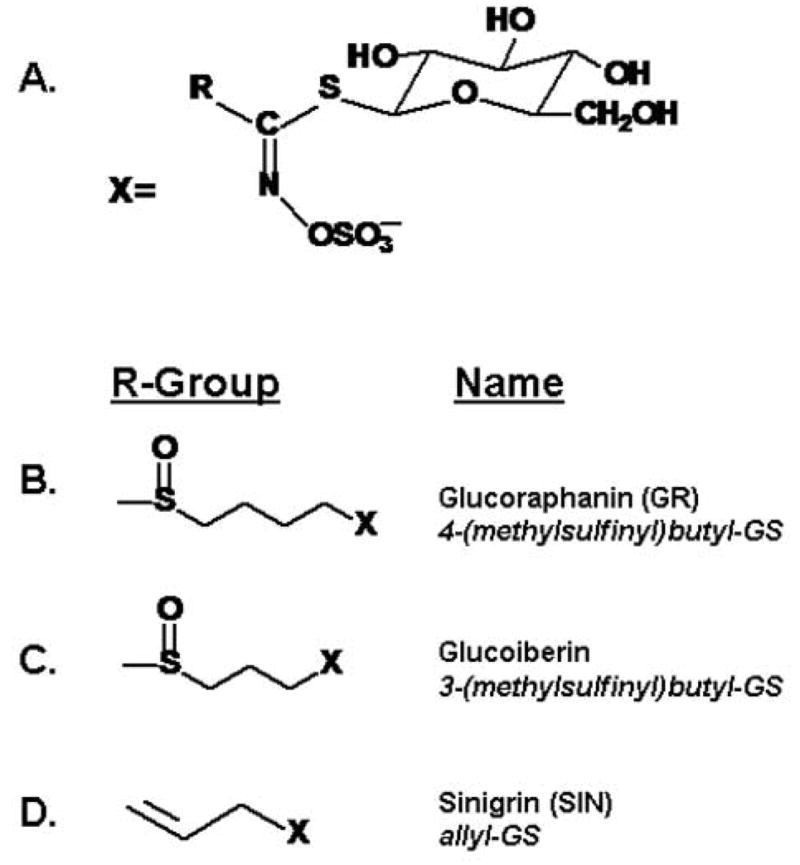
A. General structure of glucosinolates (GS; β-thioglucoside N-hydroxysulfates); Side chains, R, can be aliphatic, sulfur-containing (thioalkyl), alcoholic, aromatic, indolyl, or additionally glycosylated. Standards used in this paper are show in (B), (C), and (D).
However, there are drawbacks to this approach when endeavoring to measure levels of the small number of such compounds present in any single plant species or closely related group of edible vegetables. The paired-ion approach is not optimal because the salts we use as ion-pairing agents to separate glucosinolates cannot be used directly for mass spectroscopy [9]. Desalting is time-consuming and inefficient at the analytical scale. Gradient HPLC is far more effective in separating large numbers of glucosinolates, one from another, but the gradients must be lengthy (typically ranging up to an hour per injection) [11] and are thus not conducive to rapid screening. However, the least attractive feature of these techniques developed for glucosinolates [9–11] is the fact that, contrary to HILIC, they elute with non-target compounds. Thus to overcome the non-selective nature of the C18 chemistry, mass spectroscopy may be necessary for detection. In addition, retention times can shift substantially due to small changes in column temperature and to anomalies in solvent mixing. Thus the use of this method is best restricted to those with the most modern HPLC equipment and should be coupled with solid phase extraction sample preparation.
We therefore developed a method for HILIC separation of glucosinolates [12] that would bypass some of the issues raised herein. We have used this method to identify and quantify glucosinolates in broccoli (Brassica oleracea var. italica), as well as in other plants [13–20]. The HILIC columns utilized (polyhydroxyethyl aspartamide) have short life-spans (only a few hundred injections) compared to C18 columns, and the mobile phase required to effect such separation does not well tolerate slight changes in composition to resolve certain pairs or groups of co-eluting glucosinolates over others. Its effective range of acetonitrile content is only 82.5% to 87.5% (with 30 mM ammonium formate, pH 5.4). An alternative method developed subsequently, utilizes a much more robust, hydrophilic, C18 column and an ammonium acetate-methanol gradient [11,21], but it has disadvantages characteristic of other gradient-based approaches and it is highly sensitive to injection solvent. This limits acceptable extracts to either aqueous, or highly diluted solvent extracts.
For development and scale-up of a novel (counter-current chromatographic) method for purifying GR (glucoraphanin) from broccoli seeds [14,22], we used HILIC to evaluate the degree of separation of GR from its most persistent “contaminant”, glucoriberin (GI). Since these separations were being performed both in Europe and in the USA, we evaluated alternative HILIC column chemistries (that were readily available from companies with worldwide distribution networks), as well as altering HPLC variables such as pH, organic acids and water activity. We ultimately developed a method which is rapid and reproducible and can be performed on the most basic of HPLC equipment (e.g. single wavelength detector, no gradient mixing, no MS detection). Critically, it is also more flexible in that it utilizes a column chemistry that permits much greater alteration of the mobile phase composition for customized separation of glucosinolates of interest across a wide spectrum of polarity.
2. Experimental
Methodology was developed using a 150 × 4.6 mm, 5 μm particle size, 200 Å pore size column with a silica support and polymeric sulfoalkylbetaine zwitterionic functional groups (ZIC-HILIC; Sequant, Umeå, Sweden). A suggested set of initial conditions is: mobile phase - isocratic 15 mM ammonium formate pH 4.5 in 70:30 (v:v) acetonitrile:water, 0.5 ml/min flow rate, and column temperature of 25°C. Detection was by photo diode array (primary monitoring at 235 nm), but is also suitable for direct mass spectroscopy. All solvents (Fisher Scientific, Fairlawn, NJ; Sigma-Aldrich,, St. Louis, MO, USA; and J. T. Baker, Phillipsburg, NJ, USA) were ACS or HPLC grade. All HPLC components were from Waters (Milford, MA, USA) (e.g. Model 616 pumps; Model 717 Plus autosampler; Model 2695 Alliance System; Model 2996 PDA detectors; Empower software). GR and GI were prepared by counter-current chromatography as described in [14] and sinigrin (SIN) was purchased from Sigma/Aldrich. HPLC columns used for comparison were: (a) polyhydroxyethyl aspartamide (100 × 4.6 mm, 3 μm particle diameter, 100 Å pore diameter) made by PolyLC, Columbia, MD, USA with mobile phase of isocratic 30 mM ammonium formate pH 5.4 in 85:15 (v:v) acetonitrile:water at 2 ml/min [12]; and (b) SunFire C18 (250 × 4.6 mm, 5 μm particle diameter, 100 Å pore diameter), made by Waters, with mobile phase step gradient from 5% to 100% methanol, with 0.1% glacial acetic acid, at 1 ml/min [11].
3. Results and Discussion
Technique validation was performed using a range of plant extracts (data not shown) and using analytical standards of three glucosinolates -- GR, GI, and SIN. Standard curves for three glucosinolates show excellent linearity over at least a three log range (81 pmol to 72 nmol) with the sulfoalkylbetaine zwitterionic HILIC system. Standard curves (n = 15 for each glucosinolate) run on three different days spanning a three week period yielded peak areas and peak heights that were strictly linear and highly reproducible (r2 > 0.999). There was no significant effect of replication date (run date) on peak area (ANOVA of peak area by concentration, p>0.05).
Retention time (tR) was highly reproducible for the three glucosinolates tested. Relative standard deviations (RSD) for SIN, GR, and GI respectively, were 0.5%, 1.7%, and 2.0% between runs and 1.1%, 0.6%, and 0.6% within runs, using 15 mM ammonium formate pH 4.5, in 70:30 (v:v) acetonitrile:water, at a flow rate of 0.5 ml/min, giving evidence to the robustness of the chromatographic system. This is only noteworthy because many other solvent systems tested had very poor tR reproducibility. For example, the RSD for single day runs using a SIN standard were as high as 20% using a C18 gradient method [11] and 1.3% using a polyhydroxy aspartamide method [12], however with the latter method the tR of GR ranged from 12.5 min to 14.5 min. Such inconsistency can be explained by changes in column temperature in situations where a column oven is not used, but in this case it is most likely a reflection of the very steep gradient of solvent effects with this system -- the effective range of acetonitrile content in the mobile phase is extremely narrow (ranging from 82.5% to 87.5%, at pH 5.4, with 30 mM ammonium formate).
This basic protocol can be varied readily by modifying either salt molarity or organic solvent strength to optimize retention of target glucosinolates. For example, at a column flow rate of 0.5 ml/min, tR shifted with increasing concentrations of acetonitrile in the mobile phase. This is exemplified in Fig. 2, which contrasts the separation of two glucosinolates (GI and GR) differing only by the presence of an additional methylene moiety in the side chain of GR (Fig. 1). Increasing acetonitrile in the mobile phase increases retention of hydrophilic compounds (as is typical of HILIC chromatography), permitting enhanced resolution of closely eluting compounds with longer retention times [23]. Using a mobile phase containing 70% acetonitrile, resolution (Rs) of GR and GI was good (Rs = 1.21). Increasing the acetonitrile concentration to 75% yields better separation (Rs = 1.58). A further increase to 80% acetonitrile yields peaks that are very well separated (Rs = 2.20) but require more than twice the elution time needed with 70% acetonitrile. Elution times for GR and GI can thus be reduced by increasing the flow rate of mobile phase containing 80% acetonitrile to 1 ml/min (Rs = 1.92) (back pressure at this flow rate was still very low [4–500 psi), but resolving power may suffer with other glucosinolate pairs. A similar strategy was employed for the separation of other polar compounds (data not presented herein).
Figure 2.
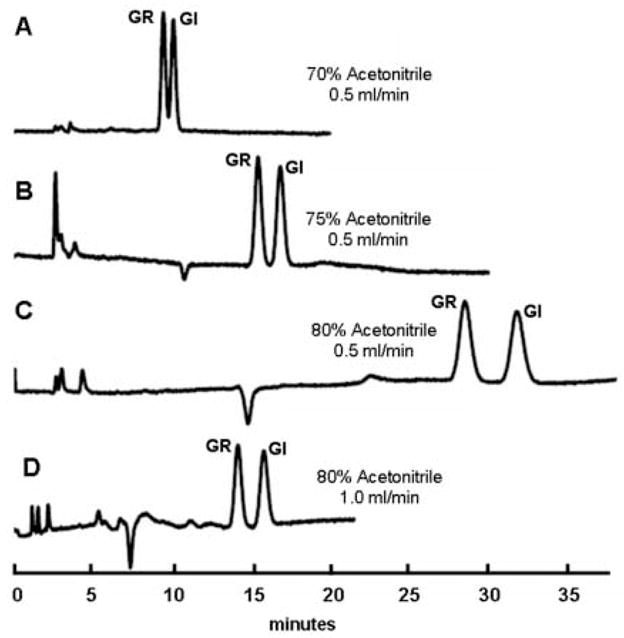
Shift in glucosinolate retention time with acetonitrile content. Equimolar quantities of glucoraphanin (GR) and glucoiberin (GI) were injected on a ZIC-HILIC column as described in the text.
The many polyhydroxyethyl aspartamide columns that we have used begin to degrade (peak geometry becomes unacceptable) after as few as 100 injections (in agreement with other reports [24]). Routine separations of GR and GI on these columns, using the most favorable conditions (85% acetonitrile, 30 mM ammonium formate, pH 5.4), only provide an Rs = 0.90. Additionally, these separations can only be achieved within a very narrow range of solvent, salt, and pH conditions [12]. Attempts to modify that system in order to accommodate more challenging glucosinolate separations than we initially reported led us to evaluate second generation HILIC columns with permanently zwitterionic stationary phases composed of sulfoalkylbetaine functional groups. Demanding separations can be more easily accommodated in this system by simple adjustment of ionic strength and pH. It is more tolerant of changes in mobile phase solvent concentration and it offers similar retention times at a lower organic solvent content, thus increasing salt solubility. The system is much more robust and flexible than previous methods, giving better separation of a greater range of glucosinolates, with much longer column life. With our oldest sulfoalkylbetaine zwitterionic column we have already made well over 500 injections with no loss in peak geometry, and others report successfully using them for as many as 3000 injections [25].
Acknowledgments
The authors thank Katherine Stephenson for technical assistance, and Paul Talalay, Derek Fisher, and Ian Sutherland for intellectual support and encouragement on this project. JWF and KLW were supported by USPHS grant CA 93780-04, and some of the HPLC equipment utilized in this report was purchased using BBSRC Research Equipment Initiative grant BB/D524583/1.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fahey JW, Zalcmann AT, Talalay P. Phytochemistry. 2001;56:5. doi: 10.1016/s0031-9422(00)00316-2. [DOI] [PubMed] [Google Scholar]
- 2.Grubb CD, Abel S. Trends Plant Sci. 2006;11:89. doi: 10.1016/j.tplants.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 3.Halkier BA, Gershenzon J. Annu Rev Plant Biol. 2006;57:303. doi: 10.1146/annurev.arplant.57.032905.105228. [DOI] [PubMed] [Google Scholar]
- 4.Holst B, Williamson G. Nat Prod Rep. 2004;21:425. doi: 10.1039/b204039p. [DOI] [PubMed] [Google Scholar]
- 5.Rosa EAS, Heaney RK, Fenwick GR, Portas CAM. Hortic Rev. 1997;19:99. [Google Scholar]
- 6.Wathelet JP. World Crops: Production, Utilization and Description. Kluwer; Dordrecht: 2002. Glucosinolates in Rapeseeds: Analytical Aspects. [Google Scholar]
- 7.ISO 9167-1. Rapeseed -- Determination of glucosinolates content. Part 1. International Organization for Standardization (ISO), Geneva, 1992, p. 8.
- 8.Betz JM, Fox WD. ACS symposium series. 546:181. [Google Scholar]
- 9.Prestera T, Fahey JW, Holtzclaw WD, Abeygunawardana C, Kachinski JL, Talalay P. Anal Biochem. 1996;239:168. doi: 10.1006/abio.1996.0312. [DOI] [PubMed] [Google Scholar]
- 10.Rangkadilok N, Nicolas ME, Bennett RN, Premier RR, Eagling DR, Taylor PWJ. Sci Hortic. 2002;96:27. [Google Scholar]
- 11.West L, Tsui I, Haas G. J Chromatogr A. 2002;966:227. doi: 10.1016/s0021-9673(02)00734-3. [DOI] [PubMed] [Google Scholar]
- 12.Troyer JK, Stephenson KK, Fahey JW. J Chromatogr A. 2001;919:299. doi: 10.1016/s0021-9673(01)00842-1. [DOI] [PubMed] [Google Scholar]
- 13.Dinkova-Kostova AT, Jenkins SN, Fahey JW, Ye L, Wehage SL, Liby KT, Stephenson KK, Wade KL, Talalay P. Cancer Lett. 2006;240:243. doi: 10.1016/j.canlet.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 14.Fahey JW, Wade KL, Stephenson KK, Chou FE. J Chromatogr A. 2003;996:85. doi: 10.1016/s0021-9673(03)00607-1. [DOI] [PubMed] [Google Scholar]
- 15.Farnham MW, Stephenson KK, Fahey JW. HortScience. 2005;40:50. [Google Scholar]
- 16.Farnham MW, Wilson PE, Stephenson KK, Fahey JW. Plant Breed. 2004;123:60. [Google Scholar]
- 17.Kensler TW, Chen JG, Egner PA, Fahey JW, Jacobson LP, Stephenson KK, Ye L, Coady JL, Wang JB, Wu Y, Sun Y, Zhang QN, Zhang BC, Zhu YR, Qian GS, Carmella SG, Hecht SS, Benning L, Gange SJ, Groopman JD, Talalay P. Cancer Epidemiol Biomarkers Prev. 2005;14:2605. doi: 10.1158/1055-9965.EPI-05-0368. [DOI] [PubMed] [Google Scholar]
- 18.Pereira FM, Rosa E, Fahey JW, Stephenson KK, Carvalho R, Aires A. J Agric Food Chem. 2002;50:6239. doi: 10.1021/jf020309x. [DOI] [PubMed] [Google Scholar]
- 19.Tang L, Zhang Y, Jobson HE, Li J, Stephenson KK, Wade KL, Fahey JW. Mol Cancer Ther. 2006;5:935. doi: 10.1158/1535-7163.MCT-05-0476. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y, Munday R, Jobson HE, Munday CM, Lister C, Wilson P, Fahey JW, Mhawech-Fauceglia P. J Agric Food Chem. 2006;54:9370. doi: 10.1021/jf062109h. [DOI] [PubMed] [Google Scholar]
- 21.West LG, Meyer KA, Balch BA, Rossi FJ, Schultz MR, Haas GW. J Agric Food Chem. 2004;52:916. doi: 10.1021/jf0307189. [DOI] [PubMed] [Google Scholar]
- 22.Fisher D, Garrard IJ, Van den Heuvel R, Sutherland IA, Chou FE, Fahey JW. J Liq Chromatogr Relat Technol. 2005;28:1913. [Google Scholar]
- 23.Felinger A. Anal Chem. 1997;69:2976. doi: 10.1021/ac970241y. [DOI] [PubMed] [Google Scholar]
- 24.Hemstrom P, Irgum K. J Sep Sci. 2006;29:1784. doi: 10.1002/jssc.200600199. [DOI] [PubMed] [Google Scholar]
- 25.Bengtsson J, Jansson B, Hammarlund-Udenaes M. Rap Commun Mass Spectrom. 2005;19:2116. doi: 10.1002/rcm.2035. [DOI] [PubMed] [Google Scholar]